Mesenchymal Stromal Cells: Emerging Roles in Bone Metastasis


Abstract
1. Introduction
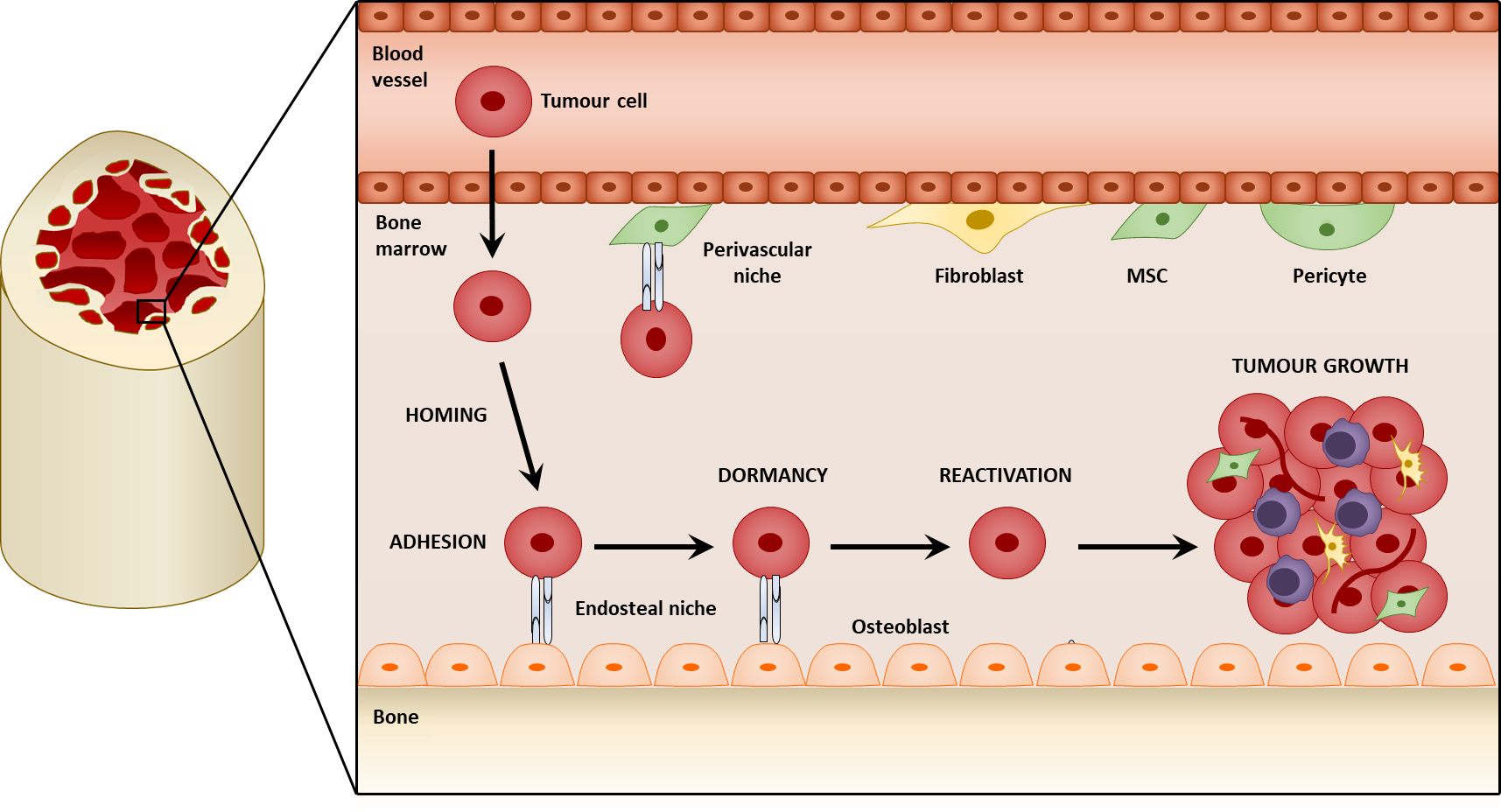
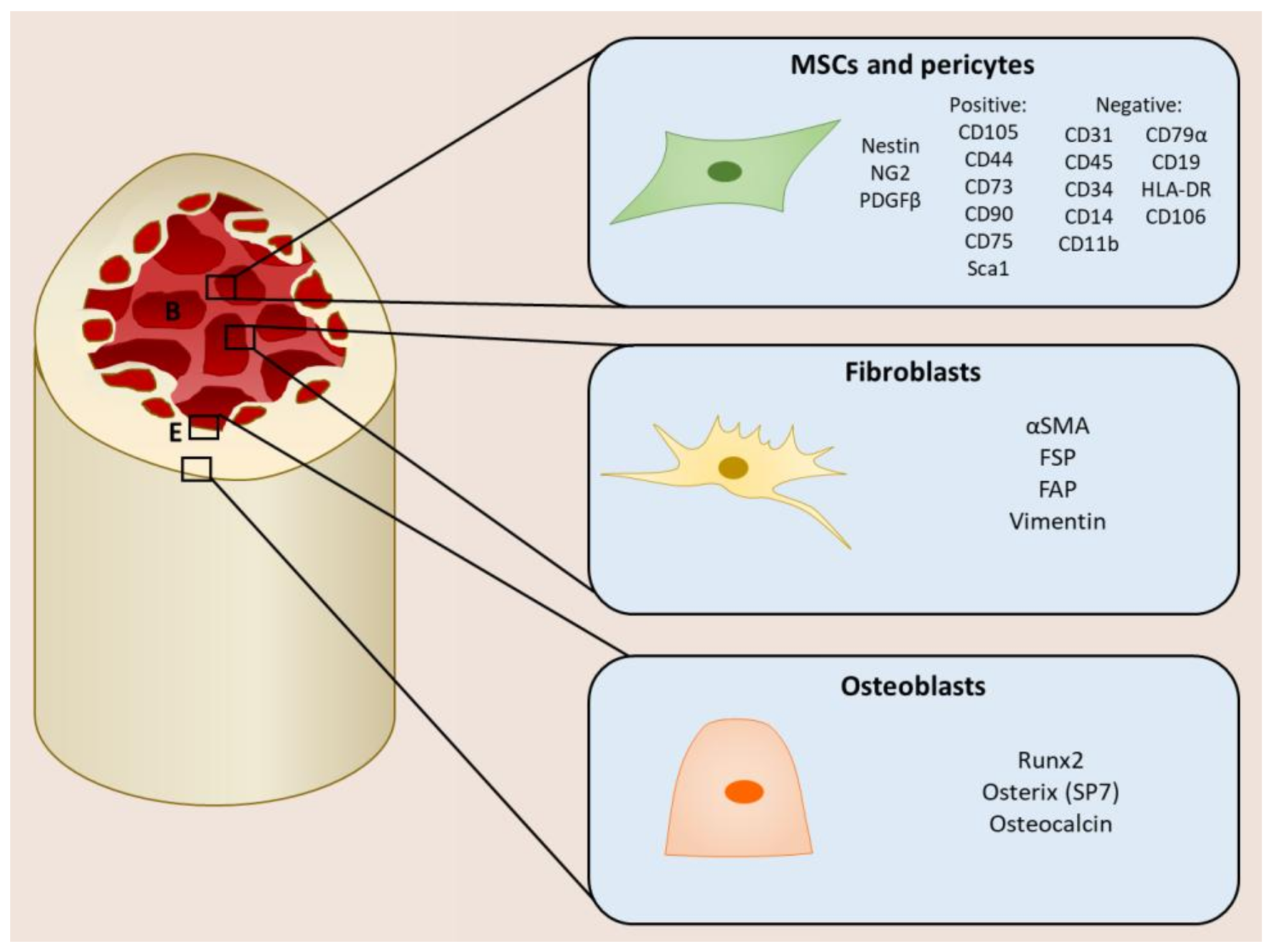
2. Mesenchymal Stromal Cells within the Tumor Microenvironment
- Adherence to plastic;
- Ability to self-renew;
- Ability to differentiate into osteoblasts, chrondocytes, and adipocytes;
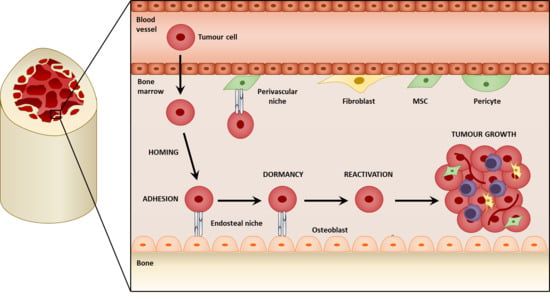
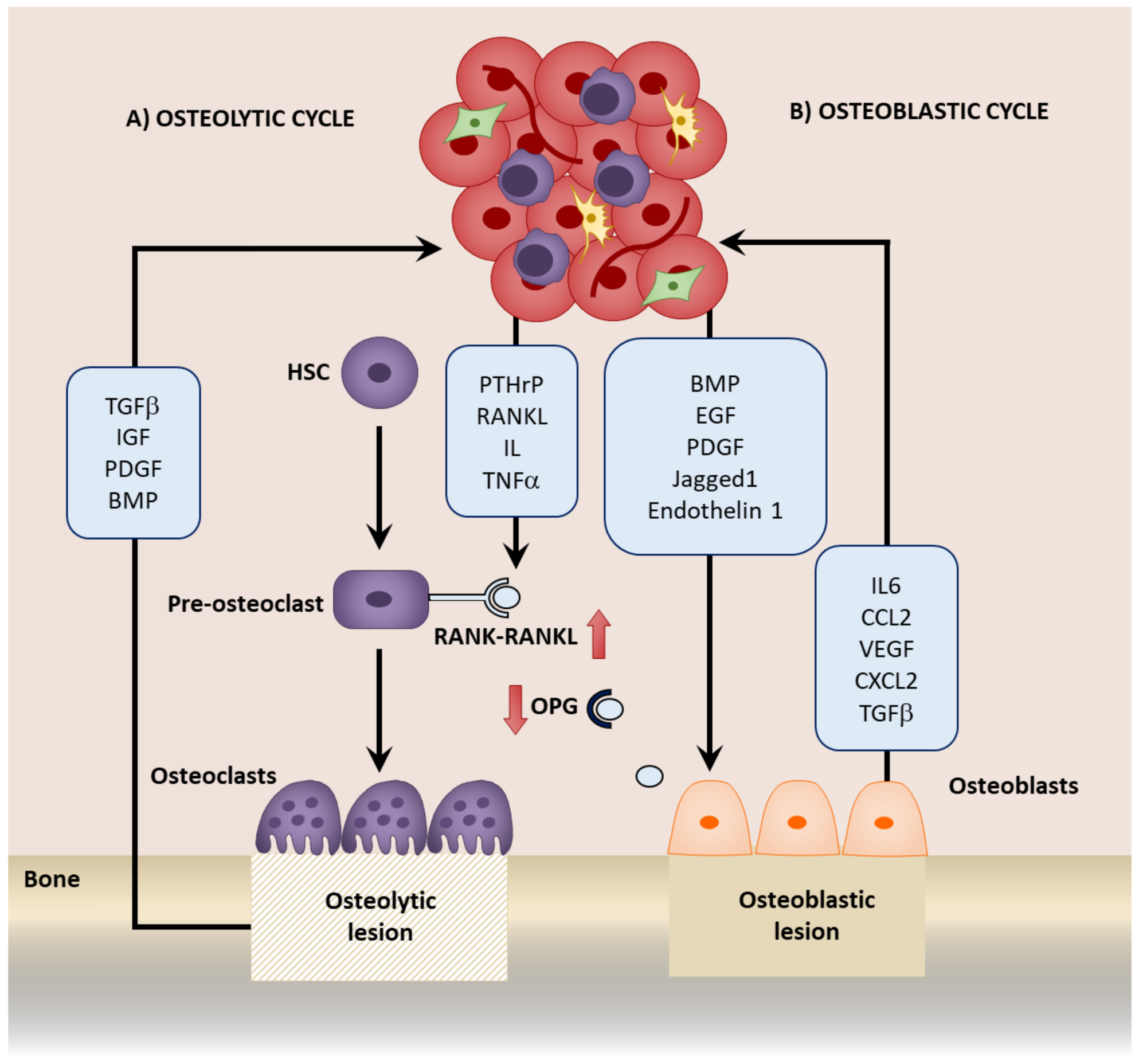
3. Mesenchymal Stromal Cells in the Colonization of the Bone
3.1. Metastatic Niches within the Bone
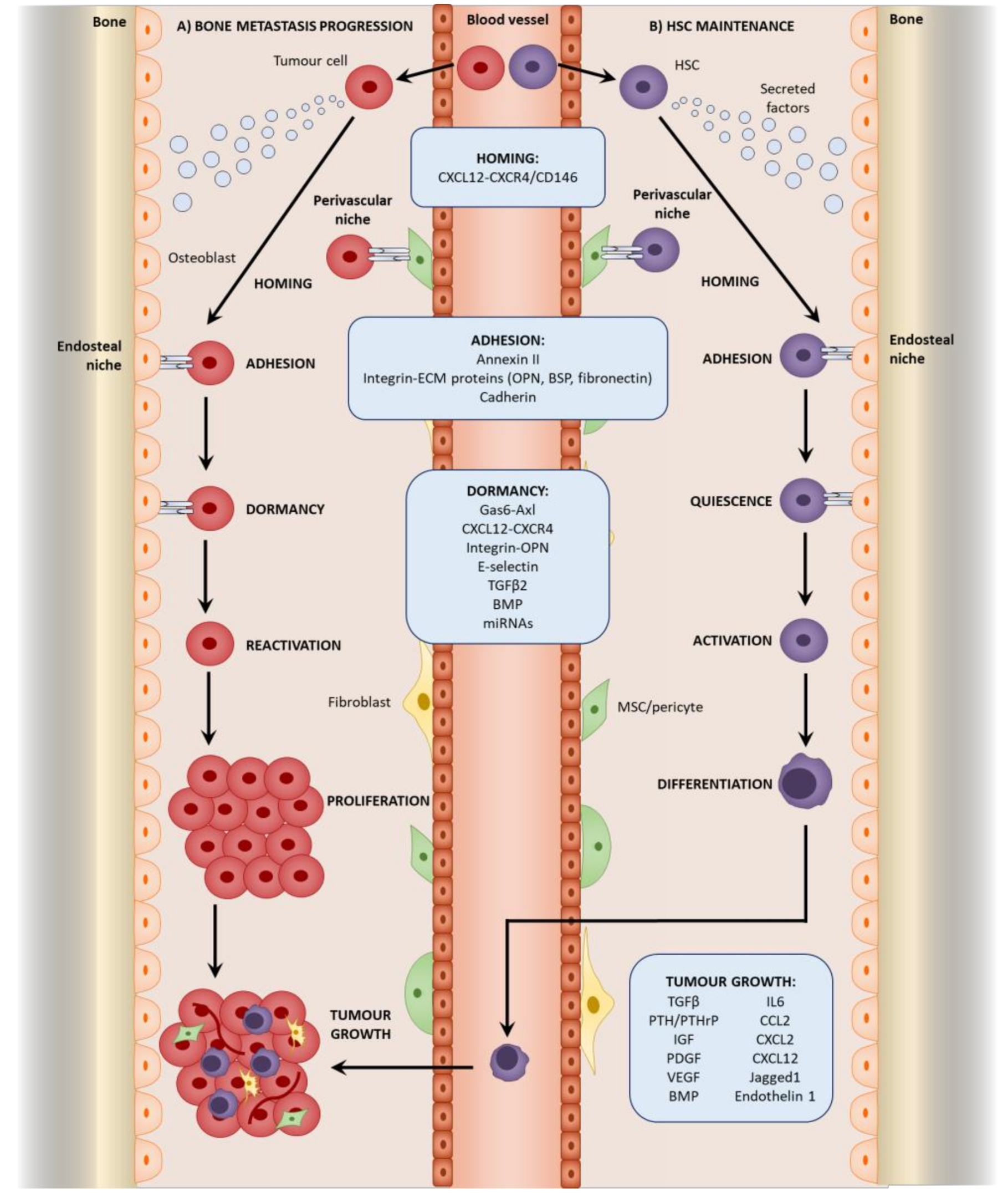
3.2. Homing to the Niche
3.3. Direct Interactions with the HSC Niche
4. Mesenchymal Stromal Cells in Tumor Cell Dormancy
4.1. Dormancy at the Endosteal Niche
4.2. Reactivation from Dormancy
5. Mesenchymal Stromal Cells in Bone Metastatic Growth
6. Mesenchymal Stromal Cells in Treatment Resistance
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| αSMA | alpha-smooth muscle actin |
| BDNF | brain-derived neurotrophic factor |
| BMP | bone morphogenetic protein |
| BSP | bone sialoprotein |
| CAF | cancer-associated fibroblasts |
| CCL | C-C motif chemokine |
| CTC | circulating tumor cell |
| CXCL | C-X-C motif chemokine 12 |
| CXCR | C-X-C chemokine receptor |
| DDR | discoidin domain receptor family member 1 |
| DKK1 | dickkopf-related protein 1 |
| DTC | disseminated tumor cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EPO | erythropoietin |
| ERK | extracellular receptor kinase |
| FAK | focal adhesion kinase |
| FAP | fibroblast-activated protein |
| FGF | fibroblast growth factor |
| FSP | fibroblast specific protein (S100A4) |
| Gas6 | growth arrest specific 6 |
| HBGF | heparin-binding growth factor |
| HGF | hepatocyte growth factor |
| HIFα | hypoxia-inducible factor |
| HPC | hematopoietic progenitor cell |
| HSC | hematopoietic stem cell |
| ICAM1 | intercellular adhesion molecule 1 |
| IGF | insulin-like growth factor |
| IL | interleukin |
| MAPK | mitogen-activated protein kinase |
| MARCKS | myristolyated alanine-rich C-kinase substrate |
| miRNA | microRNA |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stem cell |
| NF-kB | nuclear factor kB |
| NG2 | neuron-glial antigen 2 (CSPG4) |
| OPG | osteoprotegerin |
| OPN | osteopontin |
| PDGF | platelet-derived growth factor |
| PDGFRβ | platelet-derived growth factor receptor beta |
| PI3K | phosphoinositide 3-kinase |
| PTH | parathyroid hormone |
| PTHrP | parathyroid hormone-related protein |
| RANK/RANKL | receptor activator of nuclear factor kappa-B/ligand |
| Runx2 | runt-related transcription factor 2 |
| sRAGE | soluble receptor for advanced glycation end products |
| SOD2 | superoxide dismutase 2 |
| TGFβ | transforming growth factor beta |
| TME | tumor microenvironment |
| TNFα | tumor necrosis factor alpha |
| VCAM1 | vascular cell adhesion protein 1 |
| VEGF | vascular endothelial growth factor |
References
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. The role of bisphosphonates in breast cancer. Breast 2004, 13, S19–S28. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. Homing of Cancer Cells to the Bone. Cancer Microenviron. 2011, 4, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Maxson, S.; Lopez, E.A.; Yoo, D.; Danilkovitch-Miagkova, A.; LeRoux, M.A. Concise Review: Role of Mesenchymal Stem Cells in Wound Repair. Stem Cells Transl. Med. 2012, 1, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zeng, X.; Hu, J.; Chen, Q. Characterization of Nestin, a Selective Marker for Bone Marrow Derived Mesenchymal Stem Cells. Stem Cells Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The Essential Functions of Adipo-osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Rudland, P.S.; Platt-Higgins, A.; Renshaw, C.; West, C.R.; Winstanley, J.H.R.; Robertson, L.; Roger, B. Prognostic Significance of the Metastasis-inducing Protein S100A4 (p9Ka) in Human Breast Cancer. Cancer Res. 2000, 60, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers (Basel) 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Hsieh, C.; Law, A.; Zhau, H.E.; Pathak, S.; Multani, A.S.; Lim, S.; Coleman, I.M.; Wu, L.; Figg, W.D.; et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: Implications for cancer growth and metastasis. Cancer Res. 2008, 68, 9996–10003. [Google Scholar] [CrossRef] [PubMed]
- Connell, J.T.O.; Sugimoto, H.; Cooke, V.G.; Macdonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ Stromal Cells Are Important for Metastatic Colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; Crombrugghe, B. De The Novel Zinc Finger-Containing Transcription Factor Osterix Is Required for Osteoblast Differentiation and Bone Formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.X.; Aeran, R.; Liao, W.; Lu, M.; Polovin, G.; Pone, E.J.; Zhao, W. Exogenous marker-engineered mesenchymal stem cells detect cancer and metastases in a simple blood assay. Stem Cell Res. Ther. 2015, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Battula, L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct Evidence of Mesenchymal Stem Cell Tropism for Tumor and Wounding Microenvironments using In Vivo Bioluminescence Imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, F.; De, A.; Cao, Y.; Contag, C.; Gambhir, S.S.; Wu, J.C.; Chen, X. Trafficking mesenchymal stem cell engraftment and differentiation in tumor-bearing mice by bioluminescence imaging. Stem Cells 2009, 27, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Mi, Z.; Bhattacharya, S.D.; Kim, V.M.; Guo, H.; Talbotq, L.J.; Kuo, P.C. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 2011, 32, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.L.; LaMarca, H.L.; Tomchuck, S.L.; Honer zu Bentrup, K.; Danka, E.S.; Henkle, S.L.; et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, J.; Everett, A.D.; Clevenger, C.V.; Koneru, M.; Mishra, J.; Kamen, B.; Banerjee, D.; Glod, J. The isolation of novel mesenchymal stromal cell chemotactic factors from the conditioned medium of tumor cells. October 2008, 314, 3107–3117. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, S.E.; Lennard, T.W.J.; Williams, J.R.; Birch, M.A. Vascular endothelial growth factor acts as an osteolytic factor in breast cancer metastases to bone. Br. J. Cancer 2005, 92, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Chen, J.; Narizhneva, N.V.; Heston, W.; Brainard, J.; Sage, E.H.; Byzova, T.V. Molecular pathway for cancer metastasis to bone. J. Biol. Chem. 2003, 278, 39044–39050. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.; Chen, J.; Guo, Y.; Chen, Z.W.; Cai, J. Migration mechanism of mesenchymal stem cells studied by QD/NSOM. Biochim. Biophys. Acta Biomembr. 2015, 1848, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.F.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Ringe, J.; Strassburg, S.; Neumann, K.; Endres, M.; Notter, M.; Burmester, G.-R.; Kaps, C.; Sittinger, M. Towards in situ tissue repair: Human mesenchymal stem cells express chemokine receptors CXCR1, CXCR2 and CCR2, and migrate upon stimulation with CXCL8 but not CCL2. J. Cell. Biochem. 2007, 101, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Mohyeldin, A.; Garzón-Muvdi, T.; Quiñones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Mauch, P.; Vergilio, J.-A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Barbash, I.M.; Chouraqui, P.; Baron, J.; Feinberg, M.S.; Etzion, S.; Tessone, A.; Miller, L.; Guetta, E.; Zipori, D.; Kedes, L.H.; et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation 2003, 108, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Rattigan, Y.; Hsu, J.M.; Mishra, P.J.; Glod, J.; Banerjee, D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp. Cell Res. 2010, 316, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef] [PubMed]
- Rosová, I.; Dao, M.; Capoccia, B.; Link, D.; Nolta, J.A. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells 2008, 26, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- EL-Attar, H.; Sheta, M. Hepatocyte growth factor profile with breast cancer. Indian J. Pathol. Microbiol. 2011, 54, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Sheen-Chen, S.-M.; Liu, Y.-W.; Eng, H.-L.; Choi, F.-F. Serum levels of hepatocyte growth factor in patients with breast cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. HGF-MET as a breast cancer biomarker. Aging 2015, 7, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Minuti, G.; Landi, L. MET deregulation in breast cancer. Ann. Transl. Med. 2015, 3, 181. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Hirohashi, Y.; Torigoe, T.; Nojima, M.; Inoue, R.; Kitamura, H.; Tanaka, T.; Asanuma, H.; Sato, N.; Masumori, N. Expression of hepatocyte growth factor in prostate cancer may indicate a biochemical recurrence after radical prostatectomy. Anticancer Res. 2015, 35, 413–418. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Menon, L.G.; Picinich, S.; Koneru, R.; Gao, H.; Lin, S.Y.; Koneru, M.; Mayer-Kuckuk, P.; Glod, J.; Banerjee, D. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells 2007, 25, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.F.; Huang, Y.; Han, Y.Y.; Lin, L.Y.; Sun, W.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF high neutrophils. Science 2017, 358, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-Associated Fibroblast-Like Differentiation of Human Mesenchymal Stem Cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J. Pericyte—Fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.-M.; Vessella, R.L.; Morrissey, C. The role of the microenvironment-dormant prostate disseminated tumor cells in the bone marrow. Drug Discov. Today Technol. 2014, 11, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Pantel, K.; Müller, P.; Janni, W.; Hepp, F.; Kentenich, C.R.M.; Gastroph, S.; Wischnik, A.; Dimpfl, T.; Kindermann, G.; et al. Cytokeratin-Positive Cells in the Bone Marrow and Survival of Patients with Stage I, II, or III Breast Cancer. N. Engl. J. Med. 2000, 342, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone marrow cells in the “pre-metastatic niche”: Within bone and beyond. Cancer Metastasis Rev. 2006, 25, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; Macdonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk. Lymphoma 2002, 43, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Golan, K.; Kollet, O.; Lapidot, T. Dynamic cross talk between S1P and CXCL12 regulates hematopoietic stem cells migration, development and bone remodeling. Pharmaceuticals 2013, 6, 1145–1169. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Reilly, M.J.; Emerson, S.G. Osteoblasts and The Hematopoietic Microenvironment. Hematology 2000, 4, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Ema, H.; Suda, T. Two anatomically distinct niches regulate stem cell activity. Blood 2012, 120, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Neiva, K.; Sun, Y.-X.; Taichman, R.S. The role of osteoblasts in regulating hematopoietic stem cell activity and tumor metastasis. Braz. J. Med. Biol. Res. 2005, 38, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.T.; Holen, I.; Dear, T.N.; Hunter, K.; Brown, H.K. Modifying the osteoblastic niche with zoledronic acid in vivo-Potential implications for breast cancer bone metastasis. Bone 2014, 66, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, L.; Goldstein, A.; Liu, J.; Lo, H.-C.; Sheng, K.; Welte, T.; Wong, S.T.C.; Gugala, Z.; Stossi, F.; et al. Bone-in-culture array as a platform to model early-stage bone metastases and discover anti-metastasis therapies. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The Osteogenic Niche Promotes Early-Stage Bone Colonization of Disseminated Breast Cancer Cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-initiated changes in the bone metastatic niche development. Cell Rep. 2016, 14, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Correa, D.; Somoza, R.A.; Lin, P.; Schiemann, W.P.; Caplan, A.I. Mesenchymal stem cells regulate melanoma cancer cells extravasation to bone and liver at their perivascular niche. Int. J. Cancer 2016, 138, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Keller, J.; Zhang, J.; Lu, Y.; Yao, Z.; Keller, E.T. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 2005, 65, 8274–8285. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Shiozawa, Y.; Jung, Y.; Kim, J.K.; Pedersen, E.; Mishra, A.; Zalucha, J.L.; Wang, J.; Keller, E.T.; Pienta, K.J.; et al. Disseminated Prostate Cancer Cells Can Instruct Hematopoietic Stem and Progenitor Cells to Regulate Bone Phenotype. Mol. Cancer Res. 2012, 10, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Ottewell, P.D.; Evans, C.A.; Holen, I. Location matters: Osteoblast and osteoclast distribution is modified by the presence and proximity to breast cancer cells in vivo. Clin. Exp. Metastasis 2012, 29, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Kitagawa, Y.; Zhang, J.; Yao, Z.; Mizokami, A.; Cheng, S.; Nör, J.; McCauley, L.K.; Taichman, R.S.; Keller, E.T. Vascular Endothelial Growth Factor Contributes to the Prostate Cancer-Induced Osteoblast Differentiation Mediated by Bone Morphogenetic Protein. Cancer Res. 2004, 64, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Dai, J.; Zhang, J.; Keller, J.M.; Nor, J.; Yao, Z.; Keller, E.T. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005, 65, 10921–10929. [Google Scholar] [CrossRef] [PubMed]
- Van der Pluijm, G.; Sijmons, B.; Vloedgraven, H.; Deckers, M.; Papapoulos, S.; Löwik, C. Monitoring metastatic behavior of human tumor cells in mice with species-specific polymerase chain reaction: Elevated expression of angiogenesis and bone resorption stimulators by breast cancer in bone metastases. J. Bone Miner. Res. 2001, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, M.; Toullec, A.; Buteau-Lozano, H.; Abdelkarim, M.; Vacher, S.; Velasco, G.; Christofari, M.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. MDA-MB-231 breast cancer cells overexpressing single VEGF isoforms display distinct colonisation characteristics. Br. J. Cancer 2015, 113, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Fierabracci, P.; Pinchera, A.; Miccoli, P.; Conte, P.F.; Vignali, E.; Zaccagnini, M.; Marcocci, C.; Giani, C. Increased prevalence of primary hyperparathyroidism in treated breast cancer. J. Endocrinol. Investig. 2001, 24, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Loberg, R.; Liao, J.; Ying, C.; Snyder, L.A.; Pienta, K.J.; McCauley, L.K. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009, 69, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.; Rettig, M.P.; Uy, G.L.; Deych, E.; Holt, M.S.; Ritchey, J.K.; DiPersio, J.F. BIO5192, a small molecule inhibitor of VLA-4, mobilizes hematopoietic stem and progenitor cells. Blood 2009, 114, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulou, T.; Priestley, G.V.; Nakamoto, B.; Zafiropoulos, V.; Scott, L.M.; Harlan, J.M. Synergistic mobilization of hemopoietic progenitor cells using concurrent beta1 and beta2 integrin blockade or beta2-deficient mice. Blood 2001, 97, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Priestley, G.V.; Papayannopoulou, T. Deletion of α 4 Integrins from Adult Hematopoietic Cells Reveals Roles in Homeostasis, Regeneration, and Homing. Mol. Cell. Biol. 2003, 23, 9349–9360. [Google Scholar] [CrossRef] [PubMed]
- Craddock, C.F.; Nakamoto, B.; Andrews, R.G.; Priestley, G.V.; Papayannopoulou, T. Antibodies to VLA4 integrin mobilize long-term repopulating cells and augment cytokine-induced mobilization in primates and mice. Blood 1997, 90, 4779–4788. [Google Scholar] [PubMed]
- Sun, Y.-X.; Fang, M.; Wang, J.; Cooper, C.R.; Pienta, K.J.; Taichman, R.S. Expression and Activation of αvβ3 Integrins by SDF-1/CXC12 Increases the Aggressiveness of Prostate Cancer Cells. Prostate 2007, 67, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Wang, J.; Schneider, A.; Sun, Y.X.; Koh-Paige, A.J.; Osman, N.I.; McCauley, L.K.; Taichman, R.S. Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 2006, 38, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.A.; Cher, M.L.; Zhou, Z.; Mullins, C.; Bhagat, S.; Trikha, M. Inhibition of αvβ3 integrin reduces angiogenesis, bone turnover, and tumor cell proliferation in experimental prostate cancer bone metastases. Clin. Exp. Metastasis 2003, 20, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grünewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumor dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Taichman, R.S. GAS6/Mer axis regulates the homing and survival of the E2A/PBX1-positive B-cell precursor acute lymphoblastic leukemia in the bone marrow niche. Exp. Hematol. 2010, 38, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Lymphoid Neoplasia 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.; Azab, F.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; Alt, C.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Bodenstine, T.M.; Vaidya, K.S.; Ismail, A.; Beck, B.H.; Cook, L.M.; Diers, A.R.; Landar, A.; Welch, D.R. Homotypic gap junctional communication associated with metastasis suppression increases with PKA activity and is unaffected by PI3K inhibition. Cancer Res. 2010, 70, 10002–10011. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.-U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Juárez, P.; Guise, T.A. TGF-beta in cancer and bone: Implications for treatment of bone metastases. Bone 2011, 48, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Krzeszinski, J.Y.; Wan, Y. New therapeutic targets for cancer bone metastasis. Trends Pharmacol. Sci. 2015, 36, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Carducci, M.A.; Jimeno, A. Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin. Cancer Res. 2006, 12, 6296s–6300s. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chiao, J.W.; Moonga, B.S.; Yang, Y.M.; Kancherla, R.; Mittelman, A.; Wu-Wong, J.R.; Ahmed, T. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br. J. Cancer 2000, 83, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Á.; Merino, M.; Zamora, P.; Redondo, A.; Castelo, B.; Espinosa, E. Targeting the endothelin axis in prostate carcinoma. Tumor Biol. 2012, 33, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Gunn, W.G.; Conley, A.; Deininger, L.; Olson, S.D.; Prockop, D.J.; Gregory, C.A. A Crosstalk Between Myeloma Cells and Marrow Stromal Cells Stimulates Production of DKK1 and Interleukin-6: A Potential Role in the Development of Lytic Bone Disease and Tumor Progression in Multiple Myeloma. Stem Cells 2006, 24, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Patel, H.R.H.; McGurk, C.; Tatoud, R.; Klocker, H.; Masters, J.; Williamson, M. The importance of the CXCL12-CXCR4 chemokine ligand-receptor interaction in prostate cancer metastasis. J. Exp. Ther. Oncol. 2004, 4, 291–303. [Google Scholar] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; Mccauley, L.K. Use of the Stromal Cell-derived Factor-1/CXCR4 Pathway in Prostate Cancer Metastasis to Bone Use of the Stromal Cell-derived Factor-1/CXCR4 Pathway in Prostate Cancer Metastasis to Bone 1. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Corcoran, K.E.; Trzaska, K.A.; Fernandes, H.; Bryan, M.; Taborga, M.; Srinivas, V.; Packman, K.; Patel, P.S.; Rameshwar, P. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS ONE 2008, 3, e2563. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Kollet, O.; Ponomaryov, T.; Petit, I.; Franitza, S.; Grabovsky, V.; Slav, M.M.; Nagler, A.; Lider, O.; Alon, R.; et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34+ cells: Role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 2000, 95, 3289–3296. [Google Scholar] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Sun, Y.; Song, W.; Nor, J.E.; Wang, C.Y.; Taichman, R.S. Diverse signaling pathways through the SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to altered patterns of cytokine secretion and angiogenesis. Cell. Signal. 2005, 17, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2005, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Wang, J.; Shelburne, C.E.; Lopatin, D.E.; Chinnaiyan, A.M.; Rubin, M.A.; Pienta, K.J.; Taichman, R.S. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J. Cell. Biochem. 2003, 89, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Salvucci, O.; Bouchard, A.; Baccarelli, A.; Deschenes, J.; Sauter, G.; Simon, R.; Bianchi, R.; Basik, M. The role of CXCR4 receptor expression in breast cancer: A large tissue microarray study. Breast Cancer Res. Treat. 2006, 97, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kitayama, J.; Kazama, S.; Nagawa, H. Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 2003, 5, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Petit, I.; Kollet, O.; Magid, M.; Ponomaryov, T.; Byk, T.; Nagler, A.; Ben-Hur, H.; Many, A.; Shultz, L.; et al. Dependence of Human Stem Cell Engraftment and Repopulation of NOD/SCID Mice on CXCR4. Science 1999, 283, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.M.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Miner. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shiozawa, Y.; Wang, J.; Wang, Y.; Jung, Y.; Pienta, K.J.; Mehra, R.; Loberg, R.; Taichman, R.S. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 2008, 283, 4283–4294. [Google Scholar] [CrossRef] [PubMed]
- Templeton, Z.S.; Lie, W.-R.; Wang, W.; Rosenberg-Hasson, Y.; Alluri, R.V.; Tamaresis, J.S.; Bachmann, M.H.; Lee, K.; Maloney, W.J.; Contag, C.H.; et al. Breast Cancer Cell Colonization of the Human Bone Marrow Adipose Tissue Niche. Neoplasia 2015, 17, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Honoki, K.; Fuji, H.; Tohma, Y.; Kido, A.; Mori, T.; Tsujiuchi, T.; Tanaka, Y. Mesenchymal stem cells promote tumor engraftment and metastatic colonization in rat osteosarcoma model. Int. J. Oncol. 2012, 40, 163–169. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; McGrail, D.J.; Ravikumar, N.; Dawson, M.R. Mesenchymal Stem Cells Induce Directional Migration of Invasive Breast Cancer Cells through TGF-β. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhao, S.; Song, B.; Wei, Z.; Lu, G.; Zhou, J.; Huo, T. Effects of transforming growth factor β-1 infected human bone marrow mesenchymal stem cells on high- and low-metastatic potential hepatocellular carcinoma. Eur. J. Med. Res. 2015, 20, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Swamydas, M.; Ricci, K.; Rego, S.L.; Dréau, D. Mesenchymal stem cell-derived CCL-9 and CCL-5 promote mammary tumor cell invasion and the activation of matrix metalloproteinases. Cell Adhes. Migr. 2013, 7, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Salo, S.; Bitu, C.; Merkku, K.; Nyberg, P.; Bello, I.O.; Vuoristo, J.; Sutinen, M.; Vähänikkilä, H.; Costea, D.E.; Kauppila, J.; et al. Human Bone Marrow Mesenchymal Stem Cells Induce Collagen Production and Tongue Cancer Invasion. PLoS ONE 2013, 8, e77692. [Google Scholar] [CrossRef]
- Xu, W.; Bian, Z.; Fan, Q.; Li, G.; Tang, T. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. 2009, 281, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Dong, L.; Yan, K.; Long, H.; Yang, T.-T.; Dong, M.-Q.; Zhou, Y.; Fan, Q.-Y.; Ma, B.-A. CXCR4-mediated osteosarcoma growth and pulmonary metastasis is promoted by mesenchymal stem cells through VEGF. Oncol. Rep. 2013, 30, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, Y.; Yang, J.; Zhang, X.; Zhang, H.; Zhang, T.; Zhao, S.; Zheng, P.; Huo, J.; Wu, H. Gastric cancer-derived mesenchymal stem cells prompt gastric cancer progression through secretion of interleukin-8. J. Exp. Clin. Cancer Res. 2015, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Touboul, C.; Lis, R.; Al Farsi, H.; Raynaud, C.M.; Warfa, M.; Althawadi, H.; Mery, E.; Mirshahi, M.; Rafii, A. Mesenchymal stem cells enhance ovarian cancer cell infiltration through IL6 secretion in an amniochorionic membrane based 3D model. J. Transl. Med. 2013, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, R.A.; Gálvez, B.G.; Longo, N.; Baleux, F.; van Muijen, G.N.P.; Sanchez-Mateos, P.; Arroyo, A.G.; Teixido, J. Stromal Cell-Derived Factor-1 α Promotes Melanoma Cell Invasion across Basement Membranes Involving Stimulation of Membrane-Type 1 Matrix Metalloproteinase and Rho GTPase Activities. Cancer Res. 2004, 64, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Contag, C.H.; Lie, W.-R.; Bammer, M.C.; Hardy, J.W.; Schmidt, T.L.; Maloney, W.J.; King, B.L. Monitoring Dynamic Interactions Between Breast Cancer Cells and Human Bone Tissue in a Co-culture Model. Mol. Imaging Biol. 2014, 16, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, J.; Xu, Y.; Koch, A.E.; Cai, Z.; Chen, X.; Galson, D.L.; Taichman, R.S.; Zhang, J. CXCL16 Functions as a Novel Chemotactic Factor for Prostate Cancer Cells In vitro. Mol. Cancer Res. 2008, 6, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.K.; Lee, W.; Park, H.J.; Lee, S.D.; Lee, J.Z.; Chung, M.K. Clinical significance of CXCL16/CXCR6 expression in patients with prostate cancer. Mol. Med. Rep. 2011, 4, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhen, X.; Xiong, B.; Wang, B.; Zhang, W.; Zhou, W. CXCR6 is expressed in human prostate cancer in vivo and is involved in the in vitro invasion of PC3 and LNCap cells. Cancer Sci. 2008, 99, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.K.; Anderson, R.L. Genes involved in breast cancer metastasis to bone. Cell. Mol. Life Sci. 2002, 59, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, C.M. Adhesion receptors as regulators of the hematopoietic process. Blood 1998, 92, 2609–2612. [Google Scholar] [PubMed]
- Jung, Y.; Wang, J.; Song, J.; Shiozawa, Y.; Wang, J.; Havens, A.; Wang, Z.; Sun, Y.; Emerson, S.G.; Krebsbach, P.H.; et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 2007, 110, 82–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.R.; Koltowski, L.; Ownbey, R.T.; Tuszynski, G.P.; Sharma, M.C. Angiogenesis-associated protein annexin II in breast cancer: Selective expression in invasive breast cancer and contribution to tumor invasion and progression. Exp. Mol. Pathol. 2006, 81, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Emoto, K.; Yamada, Y.; Sawada, H.; Fujimoto, H.; Ueno, M.; Takayama, T.; Kamada, K.; Naito, A.; Hirao, S.; Nakajima, Y. Annexin II overexpression correlates with stromal tenascin-C overexpression: A prognostic marker in colorectal carcinoma. Cancer 2001, 92, 1419–1426. [Google Scholar] [CrossRef]
- Cole, S.P.; Pinkoski, M.J.; Bhardwaj, G.; Deeley, R.G. Elevated expression of annexin II (lipocortin II, p36) in a multidrug resistant small cell lung cancer cell line. Br. J. Cancer 1992, 65, 498–502. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chuthapisith, S.; Bean, B.E.; Cowley, G.; Eremin, J.M.; Samphao, S.; Layfield, R.; Kerr, I.D.; Wiseman, J.; El-Sheemy, M.; Sreenivasan, T.; et al. Annexins in human breast cancer: Possible predictors of pathological response to neoadjuvant chemotherapy. Eur. J. Cancer 2009, 45, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Vishwanatha, J.K.; Chiang, Y.; Kumble, K.D.; Hollingsworth, M.A.; Pour, P.M. Enhanced expression of annexin II in human pancreatic carcinoma cells and primary pancreatic cancers. Carcinogenesis 1993, 14, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Shiozawa, Y.; Wang, J.; Patel, L.R.; Havens, A.M.; Song, J.; Krebsbach, P.H.; Roodman, G.D.; Taichman, R.S. Annexin-2 is a regulator of stromal cell-derived factor-1/CXCL12 function in the hematopoietic stem cell endosteal niche. Exp. Hematol. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Radice, G.L.; Morrison, S.J. Lack of Evidence that Hematopoietic Stem Cells Depend on N-Cadherin-Mediated Adhesion to Osteoblasts for Their Maintenance. Cell Stem Cell 2007, 1, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Acar, M.; Radice, G.L.; Morrison, S.J. Hematopoietic Stem Cells Do Not Depend on N-Cadherin to Regulate Their Maintenance. Cell Stem Cell 2009, 4, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Wein, F.; Pietsch, L.; Saffrich, R.; Wuchter, P.; Walenda, T.; Bork, S.; Horn, P.; Diehlmann, A.; Eckstein, V.; Ho, A.D.; et al. N-Cadherin is expressed on human hematopoietic progenitor cells and mediates interaction with human mesenchymal stromal cells. Stem Cell Res. 2010, 4, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Matsugaki, A.; Sekita, A.; Nakano, T. Alteration of osteoblast arrangement via direct attack by cancer cells: New insights into bone metastasis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tamura, D.; Hiraga, T.; Myoui, A.; Yoshikawa, H.; Yoneda, T. Cadherin-11-mediated interactions with bone marrow stromal/osteoblastic cells support selective colonization of breast cancer cells in bone. Int. J. Oncol. 2008, 33, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lira, C.; Chu, K.; Bilen, M.A.; Lee, Y.; Ye, X.; Kim, S.M.; Ortiz, A.; Wu, F.L.; Logothetis, C.J.; et al. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res. 2010, 70, 4580–4589. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.; Cheng, C.J.; Ye, X.; Lee, Y.C.; Zurita, A.J.; Chen, D.T.; Yu-Lee, L.Y.; Zhang, S.; Yeh, E.T.; Hu, M.C.; et al. Cadherin-11 Promotes the Metastasis of Prostate Cancer Cells to Bone. Mol. Cancer Res. 2008, 6, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Fearon, E.R. Cadherin and catenin alterations in human cancer. Genes Chromosom. Cancer 2002, 34, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Lu, J.T.; Tan, C.C.; Wang, Q.S.; Feng, Y.M. RUNX2 promotes breast cancer bone metastasis by increasing integrin α5-mediated colonization. Cancer Lett. 2016, 380, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Rittling, S.R. Mammary tumor development in MMTV-c-myc/MMTV-v-Ha-ras transgenic mice is unaffected by osteopontin deficiency. Breast Cancer Res. Treat. 2000, 63, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Fedarko, N.S.; Jain, A.; Karadag, A.; Van Eman, M.R.; Fisher, L.W. Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin. Cancer Res. 2001, 7, 4060–4066. [Google Scholar] [PubMed]
- Tuck, A.B.; Chambers, A.F.; Allan, A.L. Osteopontin overexpression in breast cancer: Knowledge gained and possible implications for clinical management. J. Cell. Biochem. 2007, 102, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Singhal, H.; Bautista, D.S.; Tonkin, K.S.; O’Malley, F.P.; Tuck, A.B.; Chambers, A.F.; Harris, J.F. Elevated Associated Decreased Plasma with Survival Osteopontin Increased in Metastatic Tumor Burden Breast and Cancer groups. Clin. Cancer Res. 1997, 3, 605–611. [Google Scholar] [PubMed]
- Rudland, P.S.; Platt-higgins, A.; El-tanani, M.; Rudland, S.D.S.; Barraclough, R.; Winstanley, J.H.R.; Howitt, R.; West, C.R. Prognostic Significance of the Metastasis-associated Protein Osteopontin in Human Breast Cancer Prognostic Significance of the Metastasis-associated Protein Osteopontin in Human. Cancer Res. 2002, 62, 3417–3427. [Google Scholar] [PubMed]
- Denhardt, D.T.; Noda, M.; O’Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Investig. 2001, 107, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Tuck, A.B.; Arsenault, D.M.; O’Malley, F.P.; Hota, C.; Ling, M.C.; Wilson, S.M.; Chambers, A.F. Osteopontin induces increased invasiveness and plasminogen activator expression of human mammary epithelial cells. Oncogene 1999, 18, 4237–4246. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liaw, L.; Skinner, M.P.; Raines, E.W.; Ross, R.; Cheresh, D.A.; Schwartz, S.M.; Giachelli, C.M. Adhesive and Migratory Effects of Osteopontin Are Mediated via Distinct Cell-Surface Integrins—Role of αvβ3 in Smooth-Muscle Cell-Migration to Osteopontin in-vitro. J. Clin. Investig. 1995, 95, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.D.; Lin, E.C.; Kovach, N.L.; Hoyer, J.R.; Smith, J.W. A biochemical characterization of the binding of osteopontin to integrins αvβ1 and αvβ5. J. Biol. Chem. 1995, 270, 26232–26238. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.T.; Ludbrook, S.B.; Murrison, E.; Horgan, C.M.T. A regulated interaction between α5β1 integrin and osteopontin. Biochem. Biophys. Res. Commun. 2000, 267, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Green, P.M.; Ludbrook, S.B.; Miller, D.D.; Horgan, C.M.T.; Barry, S.T. Structural elements of the osteopontin SVVYGLR motif important for the interaction with alpha4 integrins. FEBS Lett. 2001, 503, 75–79. [Google Scholar] [CrossRef]
- Gupta, A.; Cao, W.; Chellaiah, M.A. Integrin αvβ3 and CD44 pathways in metastatic prostate cancer cells support osteoclastogenesis via a Runx2/Smad 5/receptor activator of NF-κB ligand signaling axis. Mol. Cancer 2012, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Michigami, T.; Shimizu, N.; Williams, P.J.; Niewolna, M.; Dallas, S.L.; Mundy, G.R.; Yoneda, T. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and α4β1-integrin enhances production of osteoclast-stimulating activity. Blood 2000, 96, 1953–1960. [Google Scholar] [PubMed]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adhes. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Cook, A.C.; Kappil, M.; Günthert, U.; Chambers, A.F.; Tuck, A.B.; Denhardt, D.T. Enhanced cell surface CD44 variant (v6, v9) expression by osteopontin in breast cancer epithelial cells facilitates tumor cell migration: Novel post-transcriptional, post-translational regulation. Clin. Exp. Metastasis 2005, 22, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Rittling, S.; Hayata, T.; Amagasa, T.; Denhardt, D.; Ezura, Y.; Nakashima, K.; Noda, M. Serum osteopontin, an enhancer of tumor metastasis to bone, promotes B16 melanoma cell migration. J. Cell. Biochem. 2007, 101, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Caers, J.; Günthert, U.; De Raeve, H.; Valckenborgh, E.V.; Menu, E.; Riet, I.V.; Van Camp, B.; Vanderkerken, K. The involvement of osteopontin and its receptors in multiple myeloma cell survival, migration and invasion in the murine 5T33MM model. Br. J. Haematol. 2006, 132, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Furger, K.A.; Allan, A.L.; Wilson, S.M.; Hota, C.; Vantyghem, S.A.; Postenka, C.O.; Al-Katib, W.; Chambers, A.F.; Tuck, A.B. Beta(3) integrin expression increases breast carcinoma cell responsiveness to the malignancy-enhancing effects of osteopontin. Mol. Cancer Res. 2003, 1, 810–819. [Google Scholar] [PubMed]
- Tuck, A.B.; Elliott, B.E.; Hota, C.; Tremblay, E.; Chambers, A.F. Osteopontin-induced, integrin-dependent migration of human mammary epithelial cells involves activation of the hepatocyte growth factor receptor (Met). J. Cell. Biochem. 2000, 78, 465–475. [Google Scholar] [CrossRef]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin αvβ3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clément-Lacroix, P.; Clézardin, P. Tumor αvβ3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.C.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Luo, X.; Brehm, S.; Carbery, K.; Chung, J.J.; Prior, J.L.; Doherty, J.; Demehri, S.; Salavaggione, L.; Piwnica-Worms, D.; et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009, 69, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Kazanecki, C.C.; Petersen, T.E.; Rittling, S.R.; Denhardt, D.T.; Sørensen, E.S. Cell type-specific post-translational modifications of mouse osteopontin are associated with different adhesive properties. J. Biol. Chem. 2007, 282, 19463–19472. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, G.N.; Sikes, R.A.; Devoll, R.E.; Kiefer, J.A.; Markwalder, R.; Klima, I.; Farach-Carson, C.M.; Studer, U.E.; Chung, L.W. Osteopontin: Possible role in prostate cancer progression. Clin. Cancer Res. 1999, 5, 2271–2277. [Google Scholar] [PubMed]
- Khodavirdi, A.C.; Song, Z.; Yang, S.; Zhong, C.; Wang, S.; Wu, H.; Pritchard, C.; Nelson, P.S.; Roy-Burman, P. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006, 66, 883–888. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic Endocrine Instigation of Indolent Tumor Growth Requires Osteopontin. Cell 2008, 133, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Anborgh, P.H.; Mutrie, J.C.; Tuck, A.B.; Chambers, A.F. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J. Cell. Mol. Med. 2010, 14, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; Leong, I.; Sanchez-Sweatman, O.; Khokha, R.; Sodek, J.; Tenenbaum, H.C.; Ganss, B.; Cheifetz, S. Expression of bone sialoprotein and osteopontin in breast cancer bone metastases. Clin. Exp. Metastasis 2000, 18, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.; Stubbs, J.T.; Fisher, L.; Aaron, A.D.; Thompson, E.W. Bone sialoprotein supports breast cancer cell adhesion proliferation and migration through differential usage of the αvβ3 and αvβ5 integrins. J. Cell. Physiol. 1998, 176, 482–494. [Google Scholar] [CrossRef]
- Chen, J.; De, S.; Brainard, J.; Byzova, T.V. Metastatic properties of prostate cancer cells are controlled by VEGF. Cell Commun. Adhes. 2004, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sariisik, E.; Docheva, D.; Padula, D.; Popov, C.; Opfer, J.; Schieker, M.; Clausen-Schaumann, H.; Benoit, M. Probing the Interaction Forces of Prostate Cancer Cells with Collagen I and Bone Marrow Derived Stem Cells on the Single Cell Level. PLoS ONE 2013, 8, e57706. [Google Scholar] [CrossRef]
- Valencia, K.; Ormazábal, C.; Zandueta, C.; Luis-Ravelo, D.; Antón, I.; Pajares, M.J.; Agorreta, J.; Montuenga, L.M.; Martínez-Canarias, S.; Leitinger, B.; et al. Inhibition of collagen receptor discoidin domain receptor-1 (DDR1) reduces cell survival, homing, and colonization in lung cancer bone metastasis. Clin. Cancer Res. 2012, 18, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Janni, W.; Vogl, F.D.; Wiedswang, G.; Synnestvedt, M.; Fehm, T.; Jückstock, J.; Borgen, E.; Rack, B.; Braun, S.; Sommer, H.; et al. Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse—A European pooled analysis. Clin. Cancer Res. 2011, 17, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Diel, I.J.; Kaufmann, M.; Costa, S.D.; Holle, R.; von Minckwitz, G.; Solomayer, E.F.; Kaul, S.; Bastert, G. Micrometastatic Breast Cancer Cells in Bone Comparison With Nodal Status. J. Natl. Cancer Inst. 1996, 88, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.M.; Lange, P.H.; Porter, M.P.; Lin, D.W.; Ellis, W.J.; Gallaher, I.S.; Vessella, R.L. Disseminated Tumor Cells in Prostate Cancer Patients after Radical Prostatectomy and without Evidence of Disease Predicts Biochemical Recurrence. Clin. Cancer Res. 2009, 15, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Aft, R.; Naughton, M.; Trinkaus, K.; Watson, M.; Ylagan, L.; Zhai, J.; Kuo, S.; Shannon, W.; Diemer, K.; Dietz, J.; et al. Effect of zoledronic acid on disseminated tumour cells in women with locally advanced breast cancer: An open label, randomised, phase 2 trial. Lancet Oncol. 2010, 11, 421–428. [Google Scholar] [CrossRef]
- Pantel, K.; Muller, V.; Auer, M.; Nusser, N.; Harbeck, N.; Braun, S. Detection and clinical implications of early systemic tumor cell dissemination in breast cancer. Clin. Cancer Res. 2003, 9, 6326–6334. [Google Scholar] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.; Yang, Z.H.; Kudahetti, S.; Møller, H.; Scardino, P.; Cuzick, J.; Berney, D.M. Prognostic value of Ki-67 for prostate cancer death in a conservatively managed cohort. Br. J. Cancer 2013, 108, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Rack, B.; Jückstock, J.; Genss, E.-M.; Schoberth, A.; Schindlbeck, C.; Strobl, B.; Heinrigs, M.; Rammel, G.; Zwingers, T.; Sommer, H.; et al. Effect of zoledronate on persisting isolated tumour cells in patients with early breast cancer. Anticancer Res. 2010, 30, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Hüsemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmüller, G.; et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, R. Molecular Pathways: Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.A.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. The prostate cancer bone marrow niche: More than just fertile soil. Asian J. Androl. 2012, 14, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Weinstat-saslow, D.L.; Zabrenetzky, V.S.; VanHoutte, K.; Frazier, W.A.; Roberts, D.D.; Steeg, P.S. Transfection of Thrombospondin 1 Complementary DNA into a Human Breast Carcinoma Cell Line Reduces Primary Tumor Growth, Metastatic Potential, and Angiogenesis. Cancer Res. 1994, 54, 6504–6511. [Google Scholar] [PubMed]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Lyerly, H.K.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, ra73. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dormady, S.P.; Zhang, X.-M.; Basch, R.S. Hematopoietic progenitor cells grow on 3T3 fibroblast monolayers that overexpress growth arrest-specific gene-6 (GAS6). Proc. Natl. Acad. Sci. USA 2000, 97, 12260–12265. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, L.; Bellido-Martín, L.; De Frutos, P.G. Growth arrest-specific gene 6 (GAS6): An outline of its role in haemostasis and inflammation. Thromb. Haemost. 2008, 100, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes Gas6/AXl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 Receptor Status Is Associated with Dormancy and Bone Metastatic Tumor Formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ma, Z.; Hu, W.; Wang, D.; Gong, B.; Fan, C.; Jiang, S.; Li, T.; Gao, J.; Yang, Y. Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 2017, 8, e2700. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Taichman, R.S. Cancer stem cells and the bone marrow microenvironment. Bonekey Rep. 2012, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Carcereri de Prati, A.; Butturini, E.; Rigo, A.; Oppici, E.; Rossin, M.; Boriero, D.; Mariotto, S. Metastatic Breast Cancer Cells Enter Into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia. J. Cell. Biochem. 2017, 118, 3237–3248. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, P.; Jönsson, J.-I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell. Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Cheng, J.; Tang, Y.; Liu, Z.; Xu, C.; Liu, Y.; Sun, Y. Human bone marrow-derived mesenchymal stem cells produced TGFbeta contributes to progression and metastasis of prostate cancer. Cancer Investig. 2012, 30, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Marlow, R.; Honeth, G.; Lombardi, S.; Cariati, M.; Hessey, S.; Pipili, A.; Mariotti, V.; Buchupalli, B.; Foster, K.; Bonnet, D.; et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 2013, 73, 6886–6899. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rubin, M.; Fenig, E.; DeBlasio, A.; Mendelsohn, J.; Yahalom, J.; Wieder, R. Basic Fibroblast Growth Factor Causes Growth Arrest in MCF-7 Human Breast Cancer Cells while Inducing both Mitogenic and Inhibitory G1 Events. Cancer Res. 1997, 57, 1750–1757. [Google Scholar] [PubMed]
- Walker, N.D.; Patel, J.; Munoz, J.L.; Hu, M.; Guiro, K.; Sinha, G.; Rameshwar, P. The bone marrow niche in support of breast cancer dormancy. Cancer Lett. 2016, 380, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Moharita, A.L.; Taborga, M.; Corcoran, K.E.; Bryan, M.; Patel, P.S.; Rameshwar, P. SDF-1alpha regulation in breast cancer cells contacting bone marrow stroma is critical for normal hematopoiesis. Blood 2006, 108, 3245–3252. [Google Scholar] [CrossRef] [PubMed]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Kan, C.; Clézardin, P. Tumour-derived miRNAs and bone metastasis. Bonekey Rep. 2015, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zoni, E.; van der Pluijm, G. The role of microRNAs in bone metastasis. J. Bone Oncol. 2016, 5, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Schuettpelz, L.G.; Link, D.C. Niche competition and cancer metastasis to bone. J. Clin. Investig. 2011, 121, 1253–1255. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Isabel, C.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormancy cells induced by a Col-I enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Käkönen, S.-M.; Selander, K.S.; Chirgwin, J.M.; Yin, J.J.; Burns, S.; Rankin, W.A.; Grubbs, B.G.; Dallas, M.; Cui, Y.; Guise, T.A. Transforming Growth Factor-β Stimulates Parathyroid Hormone-related Protein and Osteolytic Metastases via Smad and Mitogen-activated Protein Kinase Signaling Pathways. J. Biol. Chem. 2002, 277, 24571–24578. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Agyin, J.K.; Wang, L.; Tang, Y.; Lei, X.; Story, B.M.; Cornell, J.E.; Pollock, B.H.; Mundy, G.R.; Sun, L.-Z. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006, 66, 6714–6721. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, K.S.; Javelaud, D.; Fournier, P.G.J.; Niewolna, M.; McKenna, C.R.; Peng, X.H.; Duong, V.; Dunn, L.K.; Mauviel, A.; Guise, T.A. TGF-β-RI Kinase Inhibitor SD-208 Reduces the Development and Progression of Melanoma Bone Metastases. Cancer Res. 2011, 71, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, A.; Ling, W.; Li, X.; Khan, S.; Wang, Y.; Barlogie, B.; Shaughnessy, J.D.; Yaccoby, S. Consequences of daily administered parathyroid hormone on myeloma growth, bone disease, and molecular profiling of whole myelomatous bone. PLoS ONE 2010, 5, e15233. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G. Prostate cancer, serum parathyroid hormone, and the progression of skeletal metastases. Cancer Epidemiol. Biomark. Prev. 2008, 17, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2012, 71, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sun, R.; Origuchi, M.; Kanehira, M.; Takahata, T.; Itoh, J.; Umezawa, A.; Kijima, H.; Fukuda, S.; Saijo, Y. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol. Med. 2011, 17, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Djouad, F.; Plence, P.; Bony, C.; Tropel, P.; Apparailly, F.; Sany, J.; Noël, D.; Jorgensen, C. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 2003, 102, 3837–3844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent Bone Metastasis in Breast Cancer Tied to Src-Dependent Survival Signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Sasser, A.K.; Sullivan, N.J.; Studebaker, A.W.; Hendey, L.F.; Axel, A.E.; Hall, B.M. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007, 21, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Muehlberg, F.L.; Song, Y.-H.; Krohn, A.; Pinilla, S.P.; Droll, L.H.; Leng, X.; Seidensticker, M.; Ricke, J.; Altman, A.M.; Devarajan, E.; et al. Tissue-resident stem cells promote breast cancer growth and metastasis. Carcinogenesis 2009, 30, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.V.; Antoon, J.W.; Muir, S.E.; Elliott, S.; Beckman, B.S.; Burow, M.E. Effects of human mesenchymal stem cells on ER-positive human breast carcinoma cells mediated through ER-SDF-1/CXCR4 crosstalk. Mol. Cancer 2010, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Martin, E.; Anwar, T.; Arellano-garcia, C.; Medhora, N.; Lama, A.; Chen, Y.; Tanager, K.S.; Yoon, E.; Kidwell, K.; et al. Mesenchymal stem cell induced DDR2 mediates stromal-breast cancer interactions and metastasis growth. Cell Rep. 2017, 18, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, X.; Wang, L.; Liu, G.; Li, Y.; Wu, X.; Jing, Y.; Li, H.; Wang, G. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.-Y.; Fan, Q.-M.; Li, G.; Xu, W.-T.; Tang, T.-T. Human mesenchymal stem cells promote growth of osteosarcoma: Involvement of interleukin-6 in the interaction between human mesenchymal stem cells and Saos-2. Cancer Sci. 2010, 101, 2554–2560. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.; Du, L.; Fan, Q.-M.; Tang, Z.; Tang, T.-T. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 2012, 325, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-S.; Yang, S.-H.; Lei, Y.-P.; Tsai, C.-C.; Chen, H.-W.; Hsu, C.-Y.; Chen, L.-L.; Wang, H.-W.; Miller, S.A.; Chiou, S.-H.; et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology 2011, 141, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the growth MSC niche and promote tumour growth. Cancer Cell 2012, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.L.; Jia, Y.-L.; Chen, L.; Zeng, Q.; Zhou, J.-N.; Fu, C.-J.; Chen, H.-X.; Yuan, H.-F.; Li, Z.-W.; Shi, L.; et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 2013, 57, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.P.; Badtke, M.M.; Dudevoir, M.L.; Harrell, J.C.; Jacobsen, B.M.; Horwitz, K.B. Vascular endothelial growth factor secreted by activated stroma enhances angiogenesis and hormone independent growth of estrogen receptor positive breast cancer. Cancer Res. 2011, 70, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Tyan, S.-W.; Kuo, W.-H.; Huang, C.-K.; Pan, C.-C.; Shew, J.-Y.; Chang, K.-J.; Lee, E.Y.H.P.; Lee, W.-H. Breast cancer cells induce cancer-associated fibroblasts to secrete hepatocyte growth factor to enhance breast tumorigenesis. PLoS ONE 2011, 6, e15313. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Al-Khaldi, A.; Al-Sabti, H.; Galipeau, J.; Lachapelle, K. Therapeutic angiogenesis using autologous bone marrow stromal cells: Improved blood flow in a chronic limb ischemia model. Ann. Thorac. Surg. 2003, 75, 204–209. [Google Scholar] [CrossRef]
- Chen, L.; Tredget, E.E.; Wu, P.Y.G.; Wu, Y. Paracrine Factors of Mesenchymal Stem Cells Recruit Macrophages and Endothelial Lineage Cells and Enhance Wound Healing. PLoS ONE 2008, 3, e1886. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yu, S.P.; Fraser, J.L.; Lu, Z.; Ogle, M.E.; Wang, J.A.; Wei, L. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 2008, 135, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, N.; Fujii, T.; Iwase, T.; Ohgushi, H.; Itoh, T.; Uematsu, M.; Yamagishi, M.; Mori, H.; Kangawa, K.; Kitamura, S. Intravenous administration of mesenchymal stem cells improves cardiac function in rats with acute myocardial infarction through angiogenesis and myogenesis. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2670–H2676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, L.; Scott, P.G.; Tredget, E.E. Mesenchymal Stem Cells Enhance Wound Healing Through Differentiation and Angiogenesis. Stem Cells 2007, 25, 2648–2659. [Google Scholar] [CrossRef] [PubMed]
- Arnulf, B.; Lecourt, S.; Soulier, J.; Ternaux, B.; Lacassagne, M.N.; Crinquette, A.; Dessoly, J.; Sciaini, A.K.; Benbunan, M.; Chomienne, C.; et al. Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia 2007, 21, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Thibault, B.; Mery, E.; Golzio, M.; Pasquet, M.; Hennebelle, I.; Bourin, P.; Mirshahi, M.; Delord, J.P.; Querleu, D.; et al. Ovarian ascites-derived Hospicells promote angiogenesis via activation of macrophages. Cancer Lett. 2012, 326, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Jing, Y.; Zhang, S.; Liu, Y.; Shi, Y.; Wei, L. The role of immunosuppression of mesenchymal stem cells in tissue repair and tumor growth. Cell Biosci. 2012, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Cuiffo, B.G.; Karnoub, A.E. MSC in tumour development. Emerging roles and conceptrs. Cell Adhes. Migr. 2012, 6, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; Daenen, L.G.M.; Roodhart, J.M.L.; Voest, E.E. The role of mesenchymal stem cells in anti-cancer drug resistance and tumour progression. Br. J. Cancer 2012, 106, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-R.; Lu, D.-Y.; Lin, H.-Y.; Yeh, W.-L. Mesenchymal stem cell-induced Doxorubicin resistance in triple negative breast cancer. Biomed. Res. Int. 2014, 2014, 532161. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemotactic Molecule | Cancer Type |
|---|---|
| CXCL12-CXCR4 | Melanoma [82] and breast cancer [122] bone metastasis |
| ILβ1 and leptin | Breast cancer bone metastasis [137] |
| CXCL12-CXCR4/CXCR7 | Osteosarcoma [138] |
| CXCL1- and CXCL5-CXCR4 | Breast cancer [139] |
| TGFβ | Breast [139] and hepatocellular cancer [140] |
| CCL5- and CCL9-induced MMP | Breast cancer [141] |
| CCL5-induced collagen | Tongue cancer [142] |
| CCL5 | Osteosarcoma cells [143] |
| VEGF-CXCR4 | Osteosarcoma [144] |
| IL8 | Gastric [145] |
| IL6 | Ovarian [146] |
| CXCL12 | Melanoma [147] |
| CCL5 mediated by tumor-derived OPN | Breast cancer [32] |
| Mesenchymal Stromal Cell Source | Cancer Type | Growth Factor |
|---|---|---|
| MSCs | Breast | CCL5 [32] |
| Breast | IL6 [253] | |
| Breast | CXCL7 [249] | |
| Breast | CXCL12–CXCR4 [254,255] | |
| Breast | Collagen-DDR2 [256] | |
| Prostate | TGFβ [229] | |
| Colorectal | Galectin 3 [257] | |
| Neuroblastoma, osteosarcoma, colorectal | IL6 [258,259,260,261] | |
| MSC-derived CAFs | Ovarian | IL6, HGF, EGF [262] |
| Gastric | IL6, Wnt, BMP [263] | |
| Liver | miR155 [264] | |
| Breast | CXCL12 [60] | |
| CAFs | Breast | CXCL12-CXCR4 [265,266] |
| Breast | VEGF [267] | |
| Breast | HGF [268] | |
| Osteoblast | Prostate bone metastasis | CCL2 [92] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, N.; Qian, B.-Z. Mesenchymal Stromal Cells: Emerging Roles in Bone Metastasis. Int. J. Mol. Sci. 2018, 19, 1121. https://doi.org/10.3390/ijms19041121
Graham N, Qian B-Z. Mesenchymal Stromal Cells: Emerging Roles in Bone Metastasis. International Journal of Molecular Sciences. 2018; 19(4):1121. https://doi.org/10.3390/ijms19041121
Chicago/Turabian StyleGraham, Nicola, and Bin-Zhi Qian. 2018. "Mesenchymal Stromal Cells: Emerging Roles in Bone Metastasis" International Journal of Molecular Sciences 19, no. 4: 1121. https://doi.org/10.3390/ijms19041121
APA StyleGraham, N., & Qian, B.-Z. (2018). Mesenchymal Stromal Cells: Emerging Roles in Bone Metastasis. International Journal of Molecular Sciences, 19(4), 1121. https://doi.org/10.3390/ijms19041121