Quantitative Changes in the Transcription of Phytohormone-Related Genes: Some Transcription Factors Are Major Causes of the Wheat Mutant dmc Not Tillering


Abstract
:1. Introduction
2. Results
2.1. Morphology of the dmc Mutant
2.2. Genetic Diversity between the WT and Mutant dmc
2.3. RNA Sequencing Data
2.4. Annotation and Functional Classification of the Unigenes
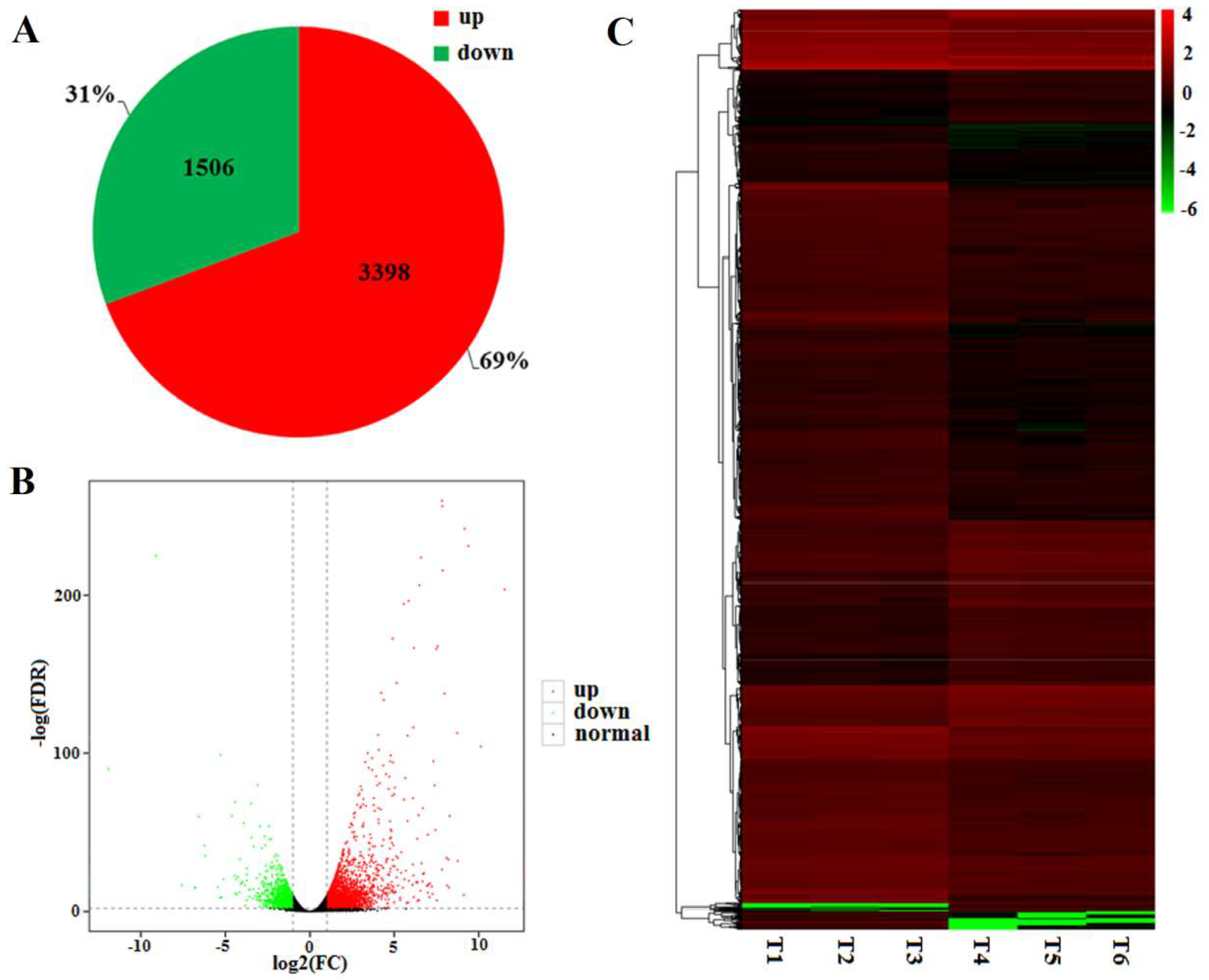
2.5. DEGs between the WT and Mutant dmc
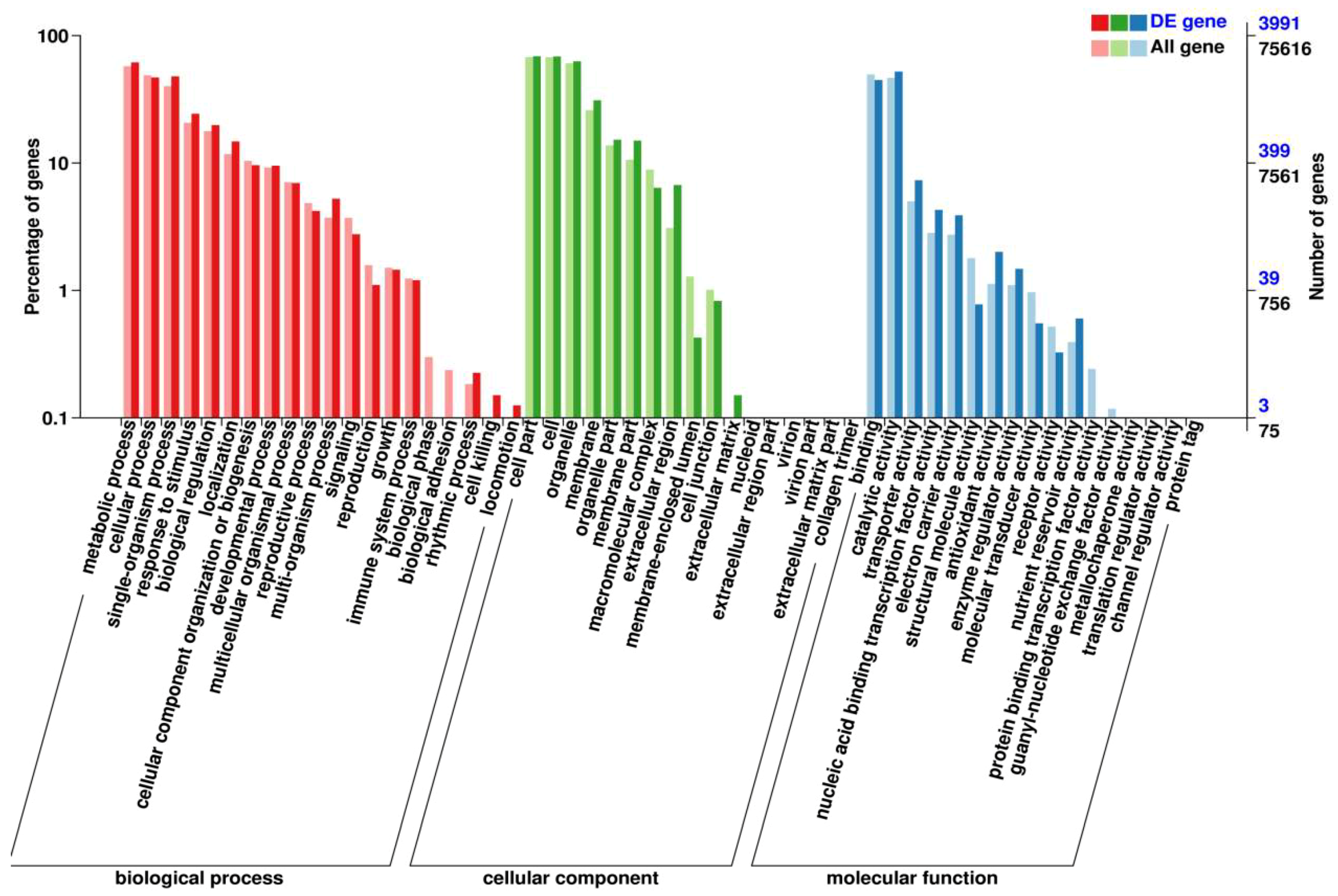
2.6. Functional Classification of the DEGs in GO
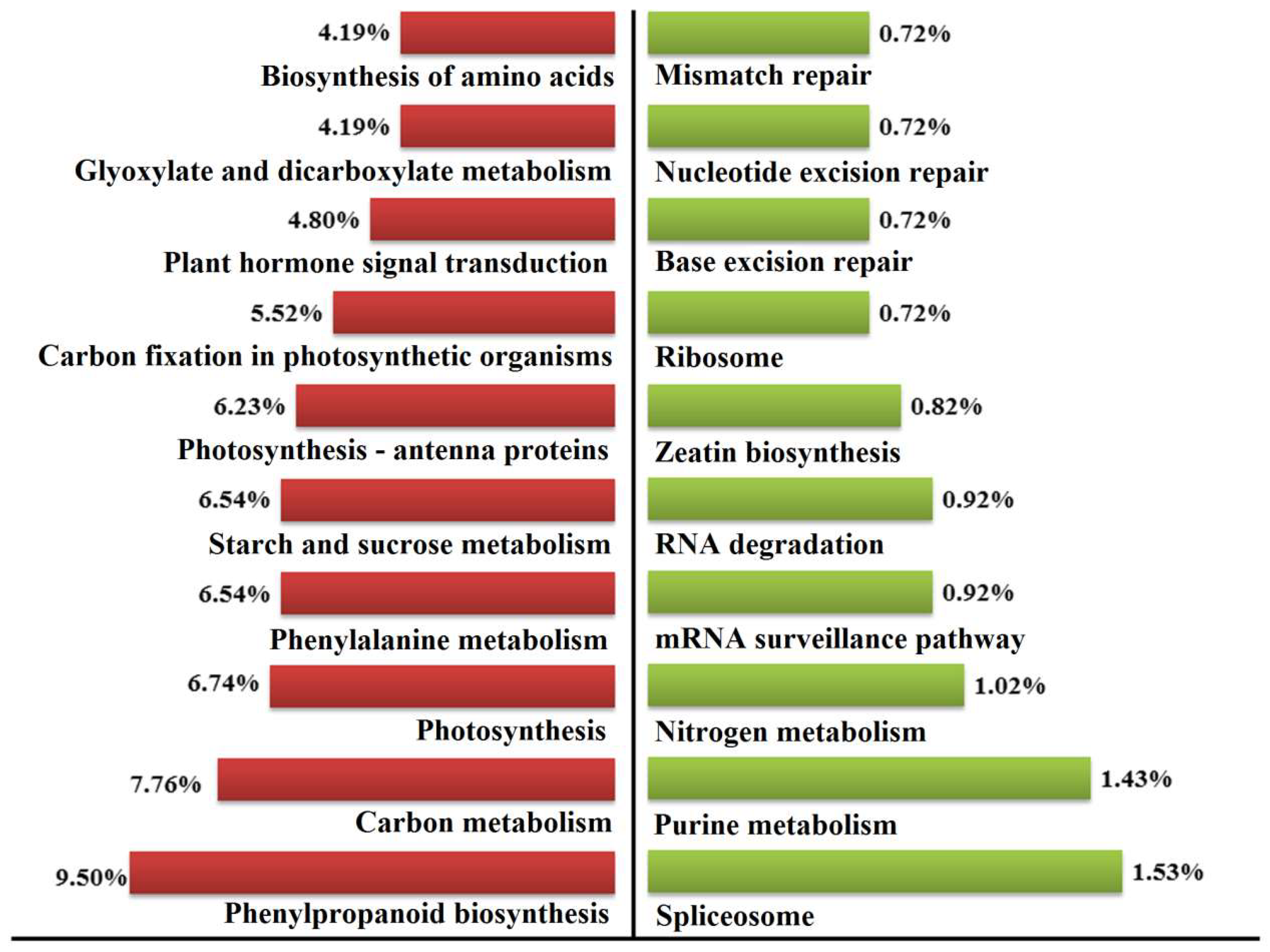
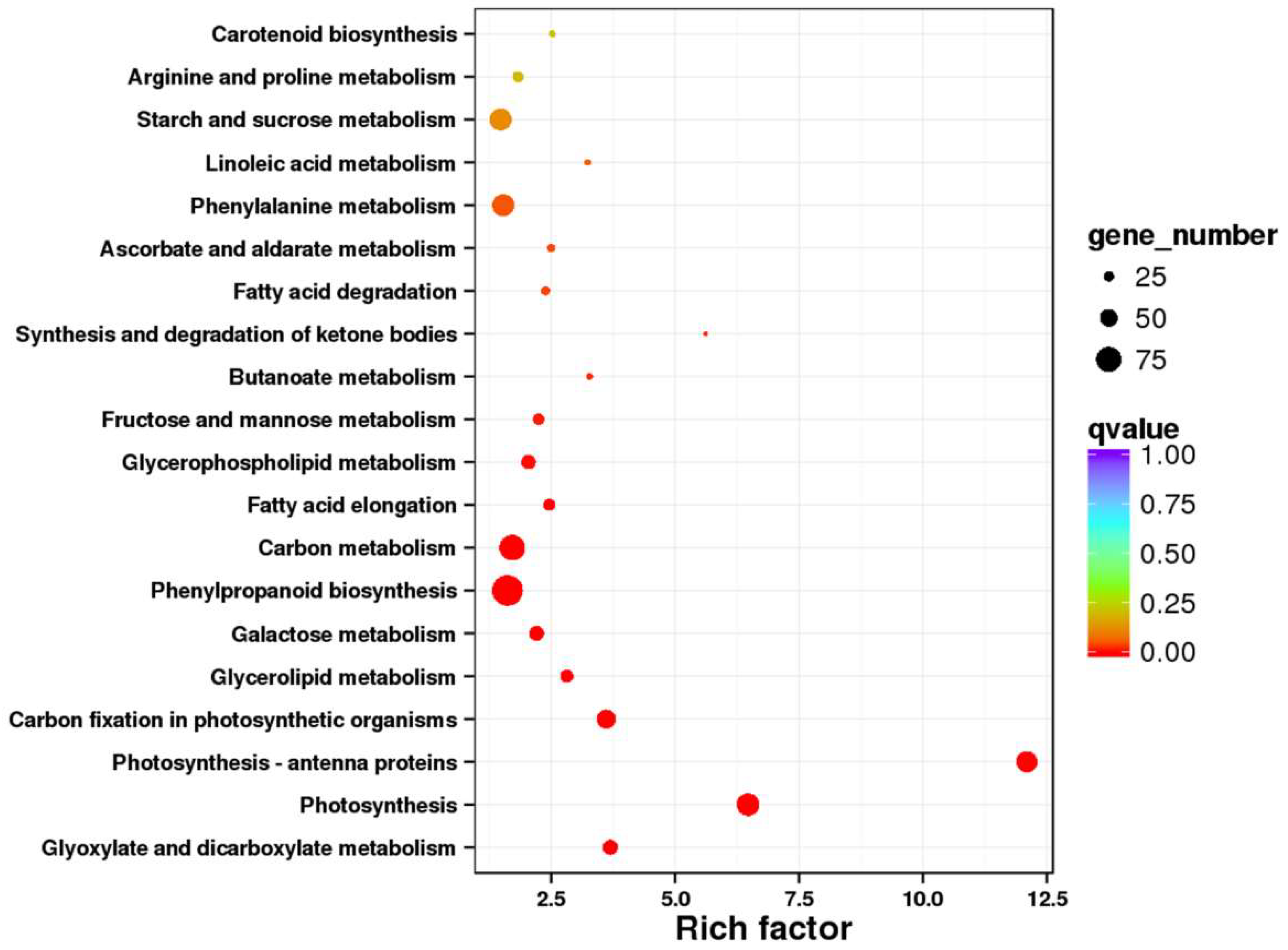
2.7. Pathway Mapping of the DEGs in KEGG
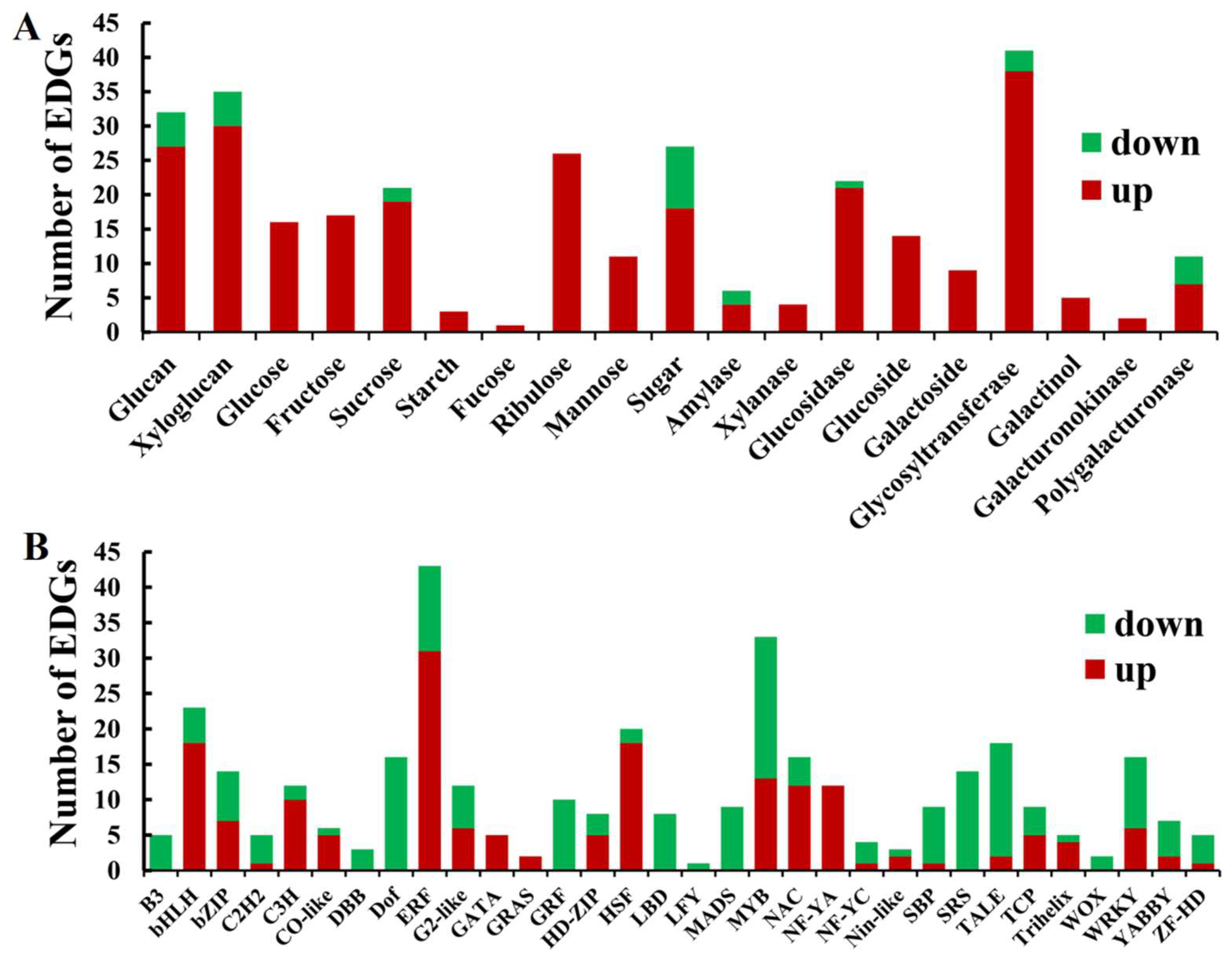
2.8. The DEGs Involved in Phytohormone Metabolisms in dmc
2.9. The DEGs Related to Carbohydrate Metabolism in the Mutant dmc
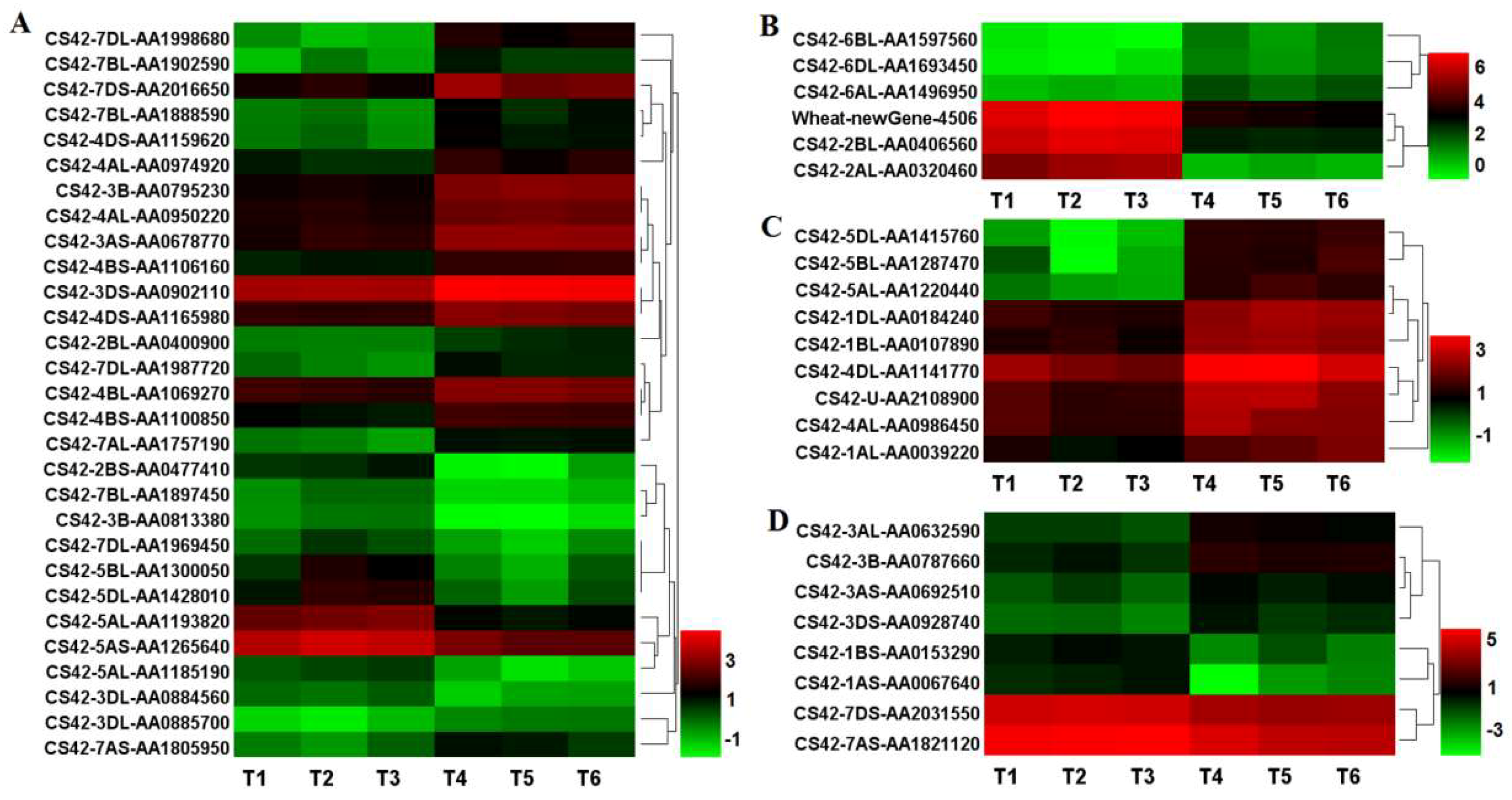
2.10. Transcription Factor Type DEGs in the Mutant dmc
2.11. The DEGs Related to Signaling Processes
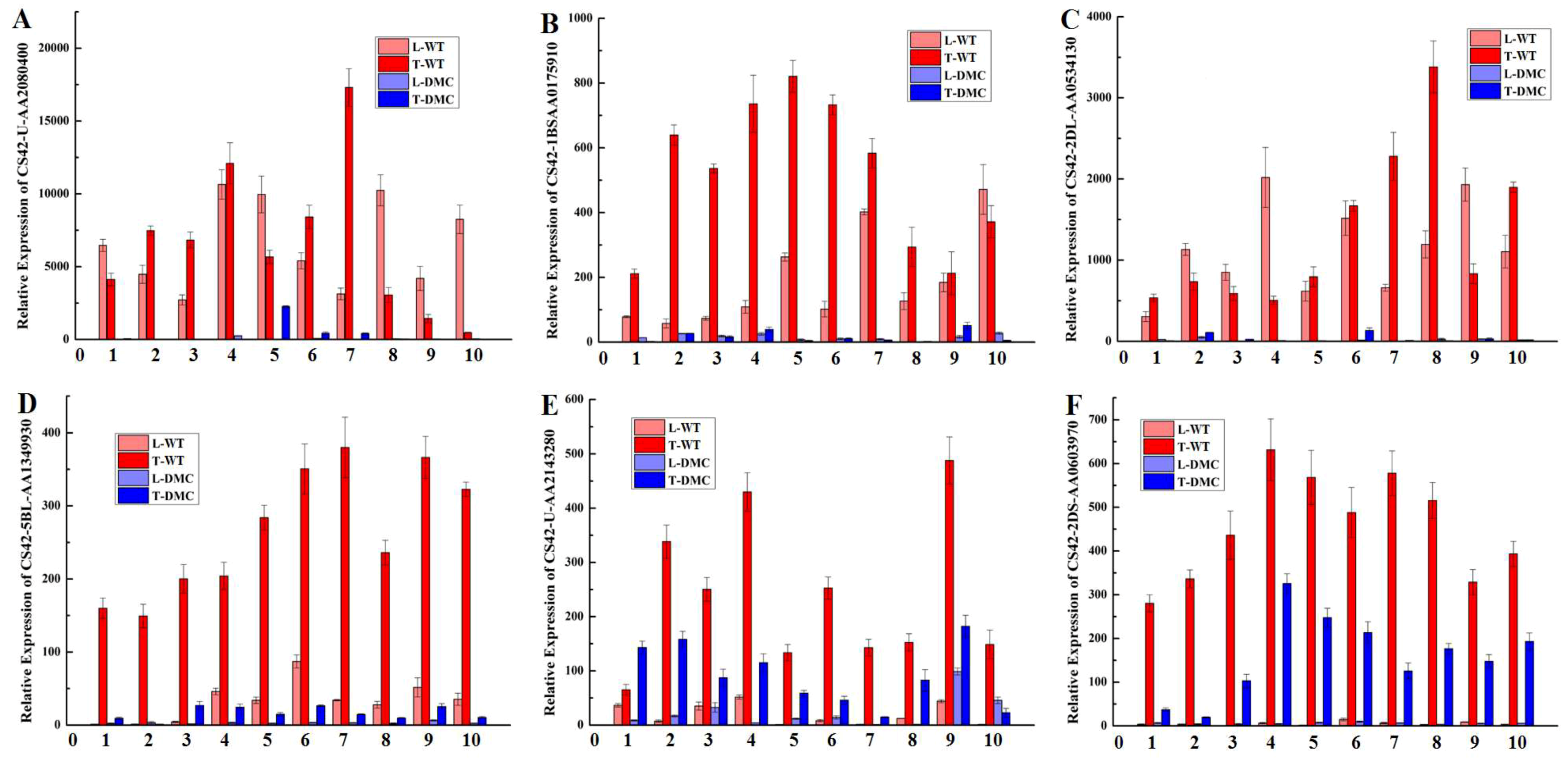
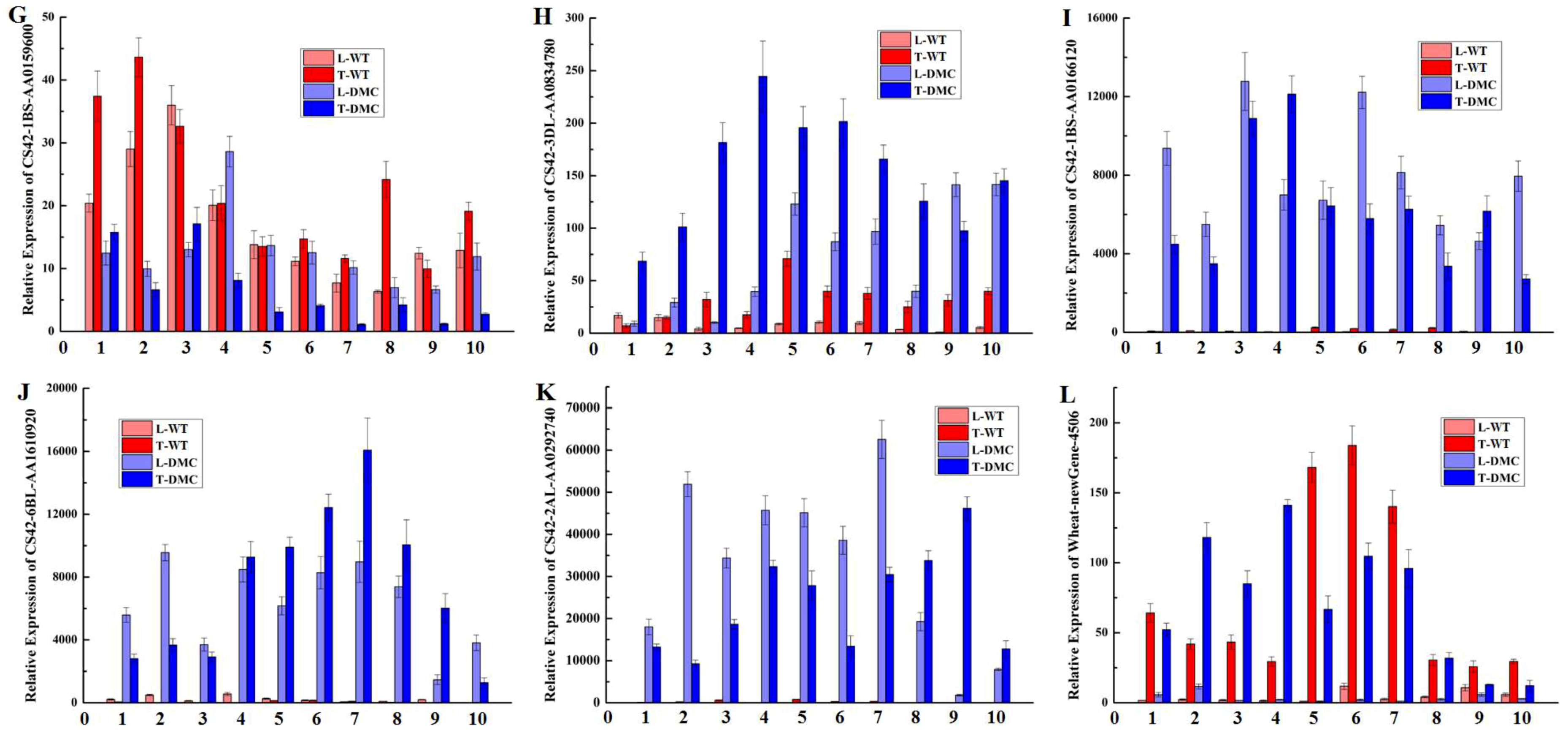
2.12. Expression Profiles of Twelve Genes in Wheat Tiller Primordia
3. Discussion
3.1. Lack of Vitality in the Mutant dmc
3.2. Auxin and Cytokinin Metabolisms Were Suppressed in dmc
3.3. Carbohydrate Metabolism and Phenylpropanoid Biosynthesis Were Active in dmc
3.4. Some Transcription Factors Are Suppressed in dmc
3.5. Signal Transduction and Photosynthesis in dmc
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Trait Measurements, Morphology Observations and SSR Analysis
4.3. RNA Extraction, Library Preparation and Sequencing
4.4. Transcriptome Analyses
4.5. qRT-PCR
4.6. Ethical Approval
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kuraparthy, V.; Sood, S.; Dhaliwal, H.S.; Chhuneja, P.; Gill, B.S. Identification and mapping of a tiller inhibition gene (tin3) in wheat. Theor. Appl. Genet. 2007, 114, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Naruoka, Y.; Talbert, L.E.; Lanning, S.P.; Blake, N.K.; Martin, J.M.; Sherman, J.D. Identification of quantitative trait loci for productive tiller number and its relationship to agronomic traits in spring wheat. Theor. Appl. Genet. 2011, 123, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Spielmeyer, W.; Richards, R.A. Comparative mapping of wheat chromosome 1AS which contains the tiller inhibition gene (tin) with rice chromosome 5S. Theor. Appl. Genet. 2004, 109, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, J.; Liu, W.; Lu, X.; Yang, X.; Gao, A.; Li, X.; Lu, Y.; Li, L. Genetic mapping of a fertile tiller inhibition gene, ftin, in wheat. Mol. Breed. 2013, 31, 441–449. [Google Scholar] [CrossRef]
- Peng, Z.S.; Yen, C.; Yang, J.L. Genetic control of oligo-culms character in common wheat. Wheat Inf. Serv. 1998, 86, 19–24. [Google Scholar]
- Xu, T.; Bian, N.; Wen, M.; Xiao, J.; Yuan, C.; Cao, A.; Zhang, S.; Wang, X.; Wang, H. Characterization of a common wheat (Triticum aestivum L.) high-tillering dwarf mutant. Theor. Appl. Genet. 2017, 130, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Dabbert, T.; Okagaki, R.J.; Cho, S.; Heinen, S.; Boddu, J.; Muehlbauer, G.J. The genetics of barley low-tillering mutants: Low number of tillers-1 (lnt1). Theor. Appl. Genet. 2010, 121, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Dabbert, T.; Okagaki, R.J.; Cho, S.; Boddu, J.; Muehlbauer, G.J. The genetics of barley low-tillering mutants: Absent lower laterals (als). Theor. Appl. Genet. 2009, 118, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Babb, S.; Muehlbauer, G.J. Genetic and morphological characterization of the barley uniculm2 (cul2) mutant. Theor. Appl. Genet. 2003, 106, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Guo, L.B.; Xue, D.W.; Zeng, D.L.; Zhang, G.H.; Dong, G.J.; Gu, M.H.; Qian, Q. Genetic analysis and gene-mapping of two reduced-culm-number mutants in rice. J. Integr. Plant Biol. 2006, 48, 341–347. [Google Scholar] [CrossRef]
- Fujita, D.; Ebron, L.A.; Araki, E.; Kato, H.; Khush, G.S.; Sheehy, J.E.; Lafarge, T.; Fukuta, Y.; Kobayashi, N. Fine mapping of a gene for low-tiller number, Ltn, in japonica rice (Oryza sativa L.) variety Aikawa 1. Theor. Appl. Genet. 2010, 120, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, J.; Li, Z.; Gu, F.; Zhang, H.; Tang, S.; Gu, M.; Liang, G. Fine mapping and cloning of MT1, a novel allele of D10. Prog. Nat. Sci. 2009, 19, 1683–1689. [Google Scholar] [CrossRef]
- Shah, M.M.; Gill, K.S.; Baenziger, P.S.; Yen, Y.; Kaeppler, S.M.; Ariyarathne, H.M. Molecular mapping of loci for agronomic traits on chromosome 3A of bread wheat. Crop Sci. 1999, 39, 1728–1732. [Google Scholar] [CrossRef]
- Kato, K.; Miura, H.; Sawada, S. Mapping QTLs controlling grain yield and its components on chromosome 5A of wheat. Theor. Appl. Genet. 2000, 101, 1114–1121. [Google Scholar] [CrossRef]
- Li, W.L.; Nelson, J.C.; Chu, C.Y.; Shi, L.H.; Huang, S.H.; Liu, D.J. Chromosomal locations and genetic relationships of tiller and spike characters in wheat. Euphytica 2002, 125, 357–366. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Shi, H.; Mo, H.; Wu, F.; Lin, Y.; Gao, S.; Wang, J.; Wei, Y.; Liu, C.; et al. Identification and validation of novel low-tiller number QTL in common wheat. Theor. Appl. Genet. 2016, 129, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Jiang, L.; Yu, C.; Zhang, W.; Wang, J.; Wan, J. The identification and mapping of a tiller angle QTL on rice chromosome 9. Crop Sci. 2008, 48, 1799–1806. [Google Scholar] [CrossRef]
- Yu, B.; Lin, Z.; Li, H.; Li, X.; Li, J.; Wang, Y.; Zhang, X.; Zhu, Z.; Zhai, W.; Wang, X.; et al. TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 2007, 52, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhu, H.; Liu, S.; Zeng, R.; Zhang, Z.; Li, W.; Ding, X.; Zhao, F.; Zhang, G. Unconditional and conditional QTL mapping for the developmental behavior of tiller number in rice (Oryza sativa L.). Genetica 2010, 138, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Maurer, A.; Pillen, K. Detection of nitrogen deficiency QTL in juvenile wild barley introgression lines growing in a hydroponic system. BMC Genet. 2012, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.A.; Arifuzzaman, M.; Muzammil, S.; Pillen, K.; Léon, J. Wild barley introgression lines revealed novel QTL alleles for root and related shoot traits in the cultivated barley (Hordeum vulgare L.). BMC Genet. 2014, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Malyshev, S.; Korzun, V.; Voylokov, A.; Smirnov, V.; Borner, A. Linkage mapping of the mutant loci in rye (Secale cereale L.). Theor. Appl. Genet. 2001, 103, 70–74. [Google Scholar] [CrossRef]
- Stirnberg, P.; Van, D.S.K.; Leyser, H.M. MAX1 and MAX2 control shoot lateral branching in Arabidopsis. Development 2002, 129, 1131–1141. [Google Scholar] [PubMed]
- Booker, J.; Sieberer, T.W.; Williamson, L.; Willett, B.; Stirnberg, P.; Turnbull, C.; Srinivasan, M.; Goddard, P.; Leyser, O. MAX1 encodes a cytochrome P450 family member that acts downstream of MAX3/4 to produce a carotenoid-derived branch-inhibiting hormone. Dev. Cell 2005, 8, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Sorefan, K.; Booker, J.; Haurogné, K.; Goussot, M.; Bainbridge, B.; Foo, E.; Chatfield, S.; Ward, S.; Beveridge, Q.C.; Rameau, C.; et al. MAX4 and RMS1 are orthologous dioxygenase-like genes that regulate shoot branching in Arabidopsis and pea. Gene Dev. 2003, 17, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Booker, J.; Auldridge, M.; Wills, S.; McCarty, D.; Klee, H.; Leyser, O. MAX3/CCD7 is a carotenoid cleavage dioxygenase required for the synthesis of a novel plant signaling molecule. Curr. Biol. 2004, 14, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, Q.; Fu, Z.; Wang, Y.; Xiong, G.; Zeng, D.; Wang, X.; Liu, X.; Teng, S.; Hiroshi, F.; et al. Control of tillering in rice. Nature 2003, 422, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhang, W.; Xiong, G.; Yan, M.; Qian, Q.; Li, J.; Wang, Y. Identification and functional analysis of the MOC1 interacting protein 1. J. Genet. Genom. 2010, 37, 69–77. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Yu, Y.; Duan, J.; Liao, Z.; Xiong, G.; Meng, X.; Liu, G.; Qian, Q.; Li, J. Degradation of MONOCULM 1 by APC/C (TAD1) regulates rice tillering. Nat. Commun. 2012, 3, 750. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Suwa, Y.; Suzuki, M.; Kitano, H.; Ueguchi-Tanaka, M.; Ashikari, M.; Matsuoka, M.; Ueguchi, C. The OsTB1 gene negatively regulates lateral branching in rice. Plant J. 2003, 33, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, L.; Comadran, J.; Druka, A.; Marshall, D.F.; Thomas, W.T.; Macaulay, M.; MacKenzie, K.; Simpson, C.; Fuller, J.; Bonar, N.; et al. INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat. Genet. 2011, 43, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Chono, M.; Honda, I.; Zeniya, H.; Yoneyama, K.; Saisho, D.; Takeda, K.; Takatsuto, S.; Hoshino, T.; Watanabe, Y. A semidwarf phenotype of barley uzu results from a nucleotide substitution in the gene encoding a putative brassinosteroid receptor. Plant Physiol. 2003, 133, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, X.; Liu, P.; Sun, J. miR156-Targeted SBP-Box transcription factors interact with DWARF53 to regulate TEOSINTE BRANCHED1 and BARREN STALK1 expression in bread wheat. Plant Physiol. 2017, 174, 1931–1948. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.M.; Mackintosh, C.A.; Shin, S.; Gilding, E.; Kravchenko, S.; Baldridge, G.; Zeyen, R.; Muehlbauer, G.J. Overexpression of the maize Teosinte Branched1 gene in wheat suppresses tiller development. Plant Cell Rep. 2008, 27, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.X.; Li, Q.Y.; Shen, C.C.; Duan, Z.B.; Yu, D.Y.; Niu, J.S.; Ni, Y.J.; Jiang, Y.M. Transcriptome analysis for abnormal spike development of the wheat mutant dms. PLoS ONE 2016, 11, e0149287. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hua, Q.; Ma, Y.; Hu, G.; Qin, Y. Comparative transcriptome analyses of a late-maturing mandarin mutant and its original cultivar reveals gene expression profiling associated with citrus fruit maturation. PeerJ. 2017, 5, e3343. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Sun, F.; Chai, G.; Wang, Y.; Shi, L.; Liu, S.; Xi, Y. De novo assembly and transcriptome analysis of two contrary tillering mutants to learn the mechanisms of tillers outgrowth in switchgrass (Panicum virgatum L.). Front. Plant Sci. 2015, 6, 749. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, X.; Peal, L.; Tang, Y.; Wu, Y.; Mahalingam, R. Transcriptome analysis of nodes and buds from high and low tillering switchgrass inbred lines. PLoS ONE 2013, 8, e83772. [Google Scholar] [CrossRef] [PubMed]
- Kebrom, T.H.; Mullet, J.E. Transcriptome profiling of tiller buds provides new insights into PhyB regulation of tillering and indeterminate growth in sorghum. Plant Physiol. 2016, 170, 2232–2250. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Yan, J.; Xie, D. Comparison of phytohormone signaling mechanisms. Curr. Opin. Plant Biol. 2012, 15, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J. Molecular basis of plant architecture. Annu. Rev. Plant Biol. 2008, 59, 253–279. [Google Scholar] [CrossRef] [PubMed]
- Mcsteen, P. Auxin and monocot development. CSH. Perspect. Biol. 2010, 2, a001479. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S. Gibberellin metabolism and its regulation. Annu. Rev. Plant Biol. 2008, 59, 225–251. [Google Scholar] [CrossRef] [PubMed]
- Hussien, A.; Tavakol, E.; Horner, D.S.; Muñoz-Amatriaín, M.; Muehlbauer, G.J.; Rossini, L. Genetics of tillering in rice and barley. Plant Genome 2014, 7, 93–113. [Google Scholar] [CrossRef]
- Janssen, B.J.; Drummond, R.S.; Snowden, K.C. Regulation of axillary shoot development. Curr. Opin. Plant Biol. 2014, 17, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Kebrom, T.H.; Spielmeyer, W.; Finnegan, E.J. Grasses provide new insights into regulation of shoot branching. Trends Plant Sci. 2013, 18, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Stirnberg, P.; Chatfield, S.P.; Leyser, H.M.O. AXR1 acts after lateral bud formation to inhibit lateral bud growth in Arabidopsis. Plant Physiol. 1999, 121, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.K.; Choi, Y.D.; Kim, J.K. OsIAA6, a member of the rice Aux/IAA gene family, is involved in drought tolerance and tiller outgrowth. Plant Sci. 2015, 236, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Booker, J.; Chatfield, S.; Leyser, O. Auxin acts in xylem-associated or medullary cells to mediate apical dominance. Plant Cell 2003, 15, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Beveridge, C.A. Roles for auxin, cytokinin, and strigolactone in regulating shoot branching. Plant Physiol. 2009, 149, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.; Shinohara, N.; Sieberer, T.; Williamson, L.; George, G.; Hepworth, J.; Müller, D.; Domagalska, M.A.; Leyser, O. Strigolactones enhance competition between shoot branches by dampening auxin transport. Development 2010, 137, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Waldie, T.; Miyawaki, K.; To, J.P.; Melnyk, C.W.; Kieber, J.J.; Kakimoto, T.; Leyser, Y. Cytokinin is required for escape but not release from auxin mediated apical dominance. Plant J. 2015, 82, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.Y.; Chen, H.W.; Ng, C.Y.; Lin, C.Y.; Tseng, T.H.; Li, W.H.; Ku, M.S. Down-regulation of cytokinin oxidase 2 expression increases tiller number and improves rice yield. Rice 2015, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.F.; Yang, S.Y.; Chen, K.T.; Hsing, Y.I.; Zeevaart, J.A.; Chen, L.J.; Yu, S.M. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 2008, 20, 2603–2618. [Google Scholar] [CrossRef] [PubMed]
- Mauriat, M.; Sandberg, L.G.; Moritz, T. Proper gibberellin localization in vascular tissue is required to control auxin-dependent leaf development and bud outgrowth in hybrid aspen. Plant J. 2011, 67, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Gao, C.; Chen, M.S.; Pan, B.Z.; Ye, K.; Xu, Z.F. Gibberellin promotes shoot branching in the perennial woody plant Jatropha curcas. Plant Cell Physiol. 2015, 56, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Sun, T. The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, Y.; Yang, Z.; Chen, L.; Condon, A.G.; Hu, Y. Comparing the effects of GA-responsive dwarfing genes Rht13, and Rht8, on plant height and some agronomic traits in common wheat. Field Crop Res. 2015, 179, 35–43. [Google Scholar] [CrossRef]
- Perilli, S.; Moubayidin, L.; Sabatini, S. The molecular basis of cytokinin function. Curr. Opin. Plant Biol. 2010, 13, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.G.; Ross, J.J.; Babst, B.A.; Wienclaw, B.N.; Beveridge, C.A. Sugar demand, not auxin, is the initial regulator of apical dominance. Proc. Natl. Acad. Sci. USA 2014, 111, 6092–6097. [Google Scholar] [CrossRef] [PubMed]
- Kebrom, T.H.; Chandler, P.M.; Swain, S.M.; King, R.W.; Richards, R.A.; Spielmeyer, W. Inhibition of tiller bud outgrowth in the tin mutant of wheat is associated with precocious internode development. Plant Physiol. 2012, 160, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Rabot, A.; Henry, C.; Baaziz, K.B.; Mortreau, E.; Azri, W.; Lothier, J.; Hamama, L.; Boummaza, R.; Leduc, N.; Pelleschi-Travier, S.; et al. Insight into the role of sugars in bud burst under light in the rose. Plant Cell Physiol. 2012, 53, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier, F.; Péron, T.; Lecerf, M.; Perez-Garcia, M.D.; Barrière, Q.; Rolčík, J.; Boutet-Mercey, S.; Citerne, S.; Lemoine, R.; Porcheron, B.; et al. Sucrose is an early modulator of the key hormonal mechanisms controlling bud outgrowth in Rosa hybrida. J. Exp. Bot. 2015, 66, 2569–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Hu, X.; Zhu, M.; Xu, M.; Wang, L. Transcription factors NF-YA2 and NF-YA10 regulate leaf growth via auxin signaling in Arabidopsis. Sci. Rep. UK 2017, 7, 1395. [Google Scholar] [CrossRef] [PubMed]
- Theißen, G.; Melzer, R.; Rümpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, G.; Zhao, J.; Chu, H.; Lin, W.; Zhang, D.; Wang, Z.; Liang, W. Dwarf Tiller1, a Wuschel-related homeobox transcription factor, is required for tiller growth in rice. PLoS Genet. 2014, 10, e1004154. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, N.; Hake, S. The maize transcription factor KNOTTED1 directly regulates the gibberellin catabolism gene ga2ox1. Plant Cell 2009, 21, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Kuijt, S.J.; Greco, R.; Agalou, A.; Shao, J.; ‘t Hoen, C.C.J.; Overnas, E.; Osnato, M.; Curiale, S.; Meynard, D.; Gulik, R.V.; et al. Interaction between the GROWTH-REGULATING FACTOR and KNOTTED1-LIKE HOMEOBOX families of transcription factors. Plant Physiol. 2014, 164, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Mjomba, F.M.; Zheng, Y.; Liu, H.; Tang, W.; Hong, Z.; Wang, F.; Wu, W. Homeobox is pivotal for OsWUS controlling tiller development and female fertility in rice. G3 Genes Genom. Genet. 2016, 6, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Fridborg, I.; Kuusk, S.; Robertson, M.; Sundberg, E. The Arabidopsis protein SHI represses gibberellin responses in Arabidopsis and barley. Plant Physiol. 2001, 127, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.B.; Shen, C.C.; Li, Q.Y.; Lü, G.Z.; Ni, Y.J.; Yu, D.Y.; Niu, J.S. Identification of a novel male sterile wheat mutant dms conferring dwarf status and multi-pistils. J. Integr. Agric. 2015, 14, 1706–1714. [Google Scholar] [CrossRef]
- Pestsova, E.; Ganal, M.W.; Röder, M.S. Isolation and mapping of microsatellite markers specific for the D genome of bread wheat. Genome 2000, 43, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Somers, D.; Isaac, P.; Edwards, K. A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2004, 109, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Cota-Sánchez, J.H.; Remarchuk, K.; Ubayasena, K. Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 2006, 24, 161–167. [Google Scholar] [CrossRef]
- Bassam, B.J.; Caetanoanollés, G.; Gresshoff, P.M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 1991, 196, 80–83. [Google Scholar] [CrossRef]
- Schulze, S.K.; Kanwar, R.; Gölzenleuchter, M.; Therneau, T.M.; Beutler, A.S. SERE: Single-parameter quality control and sample comparison for RNA-Seq. BMC Genom. 2012, 13, 524. [Google Scholar] [CrossRef] [PubMed]
- The Triticum aestivum Genome Sequence. Available online: http://ftp.ensemblgenomes.org/pub/plants/release-32/fasta/triticum_aestivum/. (accessed on 2 October 2017).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Li, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | WT | dmc |
|---|---|---|
| Plant height/cm | 64.45 ± 3.28 | 48.00 ± 2.63 ** |
| Spike length/cm | 10.47 ± 0.53 | 6.48 ± 0.90 ** |
| Internode number of main stem | 5.00 ± 0 | 4.18 ± 0.51 ** |
| The top first internode length of the main stem/cm | 21.74 ± 2.28 | 19.46 ± 2.37 |
| The top second internode length of the main stem/cm | 13.98 ± 0.27 | 11.21 ± 1.55 * |
| The top third internode length of the main stem/cm | 9.42 ± 0.74 | 6.48 ± 0.34 ** |
| The top fourth internode length of the main stem/cm | 5.78 ± 0.29 | 4.24 ± 0.22 ** |
| The top fifth internode length of the main stem/cm | 3.19 ± 0.31 | - |
| Tiller number | 21.73 ± 2.20 | 1.11 ± 0.3 ** |
| Spike number | 16.64 ± 0.92 | 1.06 ± 0.32 ** |
| Spikelet number on the main stem | 21.73 ± 1.19 | 14.85 ± 2.09 ** |
| Seed number per spike | 64.10 ± 5.36 | 34.16 ± 4.28 ** |
| 1000-grain weight/g | 44.17 ± 4.59 | 35.41 ± 4.34 ** |
| Heading stage/week | 27 | 28 |
| Anthesis stage/week | 28 | 29 |
| Maturity stage/week | 32 | 33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, R.; Ni, Y.; Li, J.; Jiao, Z.; Zhu, X.; Jiang, Y.; Li, Q.; Niu, J. Quantitative Changes in the Transcription of Phytohormone-Related Genes: Some Transcription Factors Are Major Causes of the Wheat Mutant dmc Not Tillering. Int. J. Mol. Sci. 2018, 19, 1324. https://doi.org/10.3390/ijms19051324
He R, Ni Y, Li J, Jiao Z, Zhu X, Jiang Y, Li Q, Niu J. Quantitative Changes in the Transcription of Phytohormone-Related Genes: Some Transcription Factors Are Major Causes of the Wheat Mutant dmc Not Tillering. International Journal of Molecular Sciences. 2018; 19(5):1324. https://doi.org/10.3390/ijms19051324
Chicago/Turabian StyleHe, Ruishi, Yongjing Ni, Junchang Li, Zhixin Jiao, Xinxin Zhu, Yumei Jiang, Qiaoyun Li, and Jishan Niu. 2018. "Quantitative Changes in the Transcription of Phytohormone-Related Genes: Some Transcription Factors Are Major Causes of the Wheat Mutant dmc Not Tillering" International Journal of Molecular Sciences 19, no. 5: 1324. https://doi.org/10.3390/ijms19051324
APA StyleHe, R., Ni, Y., Li, J., Jiao, Z., Zhu, X., Jiang, Y., Li, Q., & Niu, J. (2018). Quantitative Changes in the Transcription of Phytohormone-Related Genes: Some Transcription Factors Are Major Causes of the Wheat Mutant dmc Not Tillering. International Journal of Molecular Sciences, 19(5), 1324. https://doi.org/10.3390/ijms19051324