Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System

Abstract
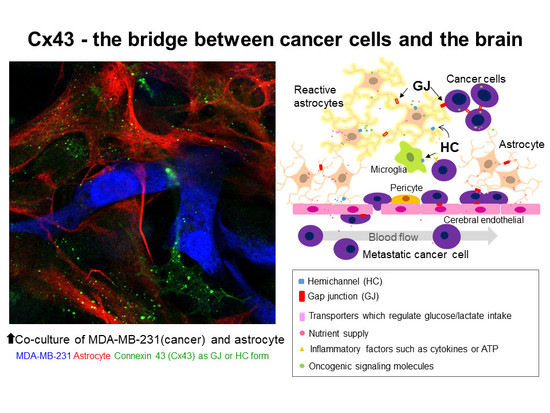
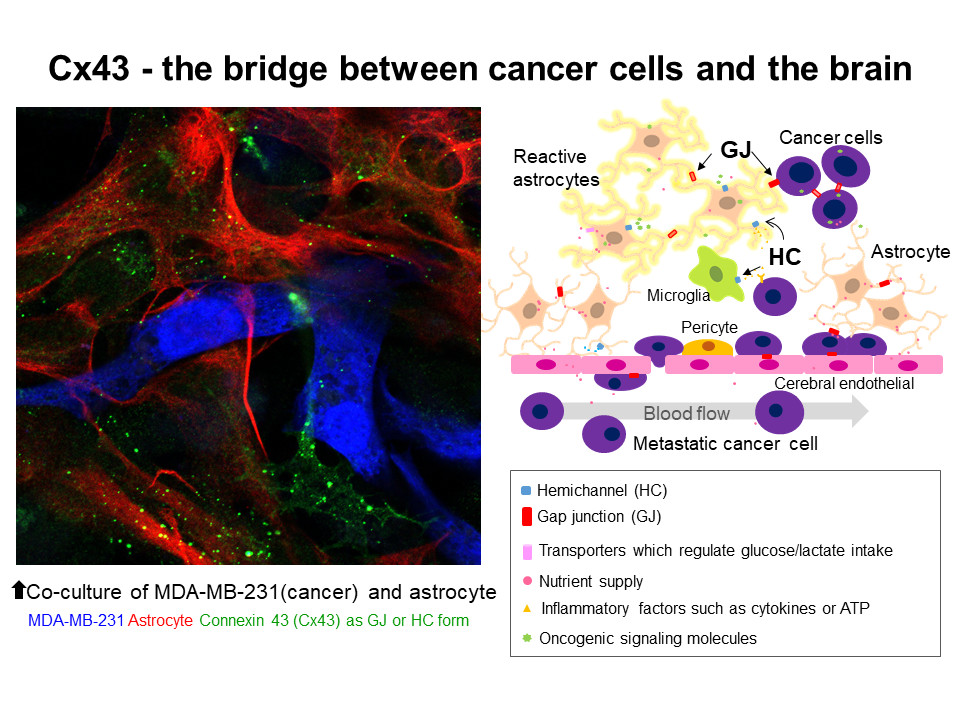
1. Introduction
2. The Role of Connexin43 (Cx43) in Tumors of the Central Nervous System (CNS)
2.1. Involvement of Cx43 in Inflammatory Responses in CNS Cancer
2.2. Opposing Roles of Cx43 in Tumor Survival and Invasion
2.3. Homocellular Junctions
2.3.1. Glioma Channels in Invasion
2.3.2. Homocellular Astrocyte Channels in Glioma Invasion
2.4. Heterocellular Junctions
2.4.1. Heterocellular GJIC in Glioma Invasion
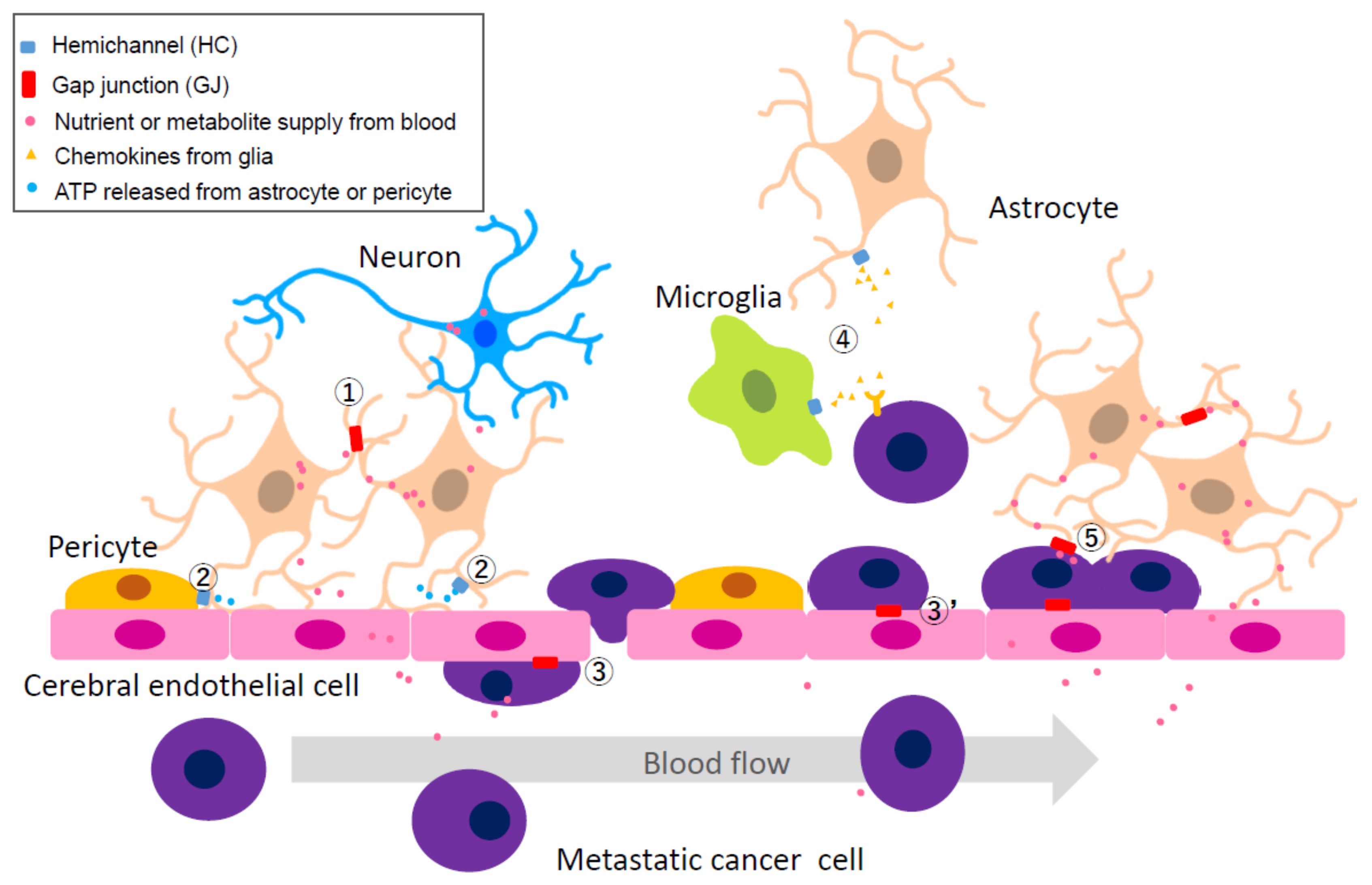
2.4.2. Heterocellular GJs or HCs: Extravascular Liberation of Metastatic Cancer—Neurovascular Unit Including Cerebral Endothelial Cells (CECs), Pericytes, Glial Cells, and Neurons
2.4.3. Heterocellular GJs: Possibility of Utilizing Metabolic Coupling between Astrocytes and Neurons
2.4.4. Heterocellular GJs: Pro-Survival Functions of GJs between Metastasized Cancer Cells and Astrocytes
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.M.; Goodenough, D.A. Diverse functions of vertebrate gap junctions. Trends Cell Biol. 1998, 8, 477–483. [Google Scholar] [CrossRef]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A.; Paul, D.L. Beyond the gap: Functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 2003, 4, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Crespin, S.; Avanzo, J.L.; Zaidan-Dagli, M.L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 2005, 1719, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Sato, H.; Virgona, N.; Hagiwara, H.; Kashiwagi, K.; Suzuki, K.; Asano, R.; Yano, T. Negative growth control of osteosarcoma cell by Bowman-Birk protease inhibitor from soybean; involvement of connexin43. Cancer Lett. 2007, 253, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Riquelme, M.A.; Gu, S.; Kar, R.; Gao, X.; Sun, L.; Jiang, J.X. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene 2016, 35, 5597–5607. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Ji, L.; Wang, H.; Wang, W.; Zheng, H.; Zou, J.; Liu, L.; Qi, X.; Liu, Z.; Du, B.; et al. Connexin43 upregulation by dioscin inhibits melanoma progression via suppressing malignancy and inducing M1 polarization. Int. J. Cancer 2017, 141, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Uzu, M.; Sato, H.; Yamada, R.; Kashiba, T.; Shibata, Y.; Yamaura, K.; Ueno, K. Effect of enhanced expression of connexin43 on sunitinib-induced cytotoxicity in mesothelioma cells. J. Pharmacol. Sci. 2015, 128, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Uzu, M.; Sato, H.; Shimizu, A.; Shibata, Y.; Ueno, K.; Hisaka, A. Connexin43 enhances Bax activation via JNK activation in sunitinib-induced apoptosis in mesothelioma cells. J. Pharmacol. Sci. 2017, 134, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Iwata, H.; Takano, Y.; Yamada, R.; Okuzawa, H.; Nagashima, Y.; Yamaura, K.; Ueno, K.; Yano, T. Enhanced effect of connexin43 on cisplatin-induced cytotoxicity in mesothelioma cells. J. Pharmacol. Sci. 2009, 110, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Y.; Si, Y.J.; Chen, X.H.; Li, Z.J.; Gao, L.; Gao, L.; Zhang, C. Effect of Cx43 gene-modified leukemic bone marrow stromal cells on the regulation of Jurkat cell line in vitro. Leuk. Res. 2012, 36, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wen, Q.; Liu, Y.; Zhang, C.; Wang, M.; Chen, G.; Gong, Y.; Zhong, J.; Chen, X.; Stucky, A.; et al. Increased expression of CX43 on stromal cells promotes leukemia apoptosis. Oncotarget 2015, 6, 44323–44331. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Naus, C.C. Gap junctions and neurological disorders of the central nervous system. Biochim. Biophys. Acta 2004, 1662, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Koulakoff, A.; Mei, X.; Orellana, J.A.; Saez, J.C.; Giaume, C. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1818, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Leybaert, L.; Naus, C.C.; Saez, J.C. Connexin and pannexin hemichannels in brain glial cells: Properties, pharmacology, and roles. Front. Pharmacol. 2013, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Bai, D. Structural analysis of key gap junction domains—Lessons from genome data and disease-linked mutants. Semin. Cell Dev. Biol. 2016, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Stoletov, K.; Strnadel, J.; Zardouzian, E.; Momiyama, M.; Park, F.D.; Kelber, J.A.; Pizzo, D.P.; Hoffman, R.; VandenBerg, S.R.; Klemke, R.L. Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 2013, 126, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Boucher, J.; Jego, G.; Pernet, N.; Cronier, L.; Hammann, A.; Solary, E.; Garrido, C. Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget 2016, 7, 73925–73934. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Aftab, Q.; Ma, N.; Chen, V.C.; Hong, X.; Lozinsky, S.; Naus, C.C.; Sin, W.C. Connexin43 confers Temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 2013, 75, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Balasubramanian, K.; Lin, Q.; Kim, S.W.; Kim, S.J. The brain microenvironment and cancer metastasis. Mol. Cells 2010, 30, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Balasubramanian, K.; Fan, D.; Kim, S.J.; Guo, L.; Wang, H.; Bar-Eli, M.; Aldape, K.D.; Fidler, I.J. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 2010, 12, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Choi, H.J.; Lee, H.J.; He, J.; Wu, Q.; Langley, R.R.; Fidler, I.J.; Kim, S.J. Role of the endothelin axis in astrocyte- and endothelial cell-mediated chemoprotection of cancer cells. Neuro Oncol. 2014, 16, 1585–1598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xu, W.Q.; Ye, M.X.; Zhang, Y.; Wang, H.Y.; Zhang, J.; Li, Y.; Wang, Y.S. Up-regulated basigin-2 in microglia induced by hypoxia promotes retinal angiogenesis. J. Cell. Mol. Med. 2017, 21, 3467–3480. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Zengotita, M.; Yachnis, A.T. Gliosis versus glioma? Don’t grade until you know. Adv. Anat. Pathol. 2012, 19, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M.; Amankulor, N.M.; Pitter, K.; Helmy, K.; Squatrito, M.; Holland, E.C. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS ONE 2012, 7, e32453. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Borboa, A.K.; Baird, A.; Eliceiri, B.P. Non-invasive quantification of brain tumor-induced astrogliosis. BMC Neurosci. 2011, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Rozental, R.; Giaume, C.; Spray, D.C. Gap junctions in the nervous system. Brain Res. Brain Res. Rev. 2000, 32, 11–15. [Google Scholar] [CrossRef]
- Dermietzel, R.; Spray, D.C. Gap junctions in the brain: Where, what type, how many and why? Trends Neurosci. 1993, 16, 186–192. [Google Scholar] [CrossRef]
- Fonseca, C.G.; Green, C.R.; Nicholson, L.F. Upregulation in astrocytic connexin43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002, 929, 105–116. [Google Scholar] [CrossRef]
- Kozoriz, M.G.; Bechberger, J.F.; Bechberger, G.R.; Suen, M.W.; Moreno, A.P.; Maass, K.; Willecke, K.; Naus, C.C. The connexin43 C-terminal region mediates neuroprotection during stroke. J. Neuropathol. Exp. Neurol. 2010, 69, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Ozog, M.A.; Bechberger, J.F.; Nakase, T. A neuroprotective role for gap junctions. Cell Commun. Adhes. 2001, 8, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Siushansian, R.; Bechberger, J.F.; Cechetto, D.F.; Hachinski, V.C.; Naus, C.C. Connexin43 null mutation increases infarct size after stroke. J. Comp. Neurol. 2001, 440, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.; Anderson, P.N.; Cook, J.E.; Green, C.R.; Becker, D.L. Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Mol. Cell. Neurosci. 2008, 39, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Lindqvist, E.; Kiehn, O.; Widenfalk, J.; Olson, L. Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. J. Comp. Neurol. 2005, 489, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Peeling, J.; Sutherland, G.R.; Hertzberg, E.L.; Nagy, J.I. Ischemia-induced cellular redistribution of the astrocytic gap junctional protein connexin43 in rat brain. Brain Res. 1994, 652, 311–322. [Google Scholar] [CrossRef]
- Kolar, K.; Freitas-Andrade, M.; Bechberger, J.F.; Krishnan, H.; Goldberg, G.S.; Naus, C.C.; Sin, W.C. Podoplanin: A marker for reactive gliosis in gliomas and brain injury. J. Neuropathol. Exp. Neurol. 2015, 74, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Fazakas, C.; Molnar, K.; Vegh, A.G.; Hasko, J.; Krizbai, I.A. Foe or friend? Janus-faces of the neurovascular unit in the formation of brain metastases. J. Cereb. Blood Flow Metab. 2018, 38, 563–587. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.V.; Garre, J.M.; Orellana, J.A.; Bukauskas, F.F.; Nedergaard, M.; Saez, J.C. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 2012, 1487, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; von Bernhardi, R.; Giaume, C.; Saez, J.C. Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev. Neurosci. 2012, 23, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta 2005, 1711, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Gulì, C.; la Torre, D.; Tomasello, C.; Angileri, F.F.; Aguennouz, M. Role of inflammation and oxidative stress mediators in gliomas. Cancers 2010, 2, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A.; Moller, T. Microglia biology in health and disease. J. Neuroimmune Pharmacol. 2006, 1, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Decrock, E.; de Vuyst, E.; Vinken, M.; van Moorhem, M.; Vranckx, K.; Wang, N.; van Laeken, L.; de Bock, M.; D’Herde, K.; Lai, C.P.; et al. Connexin43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 2009, 16, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.; Christov, C.; Guillamo, J.S.; de Bouard, S.; Palfi, S.; Venance, L.; Tardy, M.; Peschanski, M. Contribution of gap junctional communication between tumor cells and astroglia to the invasion of the brain parenchyma by human glioblastomas. BMC Cell Biol. 2005, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [PubMed]
- Zhang, W.; Couldwell, W.T.; Simard, M.F.; Song, H.; Lin, J.H.; Nedergaard, M. Direct gap junction communication between malignant glioma cells and astrocytes. Cancer Res. 1999, 59, 1994–2003. [Google Scholar] [PubMed]
- Strale, P.O.; Clarhaut, J.; Lamiche, C.; Cronier, L.; Mesnil, M.; Defamie, N. Down-regulation of connexin43 expression reveals the involvement of caveolin-1 containing lipid rafts in human U251 glioblastoma cell invasion. Mol. Carcinog. 2012, 51, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, N.; Costa, F.; Cossetti, C.; Pettinari, L.; Bassi, R.; Chiriva-Internati, M.; Cobos, E.; Gioia, M.; Pluchino, S. Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol. Rep. 2009, 22, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Lin, J.H.; Nedergaard, M. Adhesive properties of connexin hemichannels. Glia 2008, 56, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Elias, L.A.; Wang, D.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature 2007, 448, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Crespin, S.; Bechberger, J.; Mesnil, M.; Naus, C.C.; Sin, W.C. The carboxy-terminal tail of connexin43 gap junction protein is sufficient to mediate cytoskeleton changes in human glioma cells. J. Cell. Biochem. 2010, 110, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Kameritsch, P.; Wallner, S.; Pohl, U.; Pogoda, K. The carboxyl tail of Cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur. J. Cell Biol. 2010, 89, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.C.; Sin, W.C.; Aftab, Q.; Naus, C.C. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 2007, 55, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Olk, S.; Zoidl, G.; Dermietzel, R. Connexins, cell motility, and the cytoskeleton. Cell Motil. Cytoskelet. 2009, 66, 1000–1016. [Google Scholar] [CrossRef] [PubMed]
- Herve, J.C.; Bourmeyster, N.; Sarrouilhe, D. Diversity in protein-protein interactions of connexins: Emerging roles. Biochim. Biophys. Acta 2004, 1662, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Ponsaerts, R.; de Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; de Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for connexin43-hemichannel activity. FASEB J. 2010, 24, 4378–4395. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Palacios-Prado, N.; Retamal, M.A.; Shoji, K.F.; Martinez, A.D.; Saez, J.C. Connexin hemichannel composition determines the FGF-1-induced membrane permeability and free [Ca2+]i responses. Mol. Biol. Cell 2008, 19, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.A.; Zhou, S.; Bachem, M.G.; Debatin, K.M.; Fulda, S. Identification of a novel switch in the dominant forms of cell adhesion-mediated drug resistance in glioblastoma cells. Oncogene 2008, 27, 5169–5181. [Google Scholar] [CrossRef] [PubMed]
- Aftab, Q.; Sin, W.C.; Naus, C.C. Reduction in gap junction intercellular communication promotes glioma migration. Oncotarget 2015, 6, 11447–11464. [Google Scholar] [CrossRef] [PubMed]
- Baklaushev, V.P.; Yusubalieva, G.M.; Tsitrin, E.B.; Gurina, O.I.; Grinenko, N.P.; Victorov, I.V.; Chekhonin, V.P. Visualization of Connexin43-positive cells of glioma and the periglioma zone by means of intravenously injected monoclonal antibodies. Drug Deliv. 2011, 18, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Caltabiano, R.; Torrisi, A.; Condorelli, D.; Albanese, V.; Lanzafame, S. High levels of connexin43 mRNA in high grade astrocytomas. Study of 32 cases with in situ hybridization. Acta Histochem. 2010, 112, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Jansen, G.H.; Leenstra, S.; Yankaya, B.; Troost, D. Expression of connexin43 and connexin32 gap-junction proteins in epilepsy-associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathol. 2001, 101, 449–459. [Google Scholar] [PubMed]
- Jacobs, V.L.; Valdes, P.A.; Hickey, W.F.; de Leo, J.A. Current review of in vivo GBM rodent models: Emphasis on the CNS-1 tumour model. ASN Neuro 2011, 3, e00063. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, E.W.; Zagzag, D. The Murine GL261 Glioma Experimental Model to Assess Novel Brain Tumor Treatments. In CNS Cancer. Cancer Drug Discovery and Development; Meir, E.G., Ed.; Humana Press: New York, NY, USA, 2009; pp. 227–241. [Google Scholar]
- Rayes, J.; Lax, S.; Wichaiyo, S.; Watson, S.K.; Di, Y.; Lombard, S.; Grygielska, B.; Smith, S.W.; Skordilis, K.; Watson, S.P. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat. Commun. 2017, 8, 2239. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [PubMed]
- Goldberg, G.S.; Lampe, P.D.; Nicholson, B.J. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat. Cell Biol. 1999, 1, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Neijssen, J.; Herberts, C.; Drijfhout, J.W.; Reits, E.; Janssen, L.; Neefjes, J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature 2005, 434, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Polosina, Y.Y.; Miller, H.; Potapova, I.A.; Valiuniene, L.; Doronin, S.; Mathias, R.T.; Robinson, R.B.; Rosen, M.R.; Cohen, I.S.; et al. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J. Physiol. 2005, 568 Pt 2, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Wang, X.; Rogers, T.; Chopp, M. Functional microRNA is transferred between glioma cells. Cancer Res. 2010, 70, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Kizana, E.; Cingolani, E.; Marban, E. Non-cell-autonomous effects of vector-expressed regulatory RNAs in mammalian heart cells. Gene Ther. 2009, 16, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Wolvetang, E.J.; Pera, M.F.; Zuckerman, K.S. Gap junction mediated transport of shRNA between human embryonic stem cells. Biochem. Biophys. Res. Commun. 2007, 363, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.J.; Rameshwar, P. Analysis of the transfer of circulating microRNA between cells mediated by gap junction. Methods Mol. Biol. 2013, 1024, 87–96. [Google Scholar] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Mitra, A.K.; Lengyel, E.; Peter, M.E. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015, 34, 5857–5868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.L.; Sáez, J.C. Metabolic inhibition induces opening of unapposed connexin43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Patel, D.; Ochalski, P.A.; Stelmack, G.L. Connexin30 in rodent, cat and human brain: Selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience 1999, 88, 447–468. [Google Scholar] [CrossRef]
- Gordon, G.R.; Mulligan, S.J.; MacVicar, B.A. Astrocyte control of the cerebrovasculature. Glia 2007, 55, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Koehler, R.C.; Roman, R.J.; Harder, D.R. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009, 32, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, T.M.; Kuhn, B.; Gobel, W.; Huang, W.; Nakai, J.; Helmchen, F.; Flint, J.; Wang, S.S. Radially expanding transglial calcium waves in the intact cerebellum. Proc. Natl. Acad. Sci. USA 2009, 106, 3496–3501. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.; Kovacs-Oller, T.; Sagdullaev, B.T. Vascular pericyte impairment and connexin43 gap junction deficit contribute to vasomotor decline in diabetic retinopathy. J. Neurosci. 2017, 37, 7580–7594. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F. The brain metastatic niche. J. Mol. Med. 2015, 93, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Watabe, K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 2017, 22, 1805–1829. [Google Scholar] [CrossRef]
- Thuringer, D.; Jego, G.; Berthenet, K.; Hammann, A.; Solary, E.; Garrido, C. Gap junction-mediated transfer of miR-145-5p from microvascular endothelial cells to colon cancer cells inhibits angiogenesis. Oncotarget 2016, 7, 28160–28168. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [PubMed]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Tabernero, A.; Medina, J.M. Metabolic trafficking through astrocytic gap junctions. Glia 1997, 21, 114–123. [Google Scholar] [CrossRef]
- Ozog, M.A.; Siushansian, R.; Naus, C.C. Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. J. Neuropathol. Exp. Neurol. 2002, 61, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, Y.; Magistretti, P.J.; Chatton, J.Y. Astrocytes generate Na+-mediated metabolic waves. Proc. Natl. Acad. Sci. USA 2004, 101, 14937–14942. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, A.; Sylantyev, S.; Hadjihambi, A.; Hosford, P.S.; Kasparov, S.; Gourine, A.V. Hemichannel-mediated release of lactate. J. Cereb. Blood Flow Metab. 2016, 36, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cell Combination | Form of Cx | Transmitter or Partner Molecule | Effect on Malignant Behavior and Mechanisms Suggested to Be Occurred in Cancer Cells | Ref. |
|---|---|---|---|---|
| Breast cancer–Osteocyte | HCs | ATP | Released ATP from osteocyte inhibits growth, migration and invasion ability of breast cancer cells | [9] |
| Mesothelioma–Mesothelioma | Cx molecule (C-terminal) | Src, Bax, JNK | Increasing level of Cx43 in malignant mesothelioma cell enhances sensitivity against cisplatin and sunitinib treatment | [11,12,13] |
| Leukemic cell–BMSCs | GJ | - | GJ between Cx43- overexpressed BMSCs and leukemic cells induced apoptosis in leukemic cells due to caspase 3/7 activation | [14,15] |
| Glioblastoma–HMEC | GJ | miR-145-5b | miR-145-5b from HMEC is transferred to glioblastoma (U87 cells) which decrease cancer proliferation | [22] |
| Colon cancer–HMEC | GJ | miR-145-5b | miR-145-5b from HMEC is permitted to be transferred to cancer cells (SW480 cells) and up-regulated Cx43 expression, which inhibits proangiogenic effect of cancer cells | [23] |
| Glioma–Glioma | GJ, Cx molecule (extracellular loop and/or C-terminal) | miR-5096 | GJ between glioma–glioma has anti-invasive effect | [24,25] |
| Glioblastoma–HMEC | GJ | miR-5096 | mir-5096 from glioblastoma is transferred to HMEC increases proangiogenic effect of glioblastoma | [26] |
| Glioma–Glioma | GJ, Cx molecule (C-terminal) | Bcl-2, Bax | Increasing level of Cx43 in glioma cell enhances resistance against temozolomide treatmentIncreasing level of Cx43 in glioma cell enhances resistance against temozolomide treatment | [27] |
| Microglia–Astrocyte | GJ, HCs | IL-1β, TNF-α | Intercellular diffusion of glucose in CNS via GJ composed of Cx43 between astrocytes is downregulated by cytokines secreted from HCs of microglia. Oppositely, when glucose uptake in each astrocyte is increased, it switches the cell to be a reactive astrocyte. | [3] |
| Glioma–Astrocyte, Astrocyte–Astrocyte | GJ, Cx molecule (extracellular loop and/or C-terminal) | miR-5096 | Glioma–astrocyte and astrocyte–astrocyte promotes glioma invasion | [25,28] |
| Glioblastoma–HMEC | GJ | miR-5096 | mir-5096 from glioblastoma (U-87 cells) to HMEC increases proangiogenic effect | [26] |
| Melanoma–Astrocyte | GJ | - | Direct contact with astrocyte up-regulates invasion of cancer cells and drug resistance | [29] |
| Breast cancer–Astrocyte, Lung cancer–Astrocyte | GJ | GJ signaling enhances production of cytokines in cancer cells and endothelin in astrocytes, which in turn upregulate AKT/MAPK signaling in breast cancer (MDA-MB-231) and lung cancer (H226) cells to protect from cytotoxicity of chemotherapeutic drugs | [30] | |
| Breast cancer–Astrocyte, Lung cancer–Astrocyte | GJ | cGAMP | cGAMP from metastasized cancer cells to astrocytes induces STING signaling in astrocytes, which in turn stimulate cancer metastasis | [31] |
| Microglia–Retinal cerebral endothelial cell | HCs | Microgila secretes basigin via activation of PI3K/Akt signaling or IGF signaling that in turn promote angiogenesis in cerebral endothelial cell | [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzu, M.; Sin, W.C.; Shimizu, A.; Sato, H. Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1159. https://doi.org/10.3390/ijms19041159
Uzu M, Sin WC, Shimizu A, Sato H. Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System. International Journal of Molecular Sciences. 2018; 19(4):1159. https://doi.org/10.3390/ijms19041159
Chicago/Turabian StyleUzu, Miaki, Wun Chey Sin, Ayaka Shimizu, and Hiromi Sato. 2018. "Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System" International Journal of Molecular Sciences 19, no. 4: 1159. https://doi.org/10.3390/ijms19041159
APA StyleUzu, M., Sin, W. C., Shimizu, A., & Sato, H. (2018). Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System. International Journal of Molecular Sciences, 19(4), 1159. https://doi.org/10.3390/ijms19041159