Development and Characterization of Novel Genic-SSR Markers in Apple-Juniper Rust Pathogen Gymnosporangium yamadae (Pucciniales: Pucciniaceae) Using Next-Generation Sequencing

Abstract
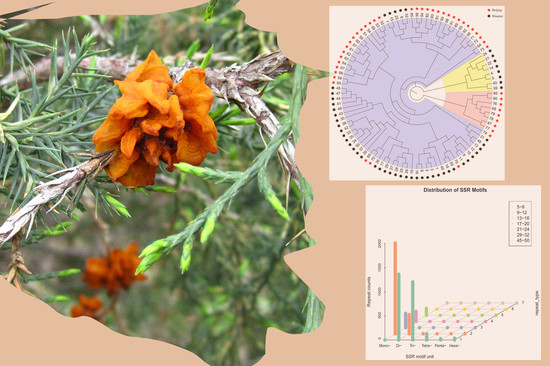
1. Introduction
2. Results
2.1. Pair-End Sequences Obtained with the Illumina de Novo Sequencing
2.2. Mining of Genic SSRs
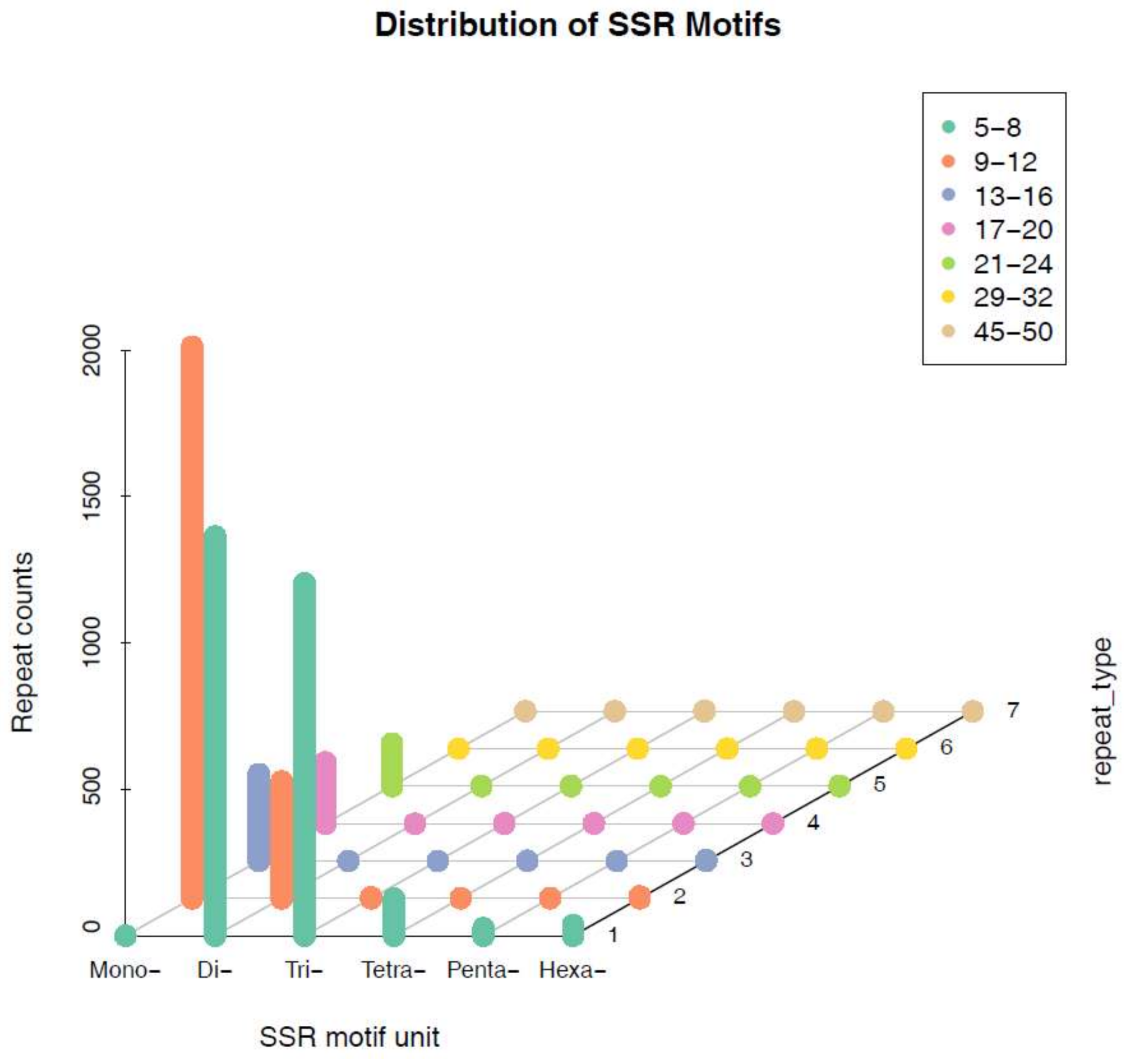
2.3. Distribution of SSR Motif
2.4. PCR Amplification and Polymorphism of Genic-SSRs
2.5. Characteristics of Validated Microsatellite Loci
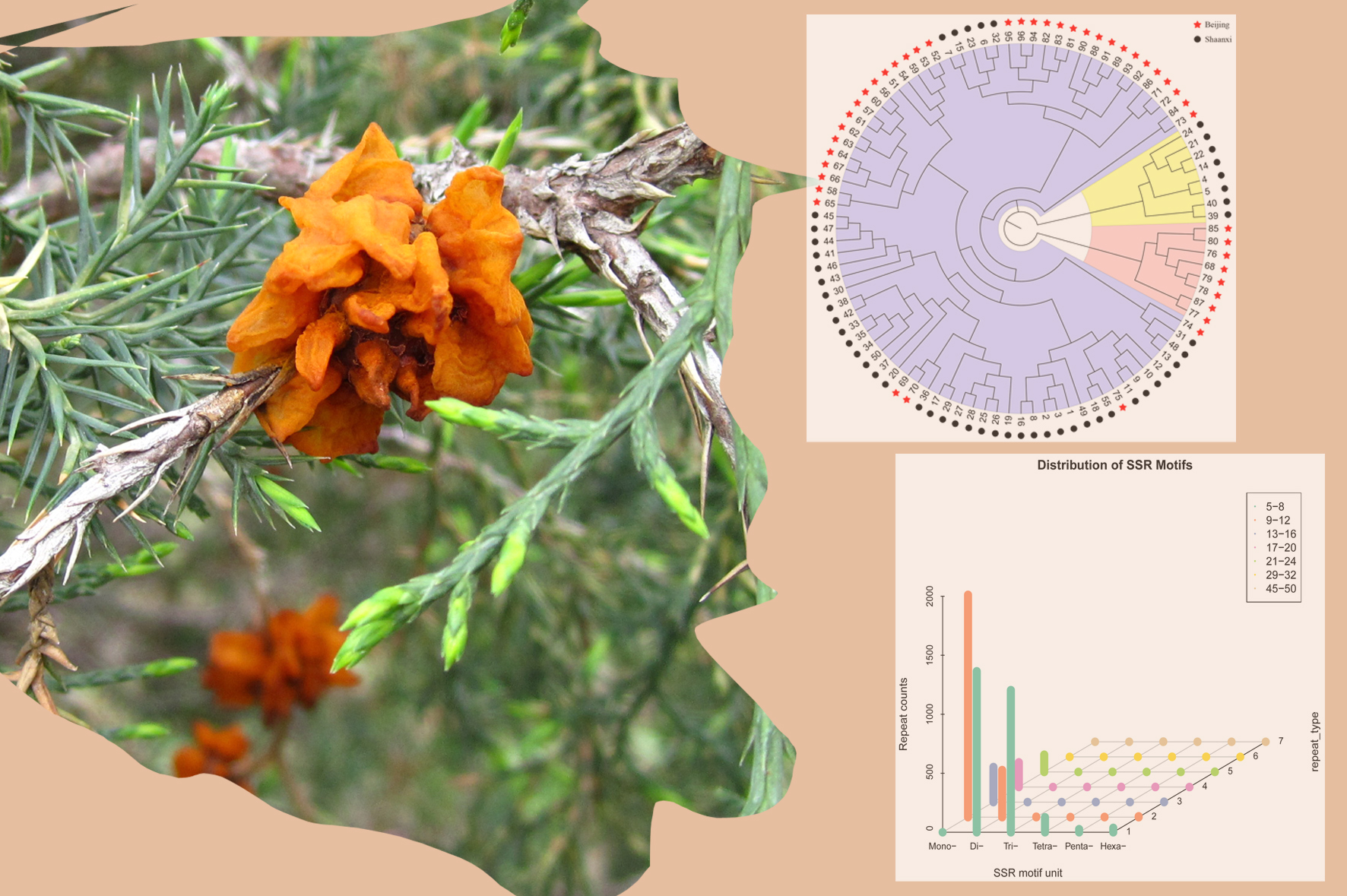
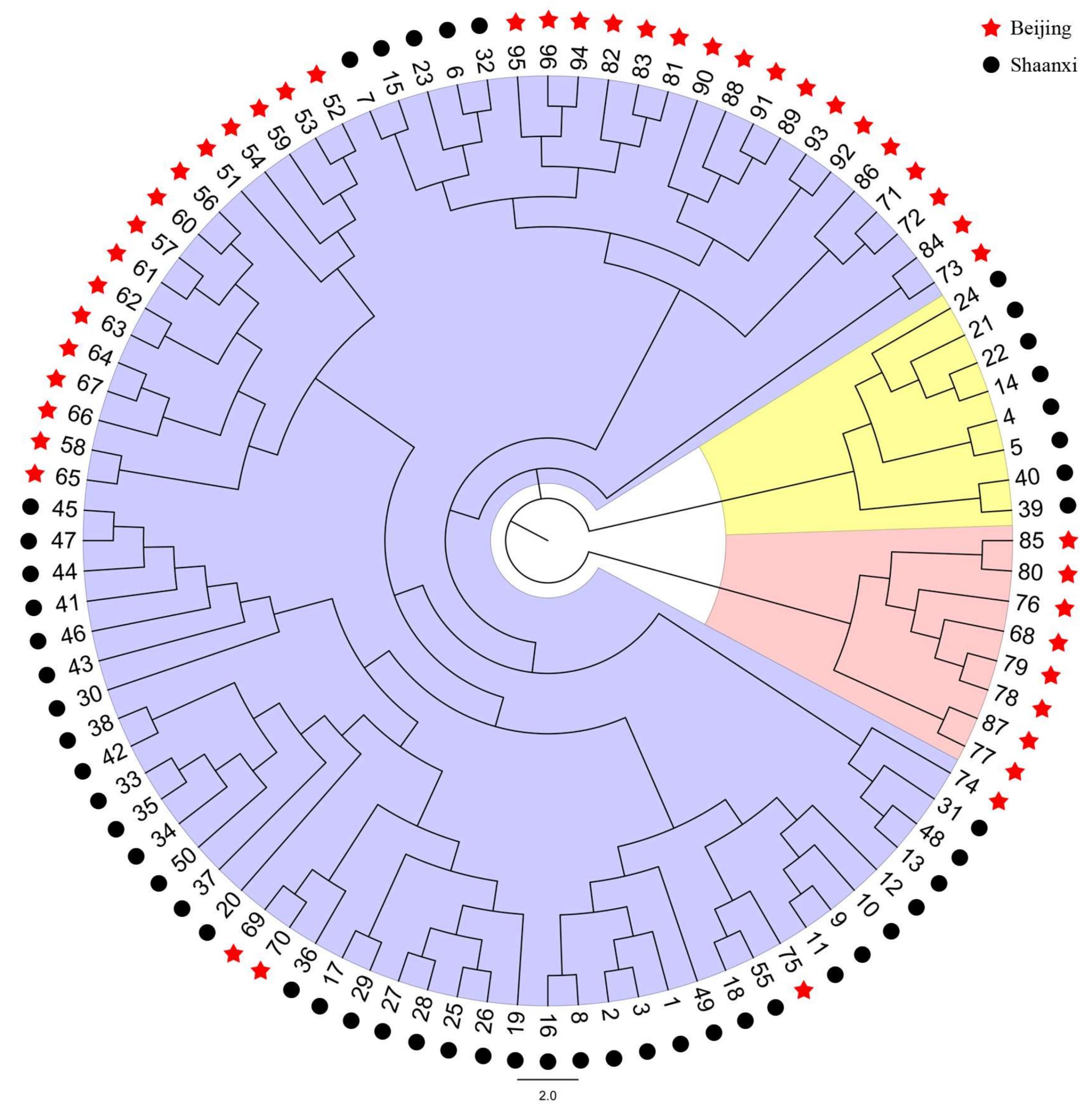
3. Discussion
4. Materials and Methods
4.1. Rust Sample Collection
4.2. RNA Isolation and Library Preparation
4.3. Reprocessing of Illumina Raw Data and de Novo Transcriptome Assembly
4.4. SSR Detection and Development of Primers
4.5. DNA Extraction, Primer Testing and Polymorphism Detection
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Velasco, R.; Zharkikh, A.J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; Salvi, S.; et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 2010, 42, 833. [Google Scholar] [CrossRef] [PubMed]
- Northland Agriculture. Available online: http://lnny.lnd.com.cn/htm/2013-07/01/content_3049863.htm (accessed on 1 July 2013).
- China Yield Information. Available online: http://www.chyxx.com/industry/201602/387126.html (accessed on 19 February 2016).
- Li, B.; Xu, X. Infection and Development of Apple Scab (Venturia inaequalis) on old leaves. J. Phytopathol. 2002, 150, 687–691. [Google Scholar] [CrossRef]
- Wang, X.; Kang, Z. Re-evaluation of pathogens causing Valsa canker on apple in China. Mycologia 2011, 103, 317. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.D. A Revised Taxonomic Account of Gymnosporangium, 3rd ed.; Pennsylvania State University Press: University Park, PA, USA, 1973; ISBN 0-271-01105-X. [Google Scholar]
- Cummins, G.B.; Hiratsuka, Y. Illustrated Genera of Rust Fungi, 3rd ed.; Burgess Pub: Minneapolis, MN, USA, 1959; ISBN 0-89054-304-6. [Google Scholar]
- European and Mediterranean Plant Protection Organization (EPPO). Available online: http://www.eppo.int/ (accessed on 19 November 2017).
- Cao, Z.M.; Li, Z.Q.; Zhuang, J.Y. Uredinales from the Qinling Mountains. Mycosystema 2000, 19, 13–23. (In Chinese) [Google Scholar]
- Guo, Y.N. The integrated management of the rust of apple (Gymnosporangium yamadae Miyabe). Bull. Agric. Sci. Technol. 1994, 19, 28–29. (In Chinese) [Google Scholar]
- Wang, S.Q.; Yang, Z.P.; Yu, J. The reasons that Apple rust happens seriously and comprehensive prevention and control measures. China Gard. Abstr. 2010, 26, 145–146. (In Chinese) [Google Scholar]
- Li, B.H.; Wang, C.X.; Dong, X.L. Research progress in apple diseases and problems in the disease management in China. Plant Prot. 2013, 39, 46–64. (In Chinese) [Google Scholar]
- Harada, Y.; Sawamura, K. Basitac (Mepronil), a promising agent for preventing basidiospore production by galls of Japanese apple rust (Gymnosporangium yamadae) on junipers. Annu. Rep. Soc. Plant Prot. N. Jpn. 1980, 31, 84. [Google Scholar] [CrossRef]
- Zambolim, L.; Vale, F.X.R.; Pereira, A.A.; Chaves, G.M. Manejo integrado das doenc_as do cafeeiro. In SIMPOSIO Internacional Sobre Café Adensado; Universidade Federal de Viçosa: Viçosa, Brazil, 1994; pp. 134–215. [Google Scholar]
- Thrall, P.H.; Laine, A.L.; Ravensdale, M.; Nemri, A.; Dodds, P.N.; Barrett, L.G.; Burdon, J.J. Rapid genetic change underpins antagonistic coevolution in a natural host-pathogen metapopulation. Ecol. Lett. 2012, 15, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Kinloch, B.B. White pine blister rust in North America: Past and prognosis. Phytopathology 2003, 93, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Kinloch, B.B.; Sniezko, R.A.; Dupper, G.E. Origin and distribution of cr2, a gene for resistance to white pine blister rust in natural populations of western white pine. Phytopathology 2003, 93, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Oelke, L.M.; Kolmer, J.A. Genetics of leaf rust resistance in spring wheat cultivars alsen and norm. Phytopathology 2005, 95, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Pinon, J.; Frey, P.; Pei, M.H.; Mccracken, A.R. Interactions between poplar clones and Melampsora populations and their implications for breeding for durable resistance. Die Keure 2005, 23, 139–154. [Google Scholar] [CrossRef]
- Harada, Y. Pear and Apple Rusts in Japan, with Special Reference to Their Life Cycles and Host Ranges; Reports; Tottori Mycological Institute: Tottori Prefecture, Japan, 1984. [Google Scholar]
- McCallum, B.D.; Roelfs, A.P.; Szabo, L.J.; Groth, J.V. Comparison of Puccinia graminis, f.sp. tritici, from South America and Europe. Plant Pathol. 2010, 48, 574–581. [Google Scholar] [CrossRef][Green Version]
- Zhong, S.; Leng, Y.; Friesen, T.; Faris, J.; Szabo, L. Development and characterization of expressed sequence tag-derived microsatellite markers for the wheat stem rust fungus Puccinia graminis f. sp. tritici. Phytopathology 2009, 99, 282–289. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, X.; Line, R.F.; Leung, H. Relationship between virulence variation and DNA polymorphism in Puccinia striiformis. Phytopathology 1993, 83, 1489–1497. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Wadl, P.A.; Dean, D.; Li, Y.; Vito, L.M.; Scheffler, B.E.; Hadziabdic, D.; Windham, M.T.; Trigiano, R.N. Development and characterization of microsatellites for switchgrass rust fungus (Puccinia emaculata). Conserv. Genet. Resour. 2011, 3, 185–188. [Google Scholar] [CrossRef]
- Queller, D.C.; Strassmann, J.E.; Hughes, C.R. Microsatellites and kinship. Ecol. Evol. 1993, 8, 285–288. [Google Scholar] [CrossRef]
- Ali, S.; Gladieux, P.; Leconte, M.; Gautier, A.; Justesen, A.F.; Hovmøller, M.S.; Enjalbert, J.; de Vallavieille-Pope, C. Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen Puccinia striiformis f.sp. tritici. PLoS Pathog. 2014, 10, e1003903. [Google Scholar] [CrossRef] [PubMed]
- Cabral, P.G.C.; Maciel-Zambolim, E.; Oliveira, S.A.S.; Caixeta, E.T.; Zambolim, L. Genetic diversity and structure of Hemileia vastatrix populations on Coffea spp. Plant Pathol. 2016, 65, 196–204. [Google Scholar] [CrossRef]
- Orquera-Tornakian, G.K.; Garrido, P.; Kronmiller, B.; Hunger, R.; Tyler, B.M.; Garzon, C.D.; Marek, S.M. Identification and characterization of simple sequence repeats (SSRs) for population studies of Puccinia novopanici. J. Microbiol. Methods 2017, 139, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bahri, B.; Leconte, M.; de Vallavieille-Pope, C.; Enjalbert, J. Isolation of ten microsatellite loci in an EST library of the phytopathogenic fungus Puccinia striiformis f. sp. tritici. Conserv. Genet. 2009, 10, 1425. [Google Scholar] [CrossRef]
- Dambroski, H.R.; Carson, M.L. Development and characterization of novel, polymorphic microsatellite markers for oat crown rust, Puccinia coronata. Mol. Ecol. Resour. 2008, 8, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Dracatos, P.M.; Dumsday, J.L.; Olle, R.S.; Cogan, N.O.; Dobrowolski, M.P.; Fujimori, M.; Roderick, H.; Stewart, A.V.; Smith, K.F.; Forster, J.W. Development and characterization of EST-SSR markers for the oat crown rust pathogen of ryegrass (Puccinia coronata f. sp. lolii). Genome 2006, 49, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Enjalbert, J.; Vautrin, D.; Solignac, M.; Giraud, T. Isolation of 12 microsatellite loci, using an enrichment protocol, in the phytopathogenic fungus Puccinia triticina. Mol. Ecol. Notes 2003, 3, 65–67. [Google Scholar] [CrossRef]
- Enjalbert, J.; Duan, X.; Giraud, T.; Vautrin, D.; Vallavieille-Pope, D.; Solignac, M. Isolation of twelve microsatellite loci, using an enrichment protocol, in the phytopathogenic fungus Puccinia striiformis f. sp. tritici. Mol. Ecol. Notes 2002, 2, 563–565. [Google Scholar] [CrossRef]
- Szabo, L.J. Development of simple sequence repeat markers for the plant pathogenic rust fungus, Puccinia graminis. Mol. Ecol. 2007, 7, 92–94. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Burke, J. EST-SSRs as a resource for population genetic analysis. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621. [Google Scholar] [CrossRef] [PubMed]
- Falk, D.A.; Holsinger, K.E. Genetics and Conservation of Rare Plants; Oxford University Press: New York, NY, USA, 1991. [Google Scholar]
- Dharmarajan, G.; Beatty, W.S.; Rhodes, O.E. Heterozygote deficiencies caused by a Wahlund effect: Dispelling unfounded expectations. J. Wildl. Manag. 2013, 77, 26–234. [Google Scholar] [CrossRef]
- Wang, J.L.; Whitlock, M.C. Estimating effective population size and migration rates from genetic samples over space and time. Genetics 2003, 163, 429–446. [Google Scholar] [PubMed]
- Liu, J.J.; Sturrock, R.N.; Sniezko, R.A.; Williams, H.; Benton, R.; Zamany, A. Transcriptome analysis of the white pine blister rust pathogen Cronartium ribicola: De novo assembly, expression profiling, and identification of candidate effectors. BMC Genom. 2015, 16, 678. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- MISA—MIcroSAtellite Identification Tool. Available online: http://pgrc.ipk-gatersleben.de/misa (accessed on 9 April 2018).
- Yeh, F.C.; Yang, R.C.; Boyle, T.B.J.; Ye, Z.H.; Mao, J.X. POPGENE, the User-Friendly Shareware for Population Genetic Analysis; Molecular Biology and Biotechnology Centre, University of Alberta: Edmonton, AB, Canada, 1997. [Google Scholar]
- Liu, K.; Muse, S.V. Powermarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephen, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DSXGY_1 | DSXGY_2 | DSXGY_3 | |
|---|---|---|---|
| Raw reads | 47,629,874 | 59,428,164 | 49,485,212 |
| Clean reads | 46,917,624 | 58,748,388 | 48,752,948 |
| Q20 (%) | 97.18 | 96.52 | 97.34 |
| Q30 (%) | 93.82 | 91.47 | 93.42 |
| Mapped reads | 38,545,026 (82.29%) | 50,102,470 (85.29%) | 41,058,330 (84.35%) |
| Transcripts | Unigenes | |
|---|---|---|
| 200~300 bp | 16,810 | 15,907 |
| 301~400 bp | 5634 | 4855 |
| 401~500 bp | 3124 | 2471 |
| 501~600 bp | 2103 | 1471 |
| 601~700 bp | 1692 | 1064 |
| 701~800 bp | 1354 | 781 |
| 801~900 bp | 1192 | 601 |
| 901~1000 bp | 1100 | 551 |
| 1001~2000 bp | 8601 | 3893 |
| 2001~3000 bp | 4516 | 2069 |
| 3001~10 kbp | 3180 | 1430 |
| >10 kbp | 13 | 9 |
| total number | 49,319 | 35,102 |
| Max length | 19,126 | 19,126 |
| Average length | 1006 | 756 |
| N50 | 1957 | 1654 |
| Total residues | 49,601,912 | 26,521,164 |
| Repeat Motif | Number of Repeat | Total Frequency (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| A/T | 1121 | 342 | 151 | 31.93% | |||||
| C/G | 131 | 92 | 47 | 5.34% | |||||
| AC/GT | 191 | 123 | 73 | 38 | 51 | 27 | 1 | 9.97% | |
| AG/CT | 198 | 92 | 64 | 43 | 33 | 38 | 3 | 9.32% | |
| AT/AT | 333 | 173 | 109 | 81 | 64 | 19 | 1 | 15.43% | |
| CG/CG | 8 | 0.16% | |||||||
| AAC/GTT | 87 | 52 | 38 | 2 | 3.54% | ||||
| AAG/CTT | 86 | 31 | 16 | 3 | 1 | 2.71% | |||
| AAT/ATT | 58 | 28 | 9 | 1 | 1 | 1.92% | |||
| ACC/GGT | 136 | 46 | 30 | 2 | 4.23% | ||||
| ACG/CGT | 14 | 13 | 7 | 2 | 1 | 0.73% | |||
| ACT/AGT | 37 | 15 | 21 | 3 | 1.50% | ||||
| AGC/CTG | 66 | 30 | 20 | 2 | 2.33% | ||||
| AGG/CCT | 67 | 16 | 14 | 2 | 1.96% | ||||
| ATC/ATG | 121 | 45 | 44 | 4 | 4.23% | ||||
| CCG/CGG | 26 | 3 | 7 | 1 | 0.73% | ||||
| AAAC/GTTT | 17 | 3 | 0.40% | ||||||
| AAAG/CTTT | 4 | 2 | 0.12% | ||||||
| AAAT/ATTT | 12 | 1 | 0.26% | ||||||
| AACC/GGTT | 5 | 2 | 0.14% | ||||||
| AACG/CGTT | 1 | 0.02% | |||||||
| AAGG/CCTT | 1 | 1 | 0.04% | ||||||
| AAGT/ACTT | 0.00% | ||||||||
| AATC/ATTG | 3 | 0.06% | |||||||
| AATG/ATTC | 6 | 1 | 0.14% | ||||||
| ACAG/CTGT | 4 | 0.08% | |||||||
| ACAT/ATGT | 27 | 5 | 0.63% | ||||||
| ACCC/GGGT | 2 | 0.04% | |||||||
| ACCG/CGGT | 1 | 0.02% | |||||||
| ACCT/AGGT | 0.00% | ||||||||
| ACGC/CGTG | 1 | 0.02% | |||||||
| ACTC/AGTG | 1 | 1 | 0.04% | ||||||
| ACTG/AGTC | 3 | 1 | 1 | 0.10% | |||||
| AGAT/ATCT | 2 | 0.04% | |||||||
| AGCC/CTGG | 2 | 2 | 1 | 0.10% | |||||
| AGCT/AGCT | 1 | 1 | 0.04% | ||||||
| AGGC/CCTG | 1 | 1 | 1 | 0.06% | |||||
| AGGG/CCCT | 3 | 1 | 0.08% | ||||||
| ATCC/ATGG | 4 | 1 | 1 | 0.12% | |||||
| OTHERS | 32 | 13 | 10 | 8 | 5 | 1 | 2 | 1 | 1.42% |
| MNR | 1252 | 434 | 198 | 37.27% | |||||
| DNR | 730 | 388 | 246 | 162 | 148 | 84 | 5 | 34.88% | |
| TNR | 698 | 279 | 206 | 21 | 2 | 1 | 1 | 23.90% | |
| TTNR | 98 | 25 | 3 | 1 | 1 | 2.53% | |||
| PNR | 17 | 4 | 5 | 2 | 0.55% | ||||
| HNR | 15 | 9 | 5 | 8 | 3 | 1 | 2 | 1 | 0.87% |
| Number of Motif Copies | Mono- | Di- | Tri- | Tetra- | Penta- | Hexa- | Total | Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.00% | |
| 5 | 0 | 0 | 698 | 98 | 17 | 15 | 828 | 14.48% |
| 6 | 0 | 730 | 279 | 25 | 4 | 9 | 1047 | 18.31% |
| 7 | 0 | 388 | 206 | 3 | 5 | 5 | 607 | 10.62% |
| 8 | 0 | 246 | 21 | 1 | 0 | 8 | 276 | 4.83% |
| 9 | 0 | 162 | 0 | 1 | 2 | 3 | 168 | 2.94% |
| 10 | 1252 | 148 | 2 | 0 | 0 | 1 | 1403 | 24.54% |
| 11 | 434 | 84 | 1 | 0 | 0 | 2 | 521 | 9.11% |
| 12 | 198 | 5 | 1 | 0 | 0 | 1 | 205 | 3.59% |
| 13 | 119 | 0 | 0 | 0 | 0 | 0 | 119 | 2.08% |
| 14 | 81 | 0 | 0 | 0 | 0 | 1 | 82 | 1.43% |
| 15 | 68 | 0 | 0 | 0 | 0 | 1 | 69 | 1.21% |
| 16 | 29 | 0 | 0 | 1 | 0 | 1 | 31 | 0.54% |
| 17 | 41 | 0 | 1 | 1 | 0 | 0 | 43 | 0.75% |
| 18 | 35 | 0 | 0 | 0 | 0 | 0 | 35 | 0.61% |
| 19 | 56 | 0 | 0 | 0 | 1 | 0 | 57 | 1.00% |
| 20 | 77 | 0 | 0 | 0 | 0 | 0 | 77 | 1.35% |
| 21 | 74 | 0 | 0 | 0 | 0 | 1 | 75 | 1.31% |
| 22 | 46 | 0 | 1 | 0 | 0 | 0 | 47 | 0.82% |
| 23 | 23 | 0 | 0 | 0 | 0 | 0 | 23 | 0.40% |
| 24 | 3 | 0 | 0 | 0 | 0 | 1 | 4 | 0.07% |
| Total | 2536 | 1763 | 1210 | 130 | 29 | 49 | 5717 | 100.00% |
| Frequency (%) | 44.36% | 30.84% | 21.16% | 2.27% | 0.51% | 0.86% | 100.00% |
| Marker | Primer Sequences 5′-3′ | Repeat Motif | Allele No. | Size Range (bp) | HWE | HO | HE | FIS | PIC | I | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shaanxi | Beijing | Shaanxi | Beijing | Shaanxi | Beijing | Shaanxi | Beijing | |||||||
| GY8 | F: AAAAAGCATCAGGGGGAGGG | (CT)6 | 5 | 261–269 | 0.272 | 0.086 | 0.489 | 0.395 | 0.555 | 0.509 | 0.120 | 0.225 | 0.454 | 0.931 |
| R: GGTTGTGTGTGGCTAAGGCT | ||||||||||||||
| GY14 | F: ACAATCAACTAGAAATTGACCTTTGT | (AT)6 | 8 | 154–180 | 0.000 | 0.000 | 0.163 | 0.205 | 0.258 | 0.772 | 0.370 | 0.737 | 0.660 | 1.521 |
| R: TGGATGCAATATTTGAACTGTCAGA | ||||||||||||||
| GY18 | F: TCCAATCACCCCTCCCATCT | (TGT)7 | 7 | 219–237 | 0.001 | 0.000 | 0.250 | 0.048 | 0.391 | 0.203 | 0.363 | 0.767 | 0.290 | 0.688 |
| R: TCGAAAGTCGCAATACCAGCT | ||||||||||||||
| GY19 | F: TTCTTGAACGCAGACAGTGT | (GA)9 | 8 | 207–223 | 0.000 | 0.000 | 0.286 | 0.318 | 0.503 | 0.562 | 0.435 | 0.437 | 0.504 | 1.169 |
| R: TTCGCTCTCCCTCCCTCTTT | ||||||||||||||
| GY20 | F: ACGAGAGTGTCAGACGAAGC | (GCAAGA)7 | 6 | 195–225 | 0.213 | 0.294 | 0.633 | 0.683 | 0.702 | 0.698 | 0.100 | 0.022 | 0.650 | 1.358 |
| R: CTGACTTGTCGAGACCGAGG | ||||||||||||||
| GY21 | F: TCGTACAGCCACACACAGAC | (AC)9 | 2 | 120–122 | 0.752 | 0.018 | 0.511 | 0.293 | 0.470 | 0.470 | −0.089 | 0.380 | 0.375 | 0.693 |
| R: AGAAGCACCATCGGTCAAGG | ||||||||||||||
| GY25 | F: GGCATGGAGAAAGCAGCAAG | (AG)8 | 3 | 141–145 | 0.000 | 0.017 | 0.300 | 0.278 | 0.496 | 0.482 | 0.432 | 0.427 | 0.376 | 0.718 |
| R: GCACCTCCAGGAAATCCCAA | ||||||||||||||
| GY27 | F: ACGTCCCTCAAATCTCATCCT | (TC)9 | 9 | 170–190 | 0.000 | 0.000 | 0.372 | 0.405 | 0.747 | 0.722 | 0.505 | 0.442 | 0.703 | 1.563 |
| R: CCGCCACGCTCAAAGAAAAT | ||||||||||||||
| GY30 | F: GATCAGGATGAGAGGCGGTG | (TC)10 | 4 | 214–230 | 0.017 | 1.000 | 0.102 | 0.231 | 0.118 | 0.207 | 0.137 | −0.118 | 0.148 | 0.333 |
| R: TTGGTATGCATGCCAGGGAG | ||||||||||||||
| GY42 | F: TGTGGTTGTGGGGTTTTGGA | (AT)9 | 9 | 145–163 | 0.000 | 0.000 | 0.143 | 0.125 | 0.820 | 0.609 | 0.827 | 0.797 | 0.782 | 1.830 |
| R: ACCACACCACATCACATCATGT | ||||||||||||||
| GY43 | F: AGTGAAAGAGAGTGGATGTGC | (GT)9 | 4 | 154–162 | 0.010 | 0.036 | 0.061 | 0.065 | 0.099 | 0.105 | 0.385 | 0.384 | 0.099 | 0.256 |
| R: TGCGTCCCATGTATGTCTGT | ||||||||||||||
| GY66 | F: CCAGCATGCCTACCTAGCTG | (TGTGAT)5 | 4 | 181–199 | 0.000 | 0.000 | 0.000 | 0.000 | 0.547 | 0.649 | 1.000 | 1.000 | 0.598 | 1.198 |
| R: CAAGGAACAACAGCAGTGGC | ||||||||||||||
| GY67 | F: CGCGGTTCCGGATTGATAGA | (CGGAGT)8 | 4 | 190–208 | 0.000 | 0.000 | 0.000 | 0.000 | 0.682 | 0.611 | 1.000 | 1.000 | 0.612 | 1.218 |
| R: GGAATTCAGTCAAGGCCCCA | ||||||||||||||
| GY68 | F: TTCTTCTCTCCCAGCTCCCA | (CACTGC)5 | 7 | 129–177 | 0.000 | 0.000 | 0.333 | 0.075 | 0.620 | 0.643 | 0.465 | 0.885 | 0.621 | 1.345 |
| R: CAGTCAGCCTGTGTCCAGAG | ||||||||||||||
| GY69 | F: CTGGAGTCCGCCAATCAACT | (CCGCAT)6 | 5 | 203–227 | 0.779 | 0.122 | 0.396 | 0.366 | 0.355 | 0.523 | −0.118 | 0.303 | 0.393 | 0.823 |
| R: ATTTTGGGACGAGCAGCTGA | ||||||||||||||
| GY70 | F: TGACCCCAATAAGACAAAAGTTGA | (ACAGCT)9 | 9 | 238–292 | 0.000 | 0.000 | 0.510 | 0.294 | 0.738 | 0.651 | 0.311 | 0.552 | 0.655 | 1.475 |
| R: GTTCAACTCCAGTCGGCTGA | ||||||||||||||
| GY72 | F: TACCACGAGTCCAGCTCTCA | (TGATCC)5 | 7 | 193–223 | 0.000 | 0.000 | 0.022 | 0.000 | 0.671 | 0.817 | 0.968 | 1.000 | 0.718 | 1.596 |
| R: AAAAGGAGTTGAGCGCGAGA | ||||||||||||||
| GY75 | F: CTCTGGCTCTGGCTTCAGTC | (CCTTAG)6 | 5 | 236–260 | 0.027 | 0.086 | 0.480 | 0.386 | 0.597 | 0.515 | 0.197 | 0.253 | 0.456 | 0.930 |
| R: AAGCCAAGCCAAGCAACTTG | ||||||||||||||
| GY79 | F: CGACAACTTTGCGCACTTGT | (TC)10 | 8 | 225–275 | 0.000 | 0.000 | 0.025 | 0.156 | 0.334 | 0.567 | 0.926 | 0.728 | 0.431 | 1.037 |
| R: ACTCCATTTTCTTGCATTTTGGGA | ||||||||||||||
| GY81 | F: ACCTCCCTGAAACACAAGCA | (CAC)7 | 5 | 245–263 | 0.004 | 0.000 | 0.265 | 0.378 | 0.372 | 0.449 | 0.289 | 0.159 | 0.352 | 0.740 |
| R: GCGTCTGAGTGGTGGAATCA | ||||||||||||||
| GY86 | F: TGTCGATTGGGATGGTTGGG | (GAAG)5 | 7 | 172–196 | 0.043 | 0.000 | 0.640 | 0.341 | 0.589 | 0.674 | −0.087 | 0.497 | 0.568 | 1.250 |
| R: GTCCTCTCTGACTCGGGTCT | ||||||||||||||
| GY87 | F: ATCAGTGGCTCCCTCTCCAT | (CAT)7 | 6 | 233–263 | 1.000 | 0.001 | 0.041 | 0.073 | 0.040 | 0.207 | −0.011 | 0.650 | 0.116 | 0.316 |
| R: GGGTAAGTGTTGGCGGAAGA | ||||||||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, S.-Q.; Cao, B.; Tian, C.-M.; Liang, Y.-M. Development and Characterization of Novel Genic-SSR Markers in Apple-Juniper Rust Pathogen Gymnosporangium yamadae (Pucciniales: Pucciniaceae) Using Next-Generation Sequencing. Int. J. Mol. Sci. 2018, 19, 1178. https://doi.org/10.3390/ijms19041178
Tao S-Q, Cao B, Tian C-M, Liang Y-M. Development and Characterization of Novel Genic-SSR Markers in Apple-Juniper Rust Pathogen Gymnosporangium yamadae (Pucciniales: Pucciniaceae) Using Next-Generation Sequencing. International Journal of Molecular Sciences. 2018; 19(4):1178. https://doi.org/10.3390/ijms19041178
Chicago/Turabian StyleTao, Si-Qi, Bin Cao, Cheng-Ming Tian, and Ying-Mei Liang. 2018. "Development and Characterization of Novel Genic-SSR Markers in Apple-Juniper Rust Pathogen Gymnosporangium yamadae (Pucciniales: Pucciniaceae) Using Next-Generation Sequencing" International Journal of Molecular Sciences 19, no. 4: 1178. https://doi.org/10.3390/ijms19041178
APA StyleTao, S.-Q., Cao, B., Tian, C.-M., & Liang, Y.-M. (2018). Development and Characterization of Novel Genic-SSR Markers in Apple-Juniper Rust Pathogen Gymnosporangium yamadae (Pucciniales: Pucciniaceae) Using Next-Generation Sequencing. International Journal of Molecular Sciences, 19(4), 1178. https://doi.org/10.3390/ijms19041178