Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides


Abstract
:1. Introduction
2. Marine Organisms that are Sources of Anticancer Peptides
2.1. Cyanobacteria
2.2. Fungi
2.3. Sponges
2.4. Tunicates and Ascidians
2.5. Mollusks and Fish
3. Bioactive Peptides with Anticancer Potential Isolated from Marine Organisms
3.1. Cyanobacteria-Derived Peptides
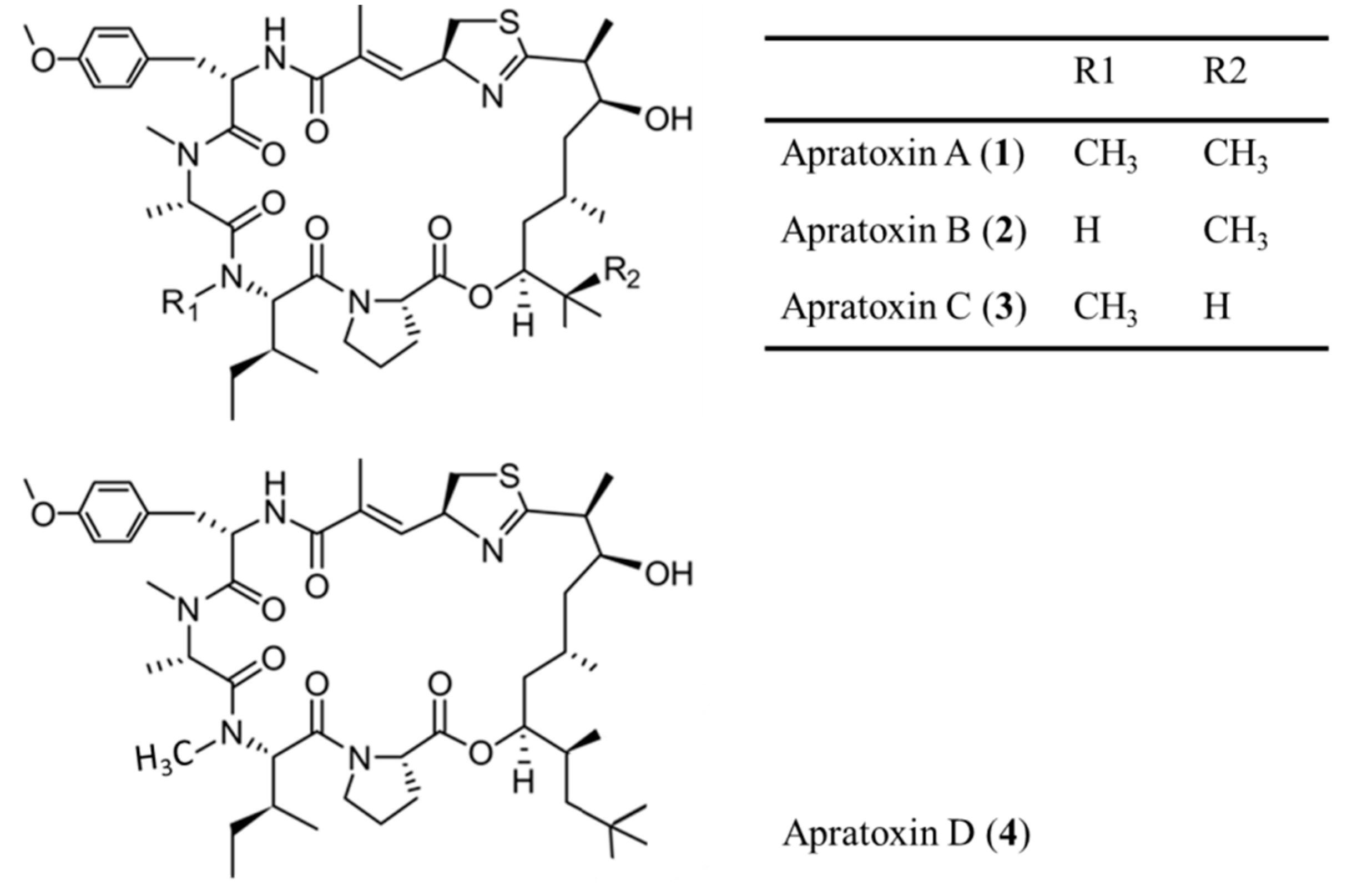
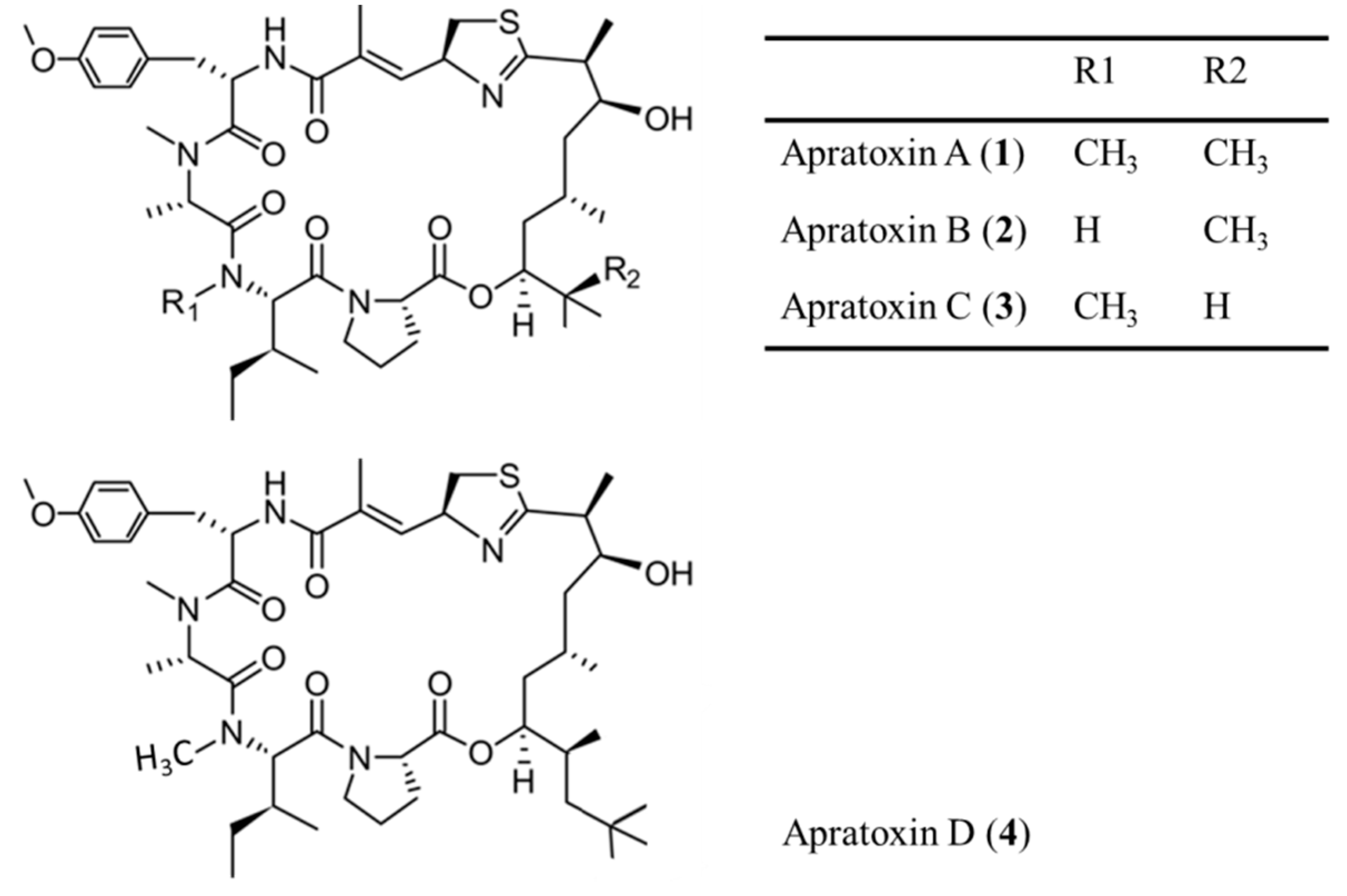
3.1.1. Apratoxin A–D
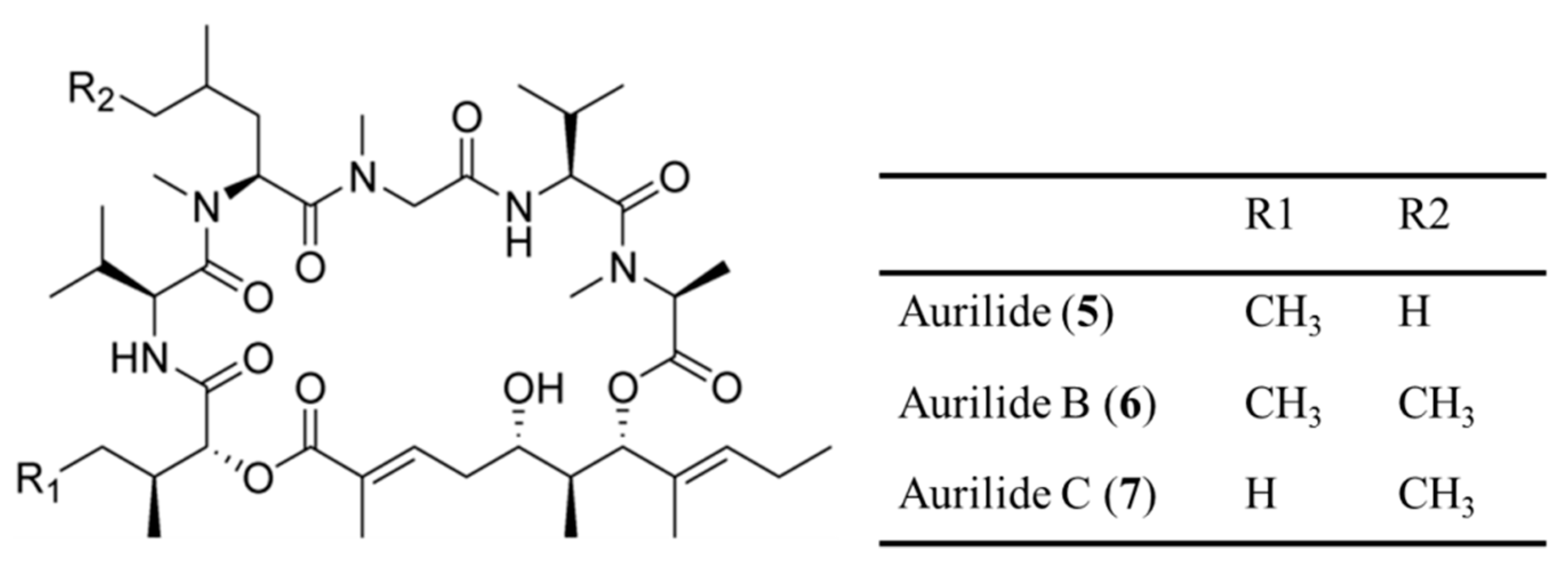
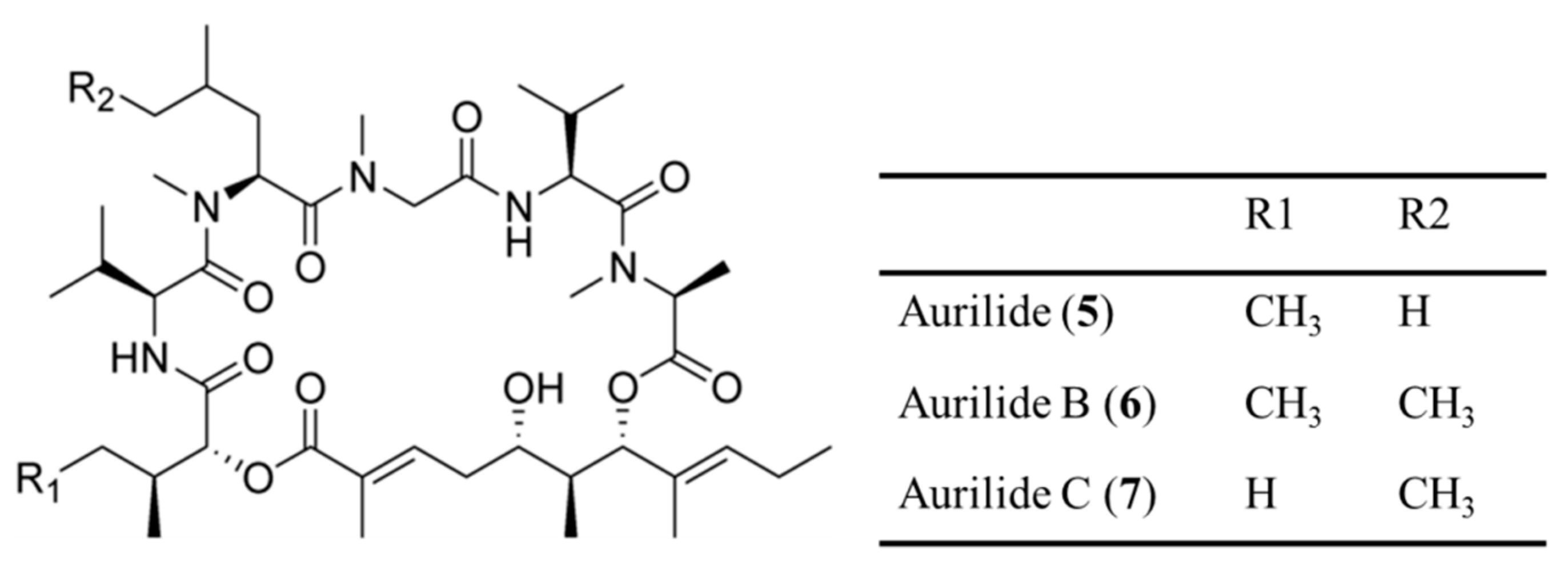
3.1.2. Aurilides
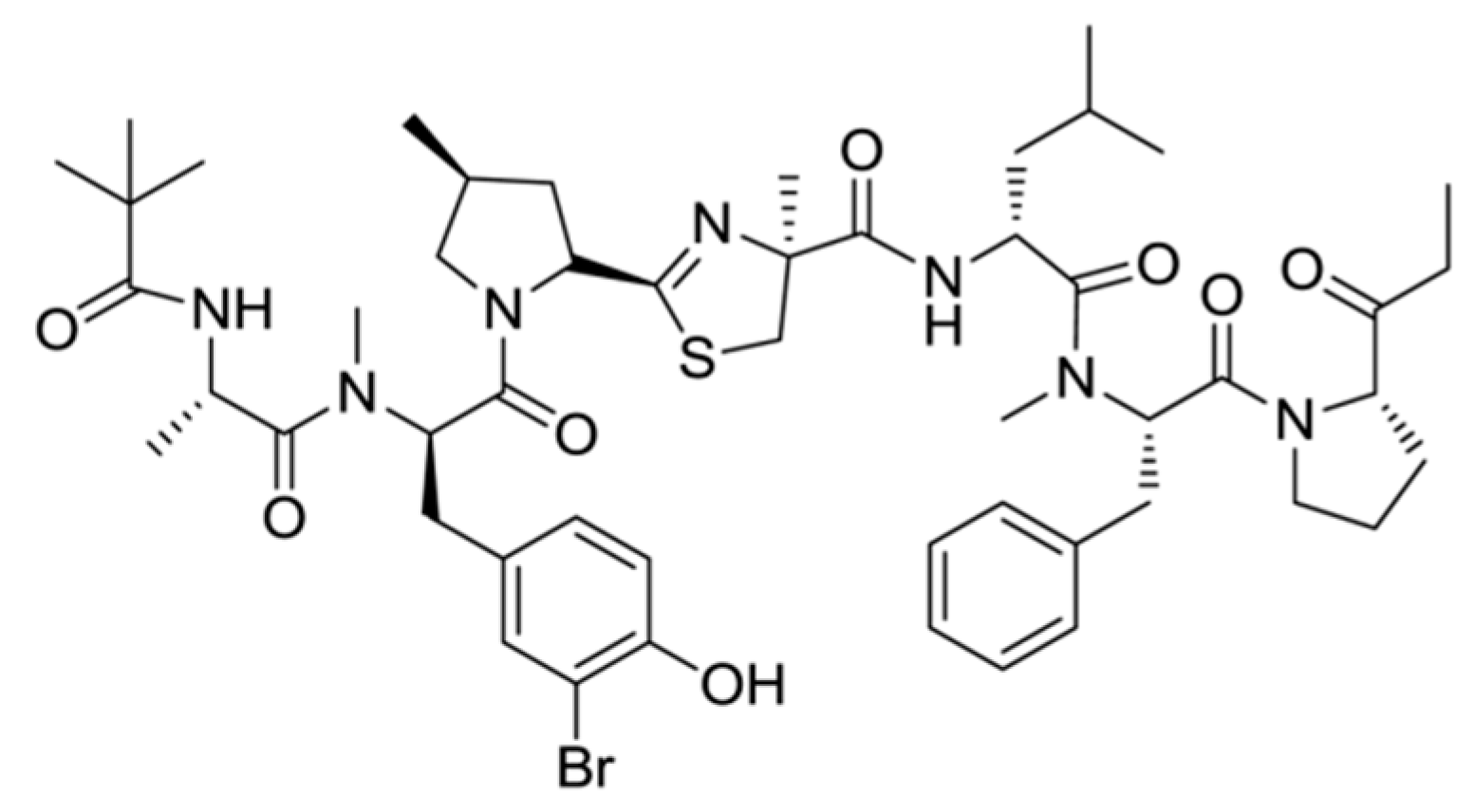
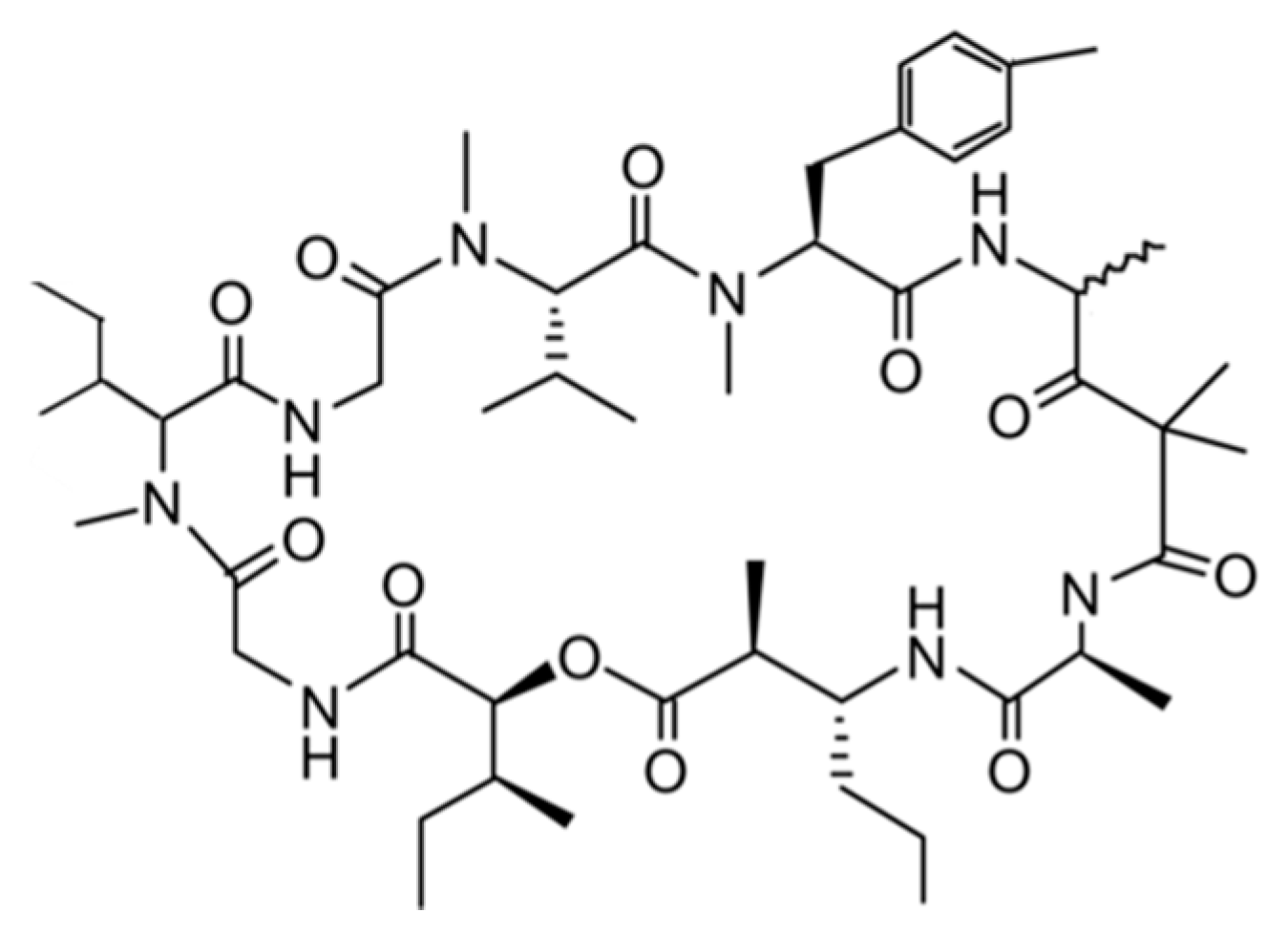
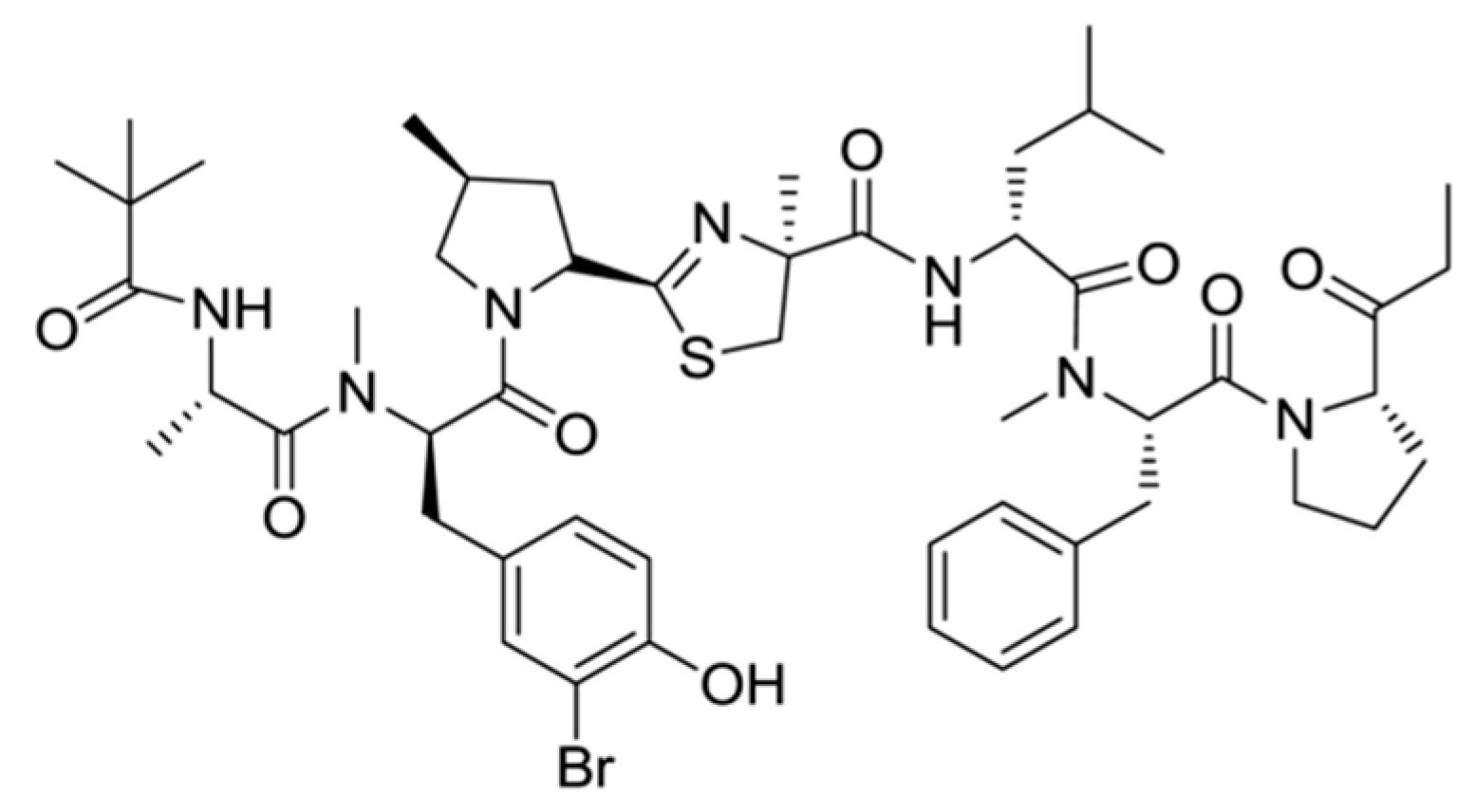
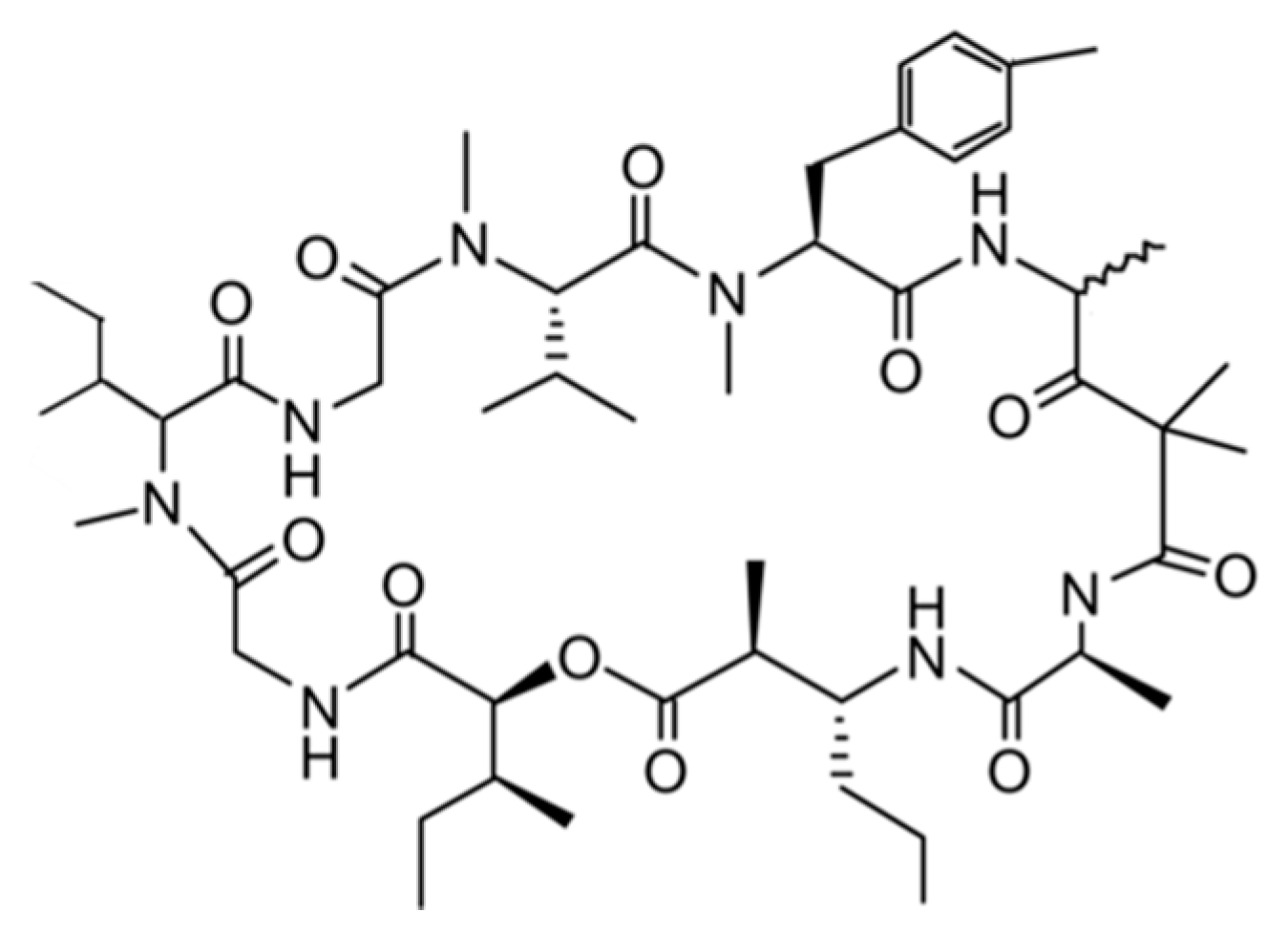
3.1.3. Bisebromoamide
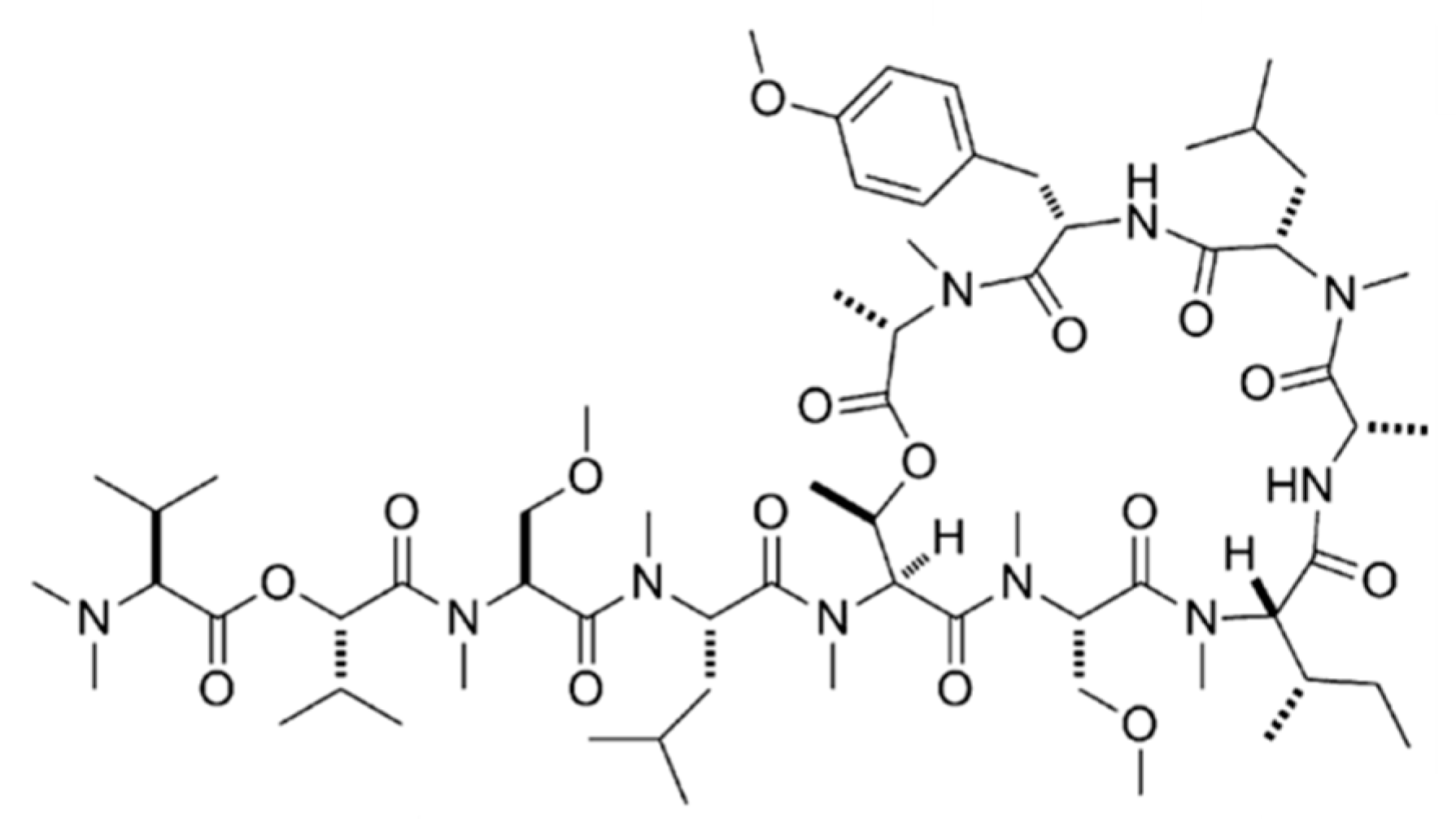
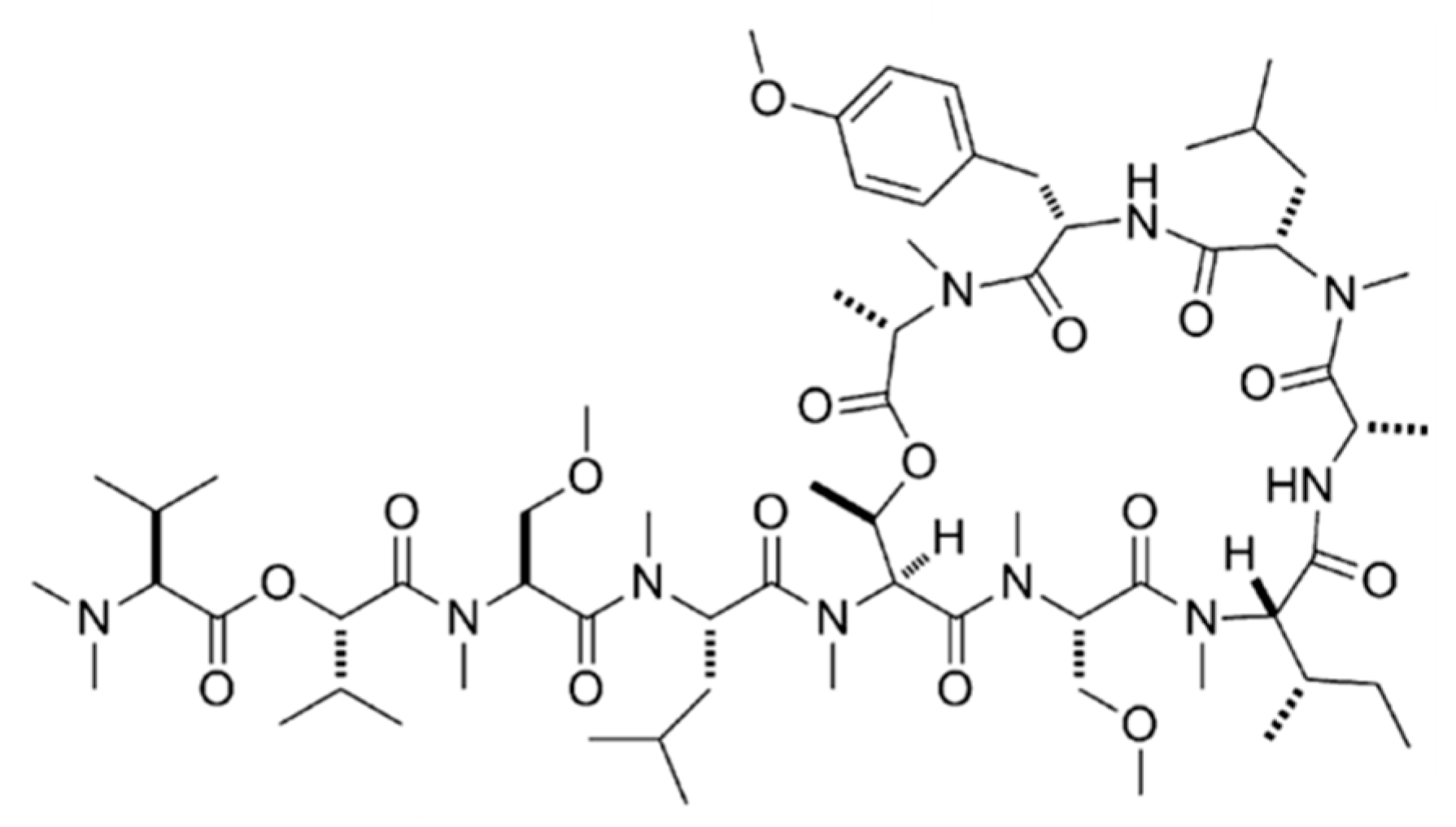
3.1.4. Coibamide A
3.1.5. Cryptophycin
3.1.6. Desmethoxymajusculamide C
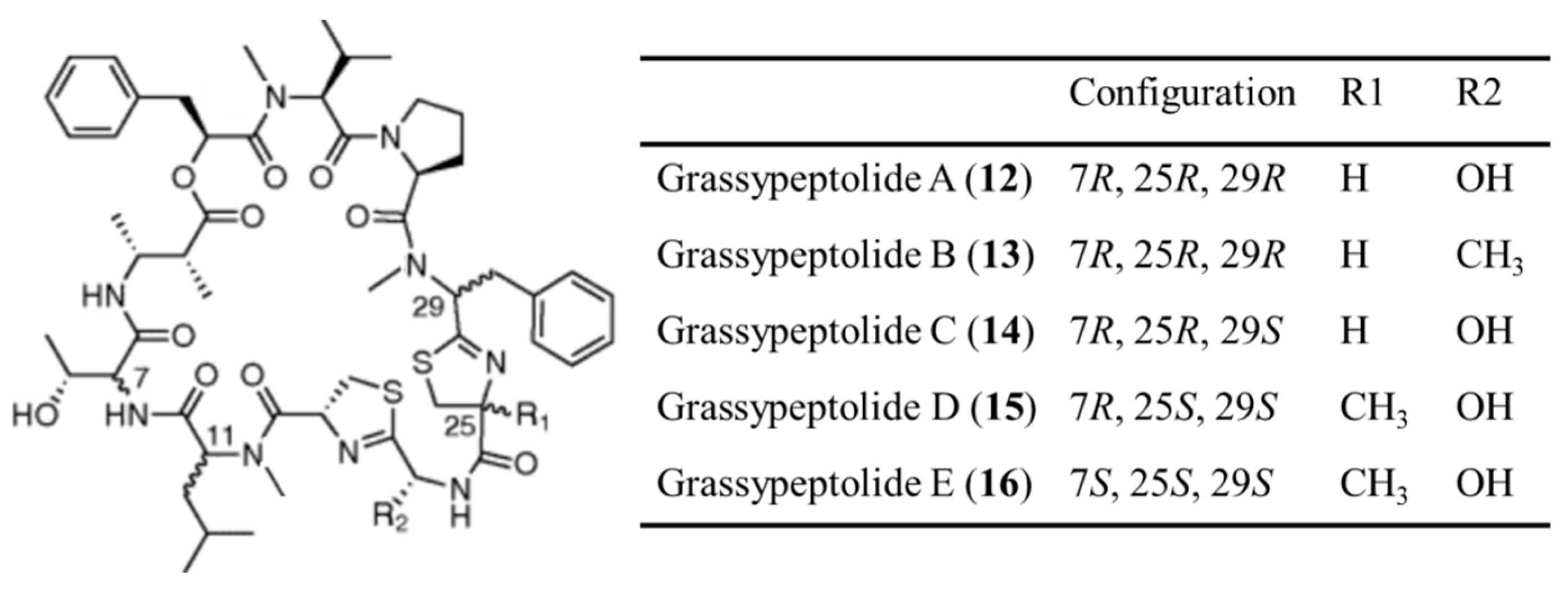
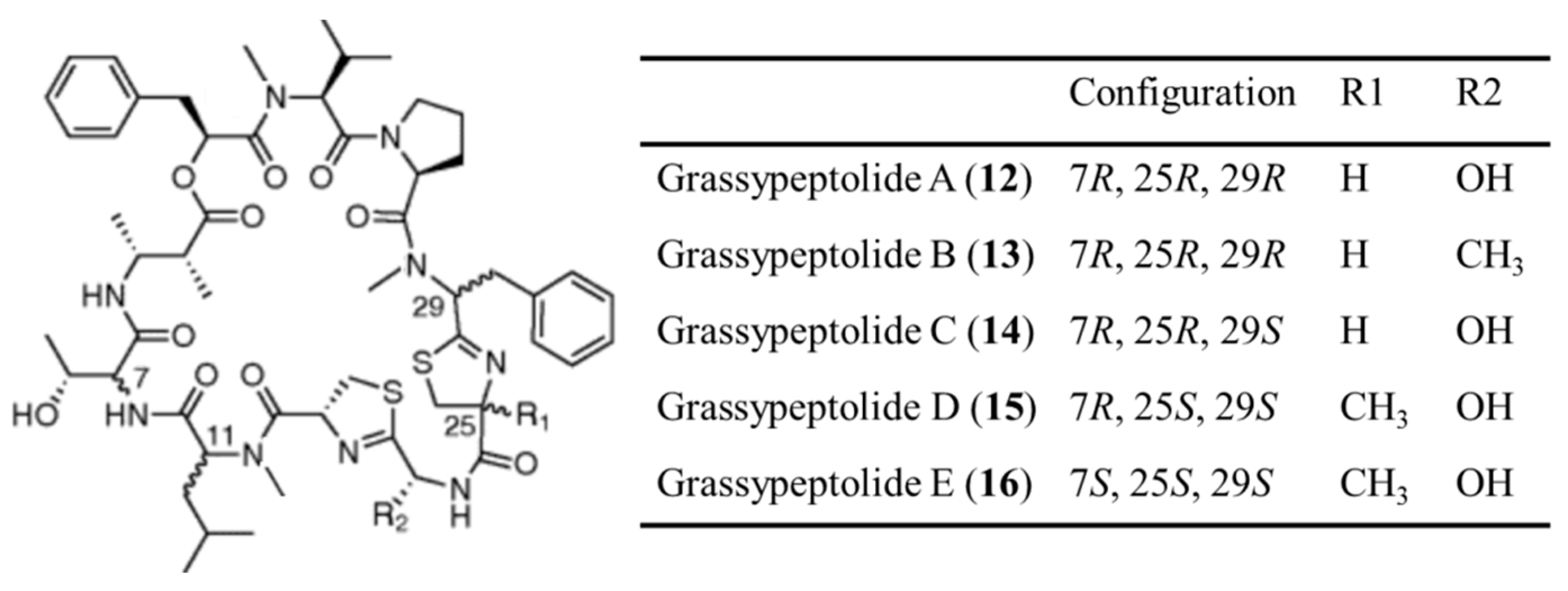
3.1.7. Grassypeptolides
3.1.8. Hantupeptin A
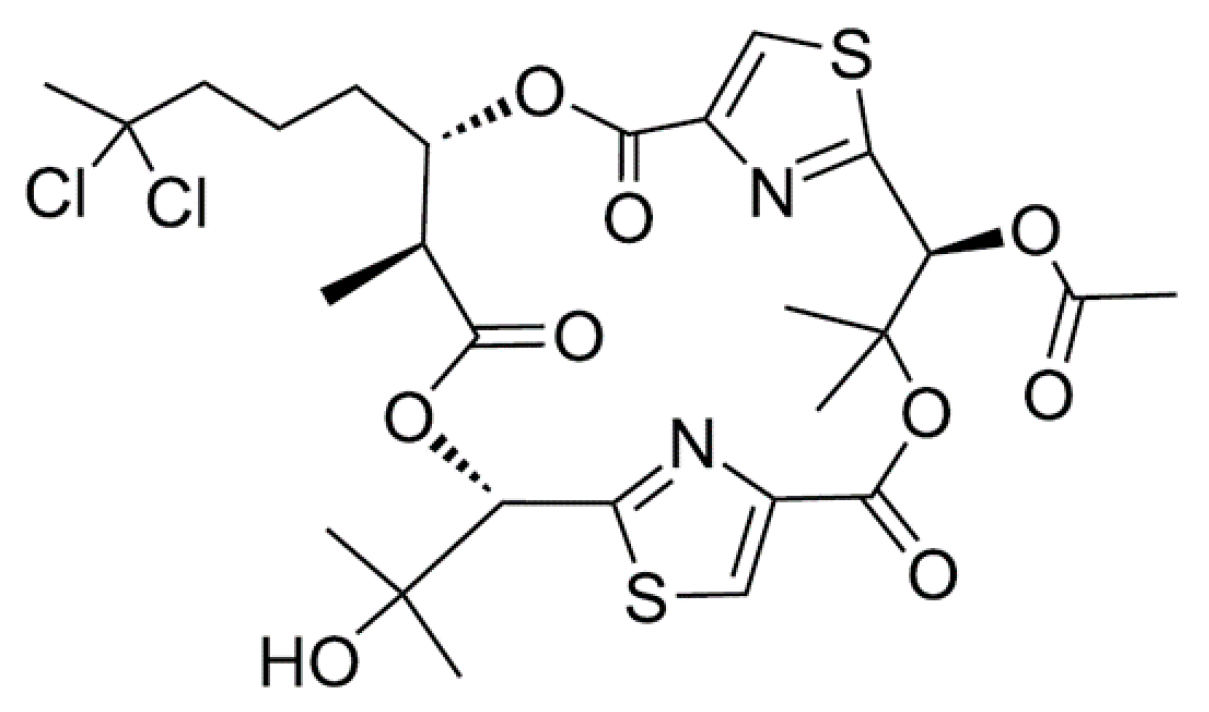
3.1.9. Hectochlorin
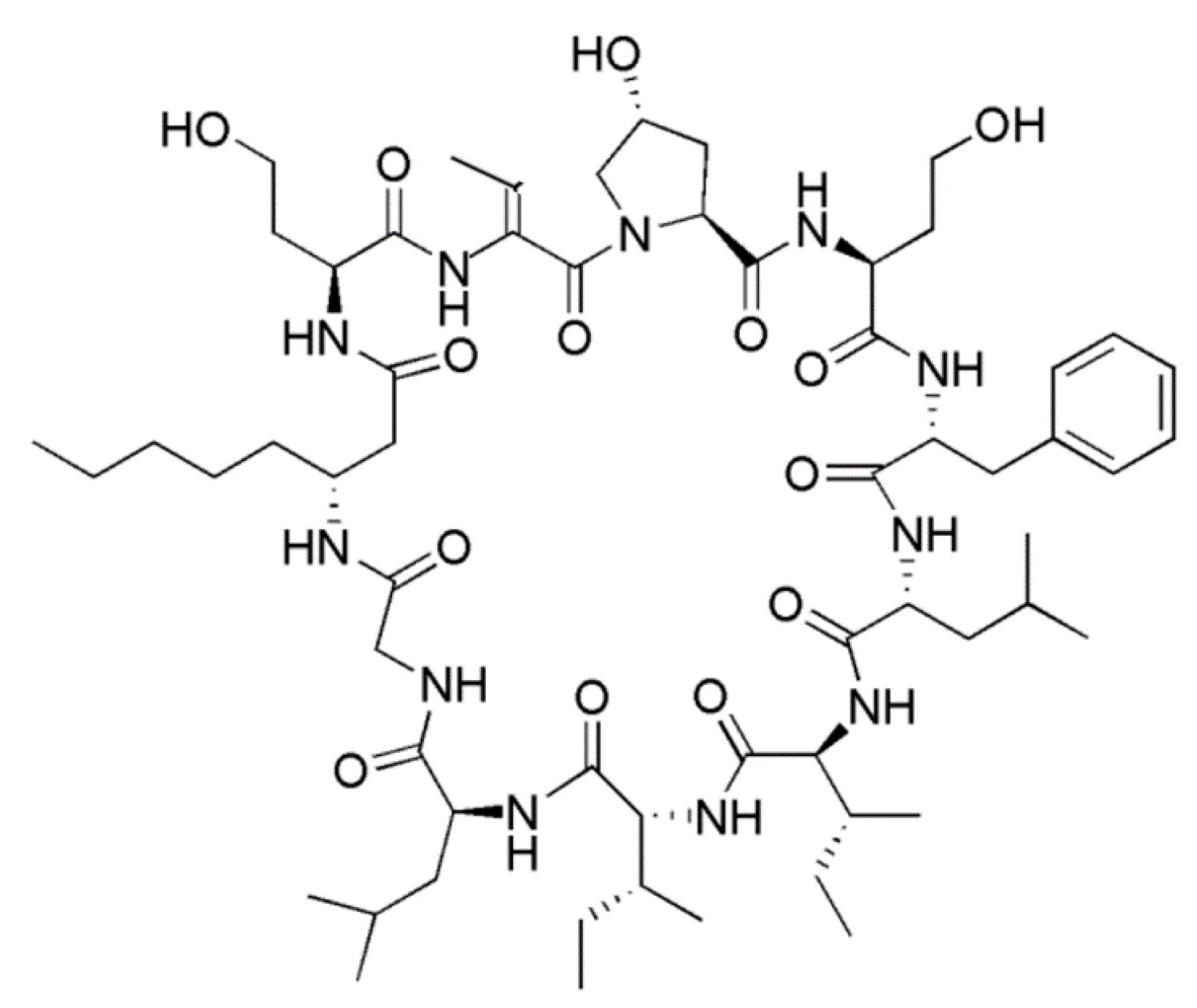
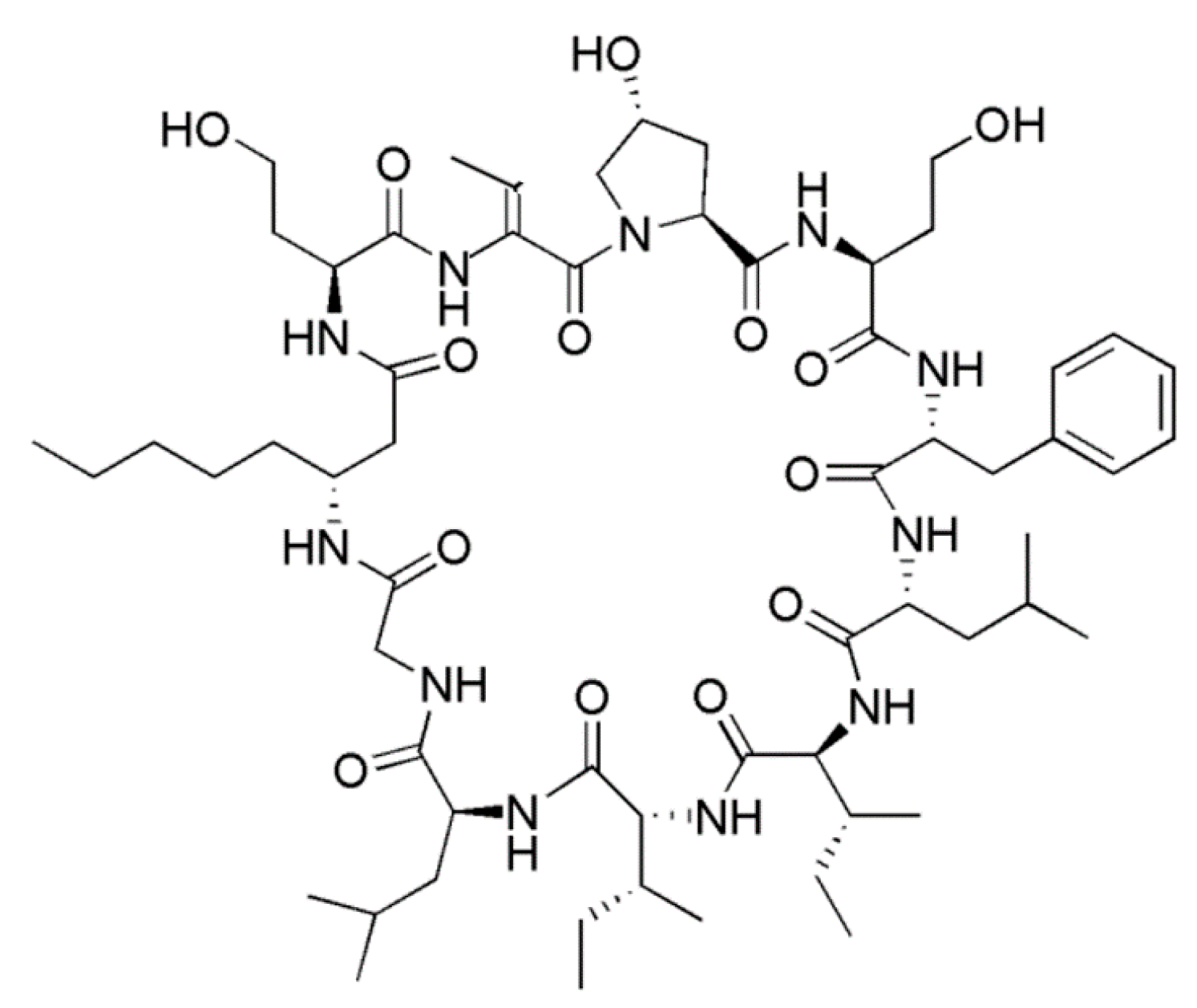
3.1.10. Hormothamnin A
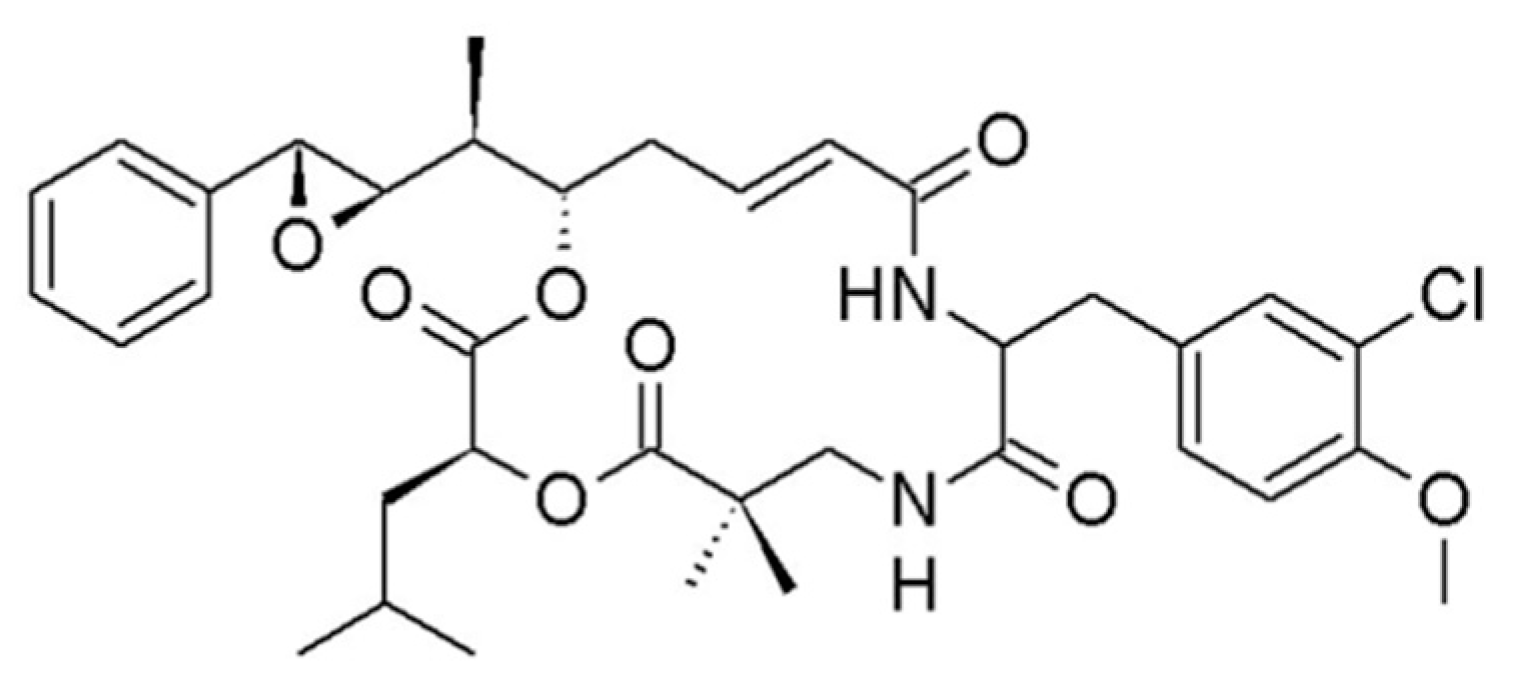
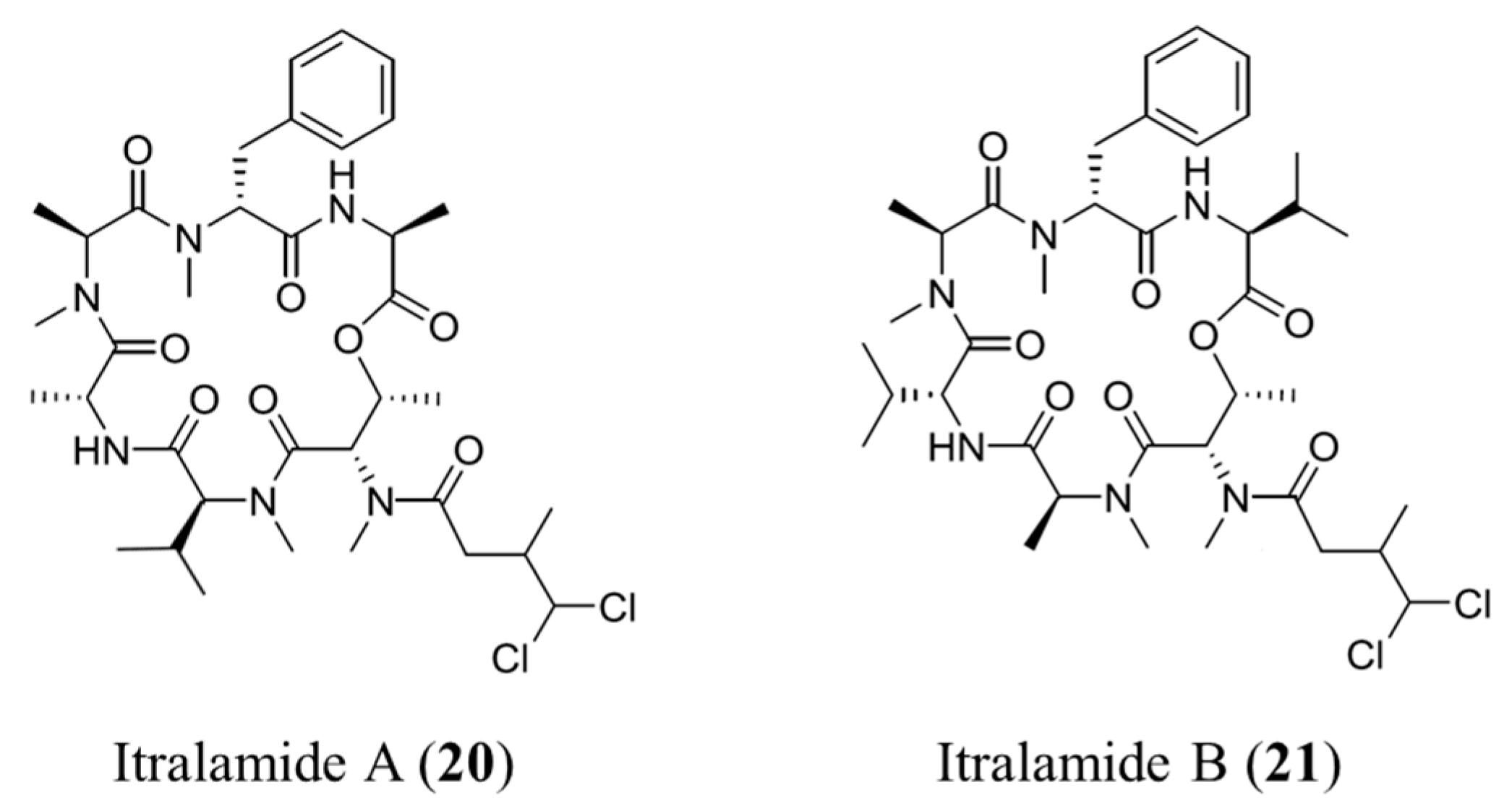
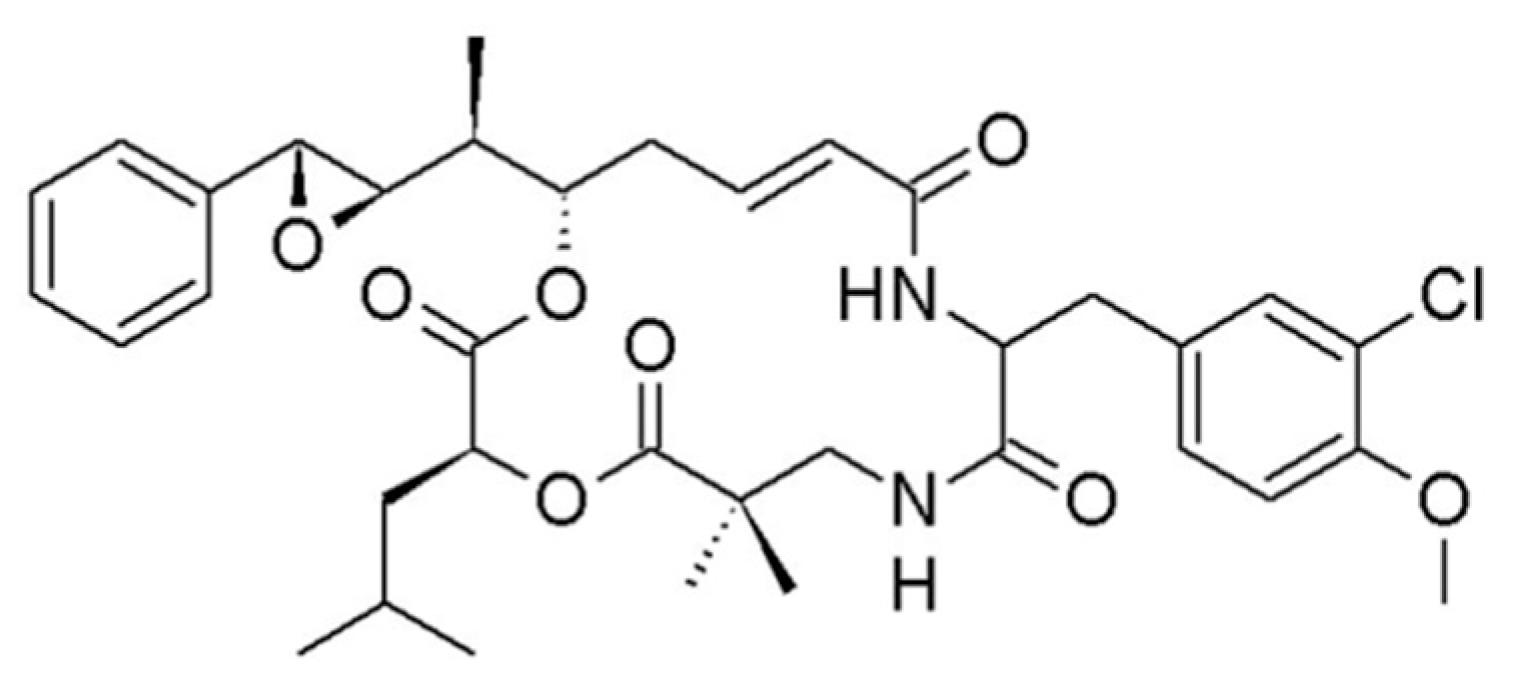
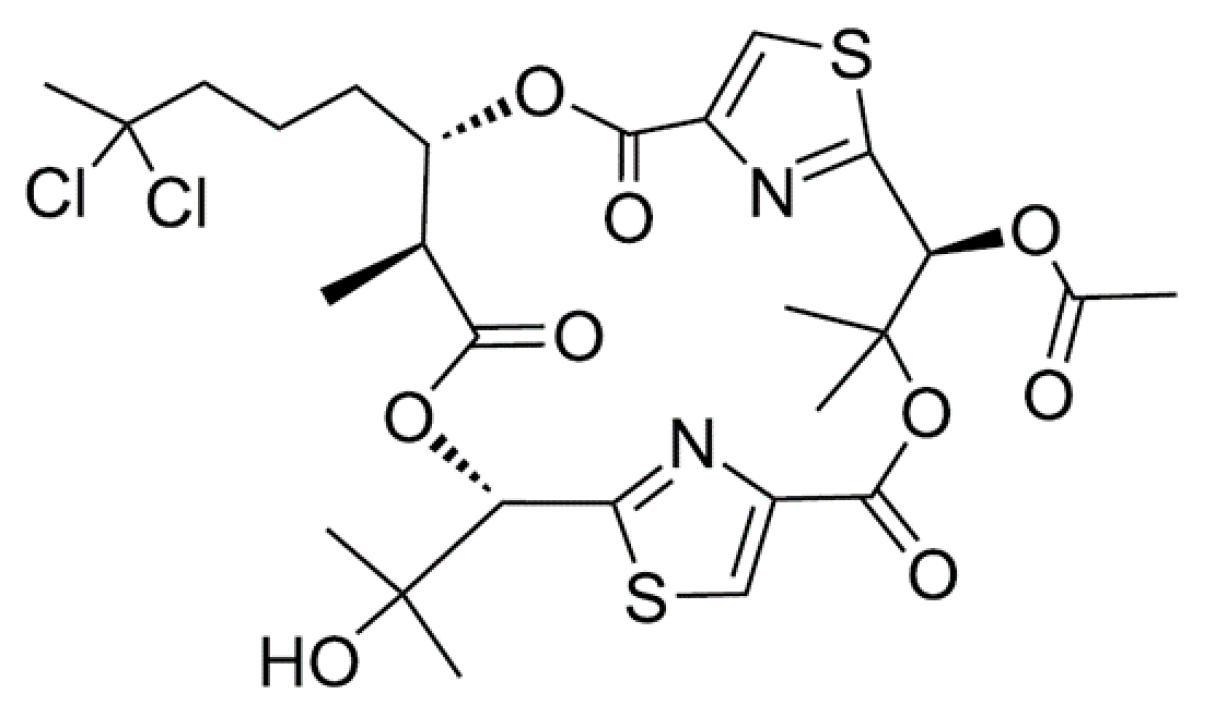
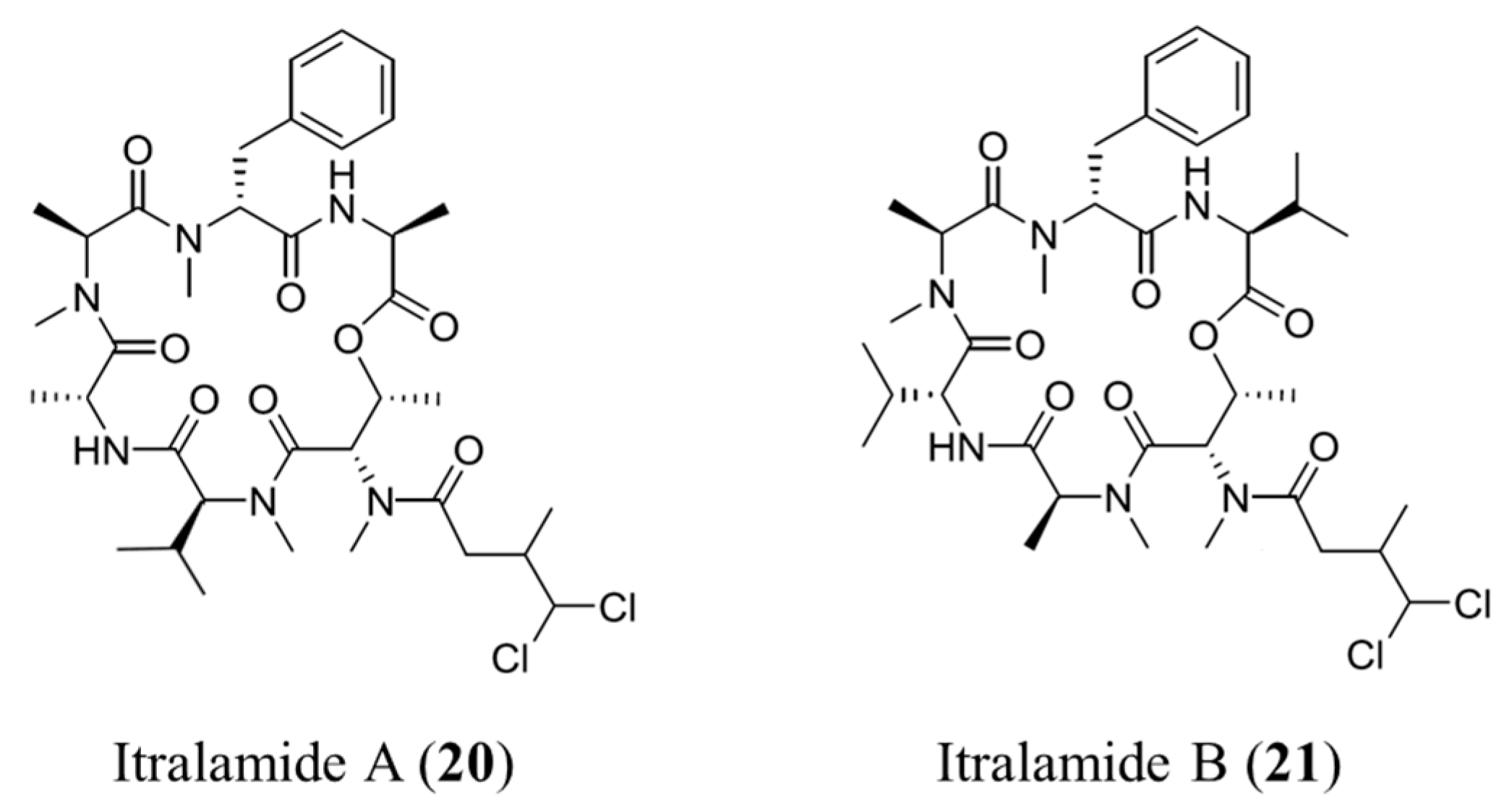
3.1.11. Itralamide A and B
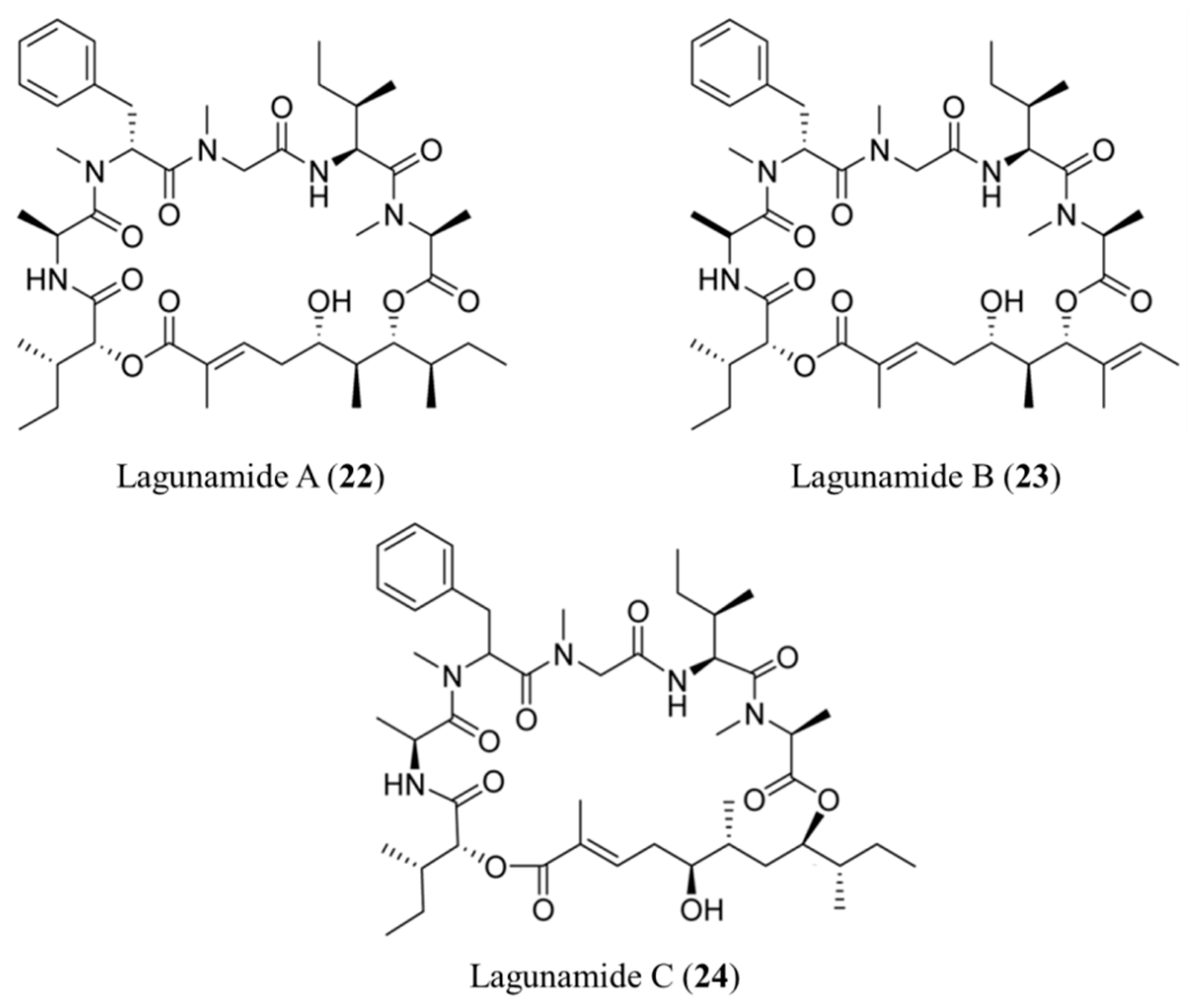
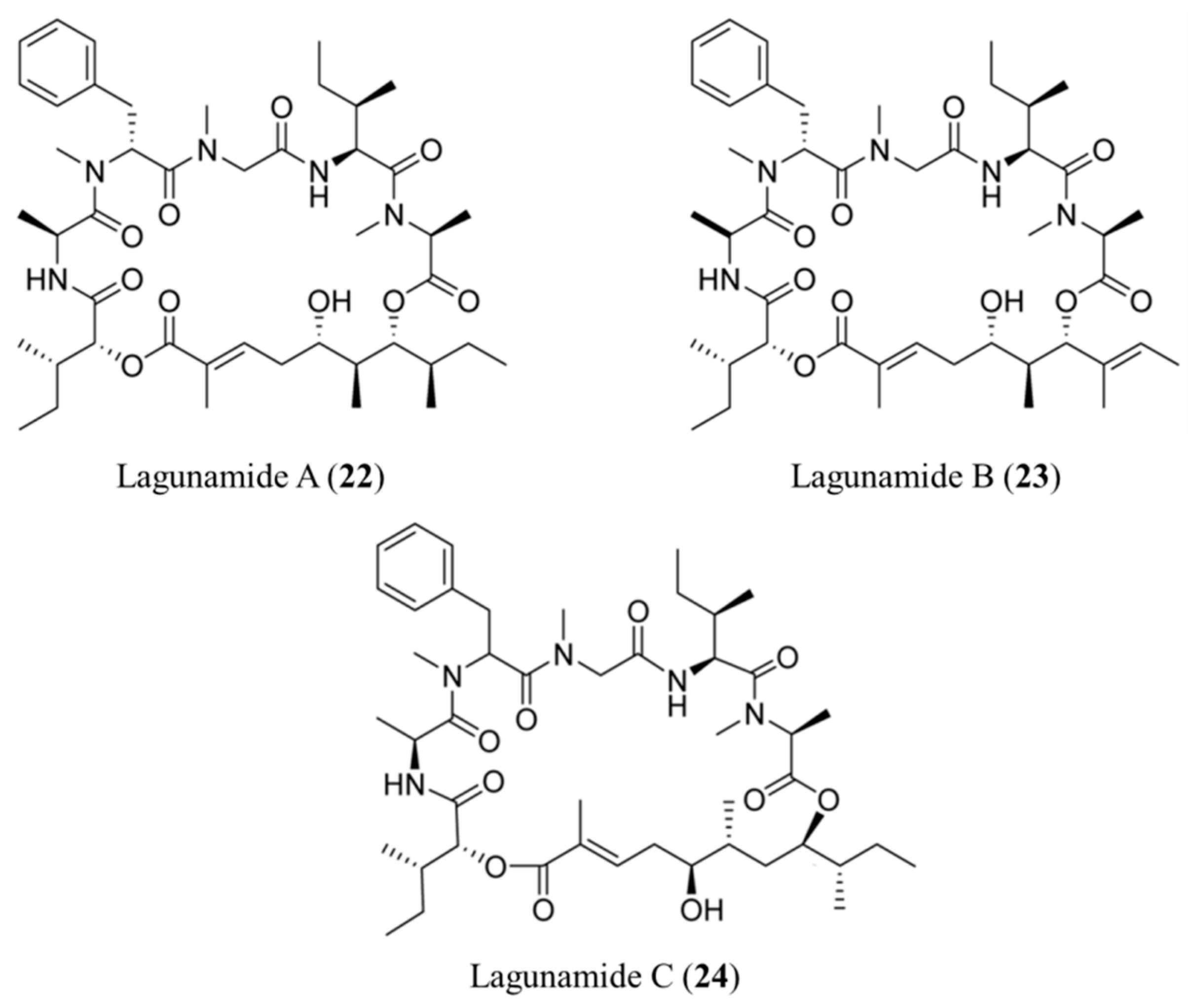
3.1.12. Lagunamides
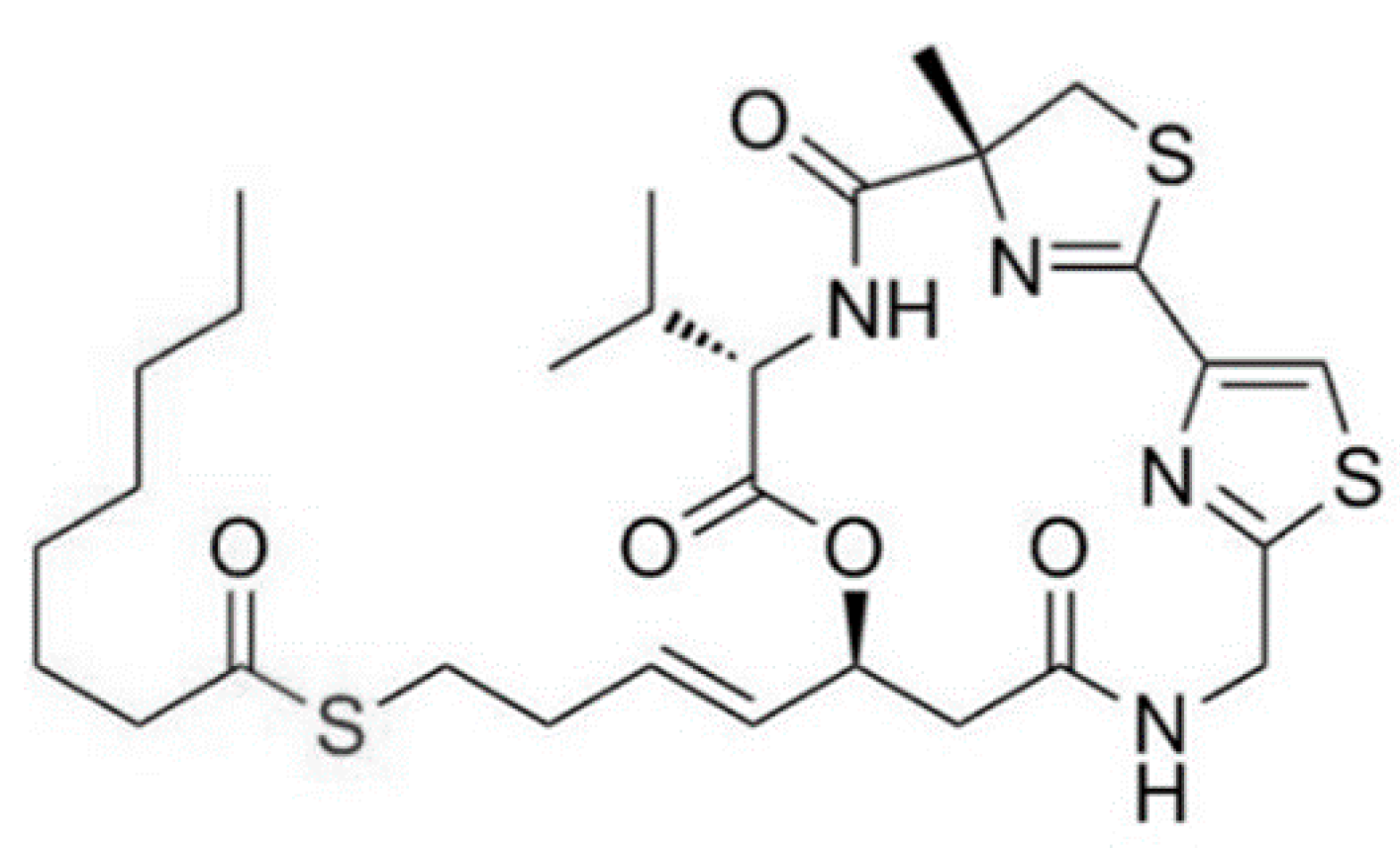
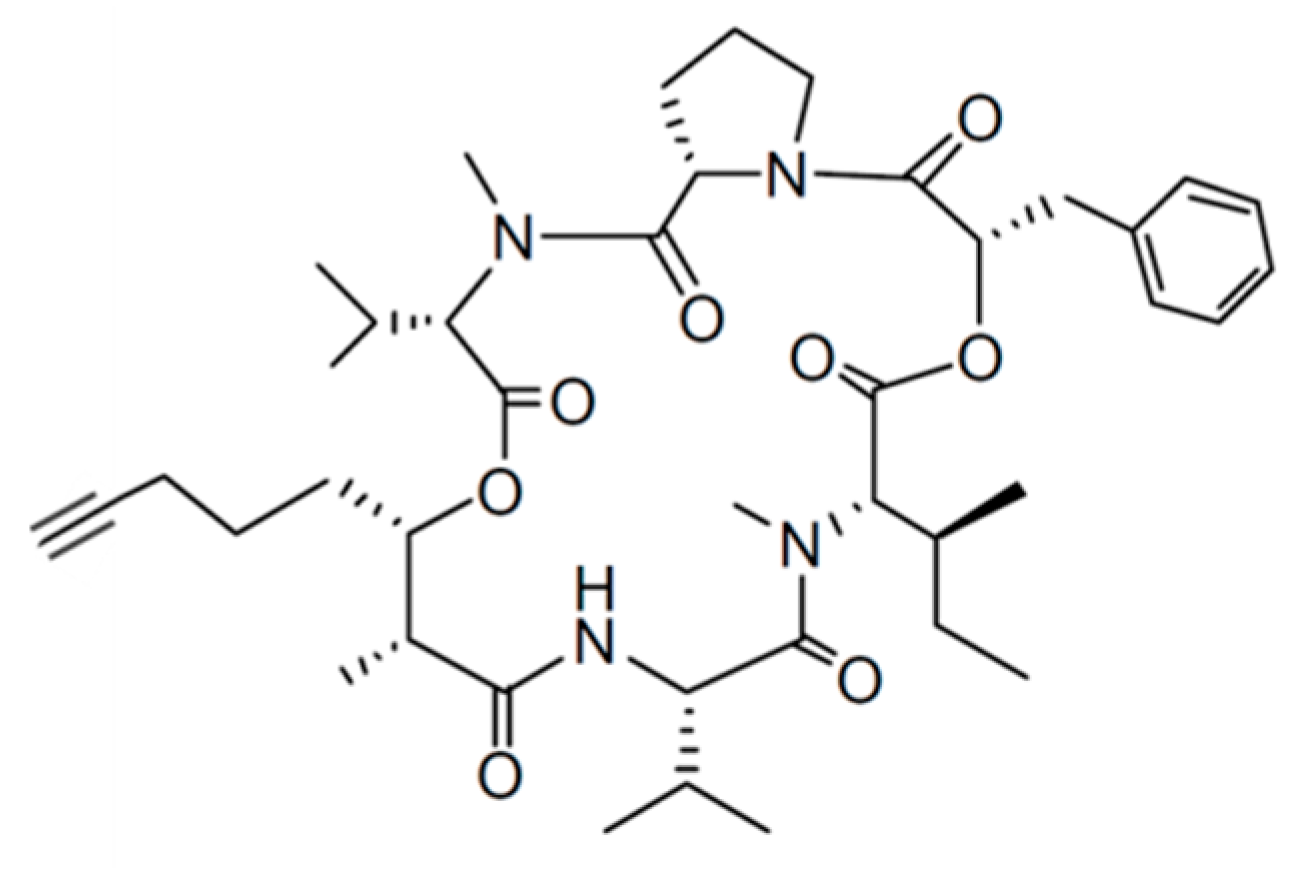
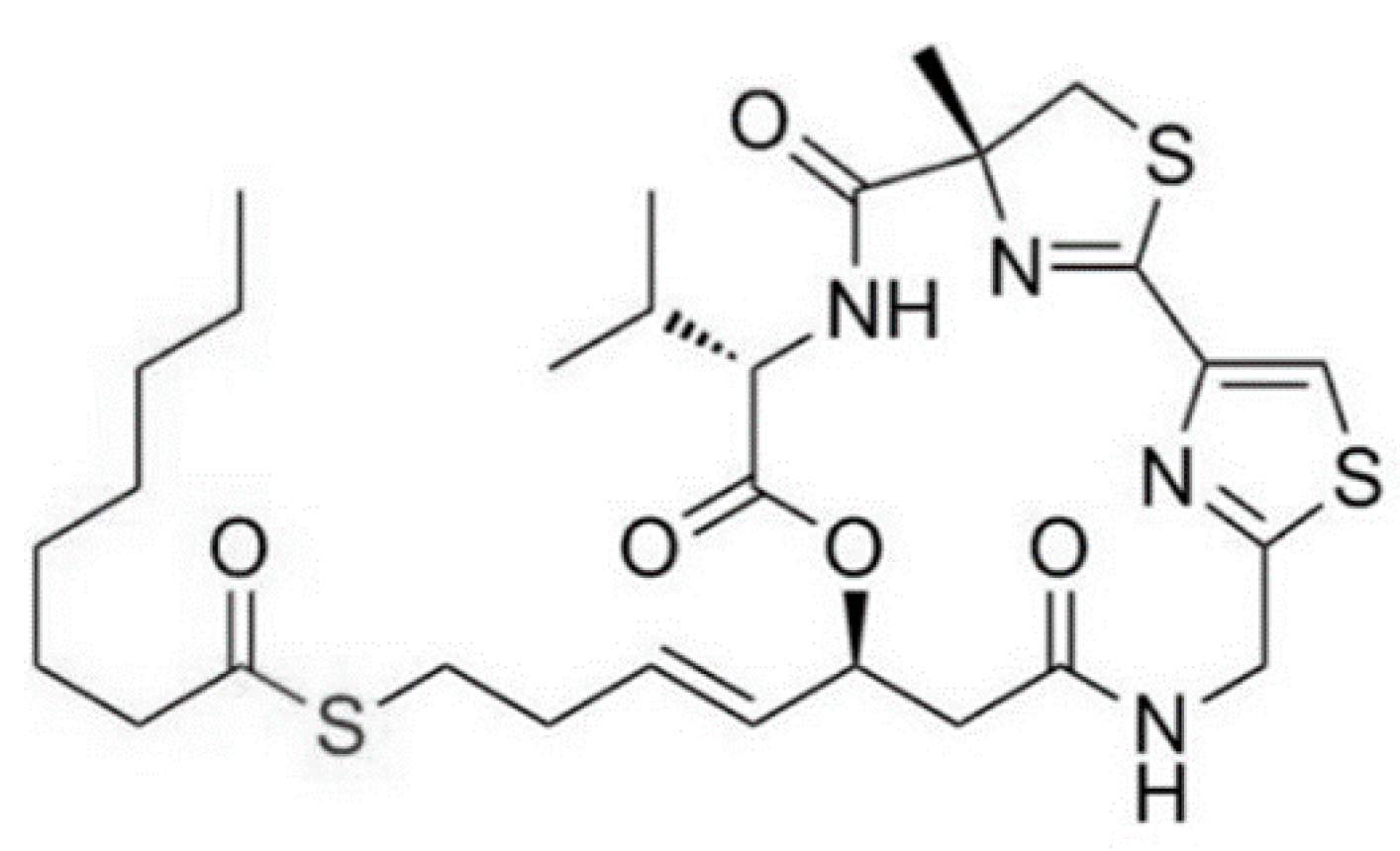
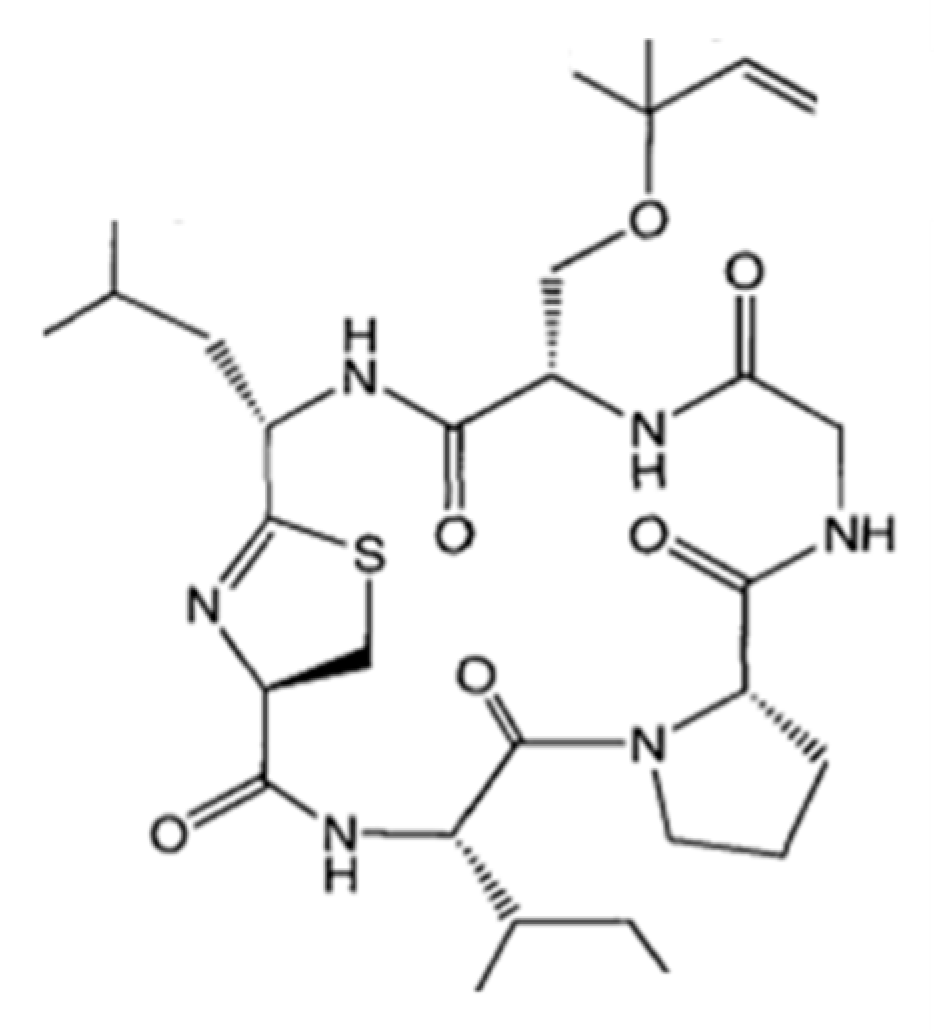
3.1.13. Largazole
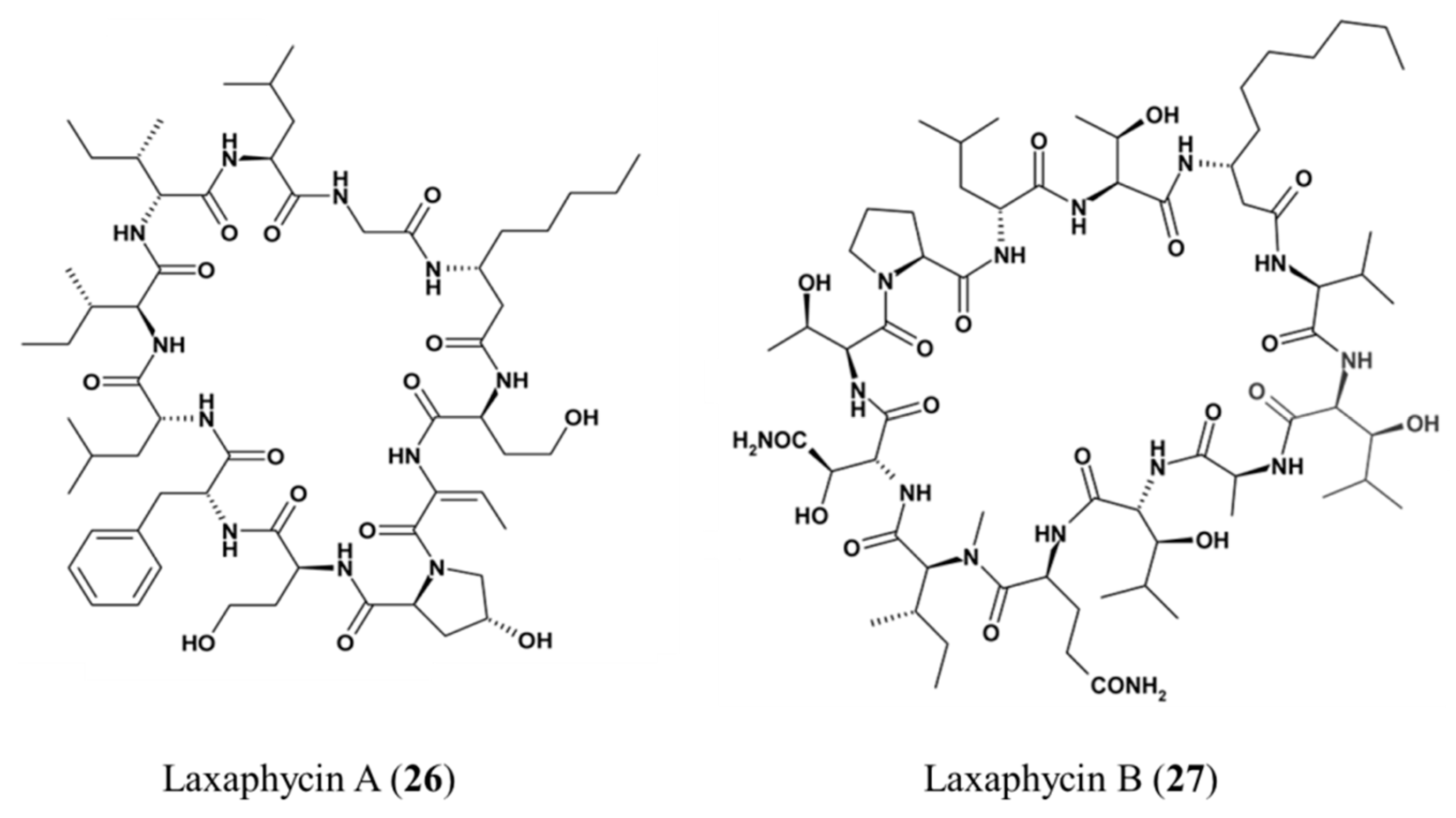
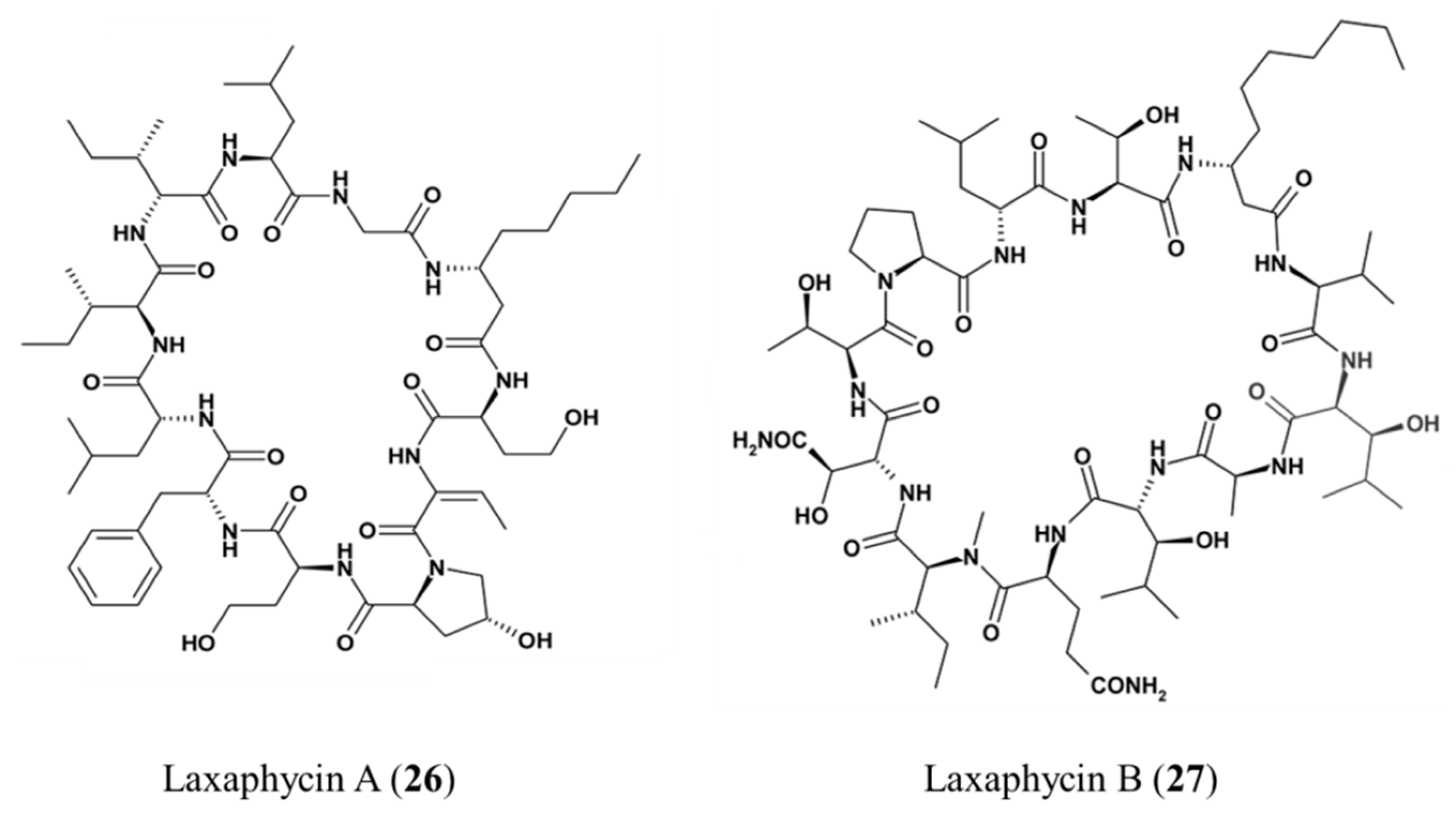
3.1.14. Laxaphycin A and B
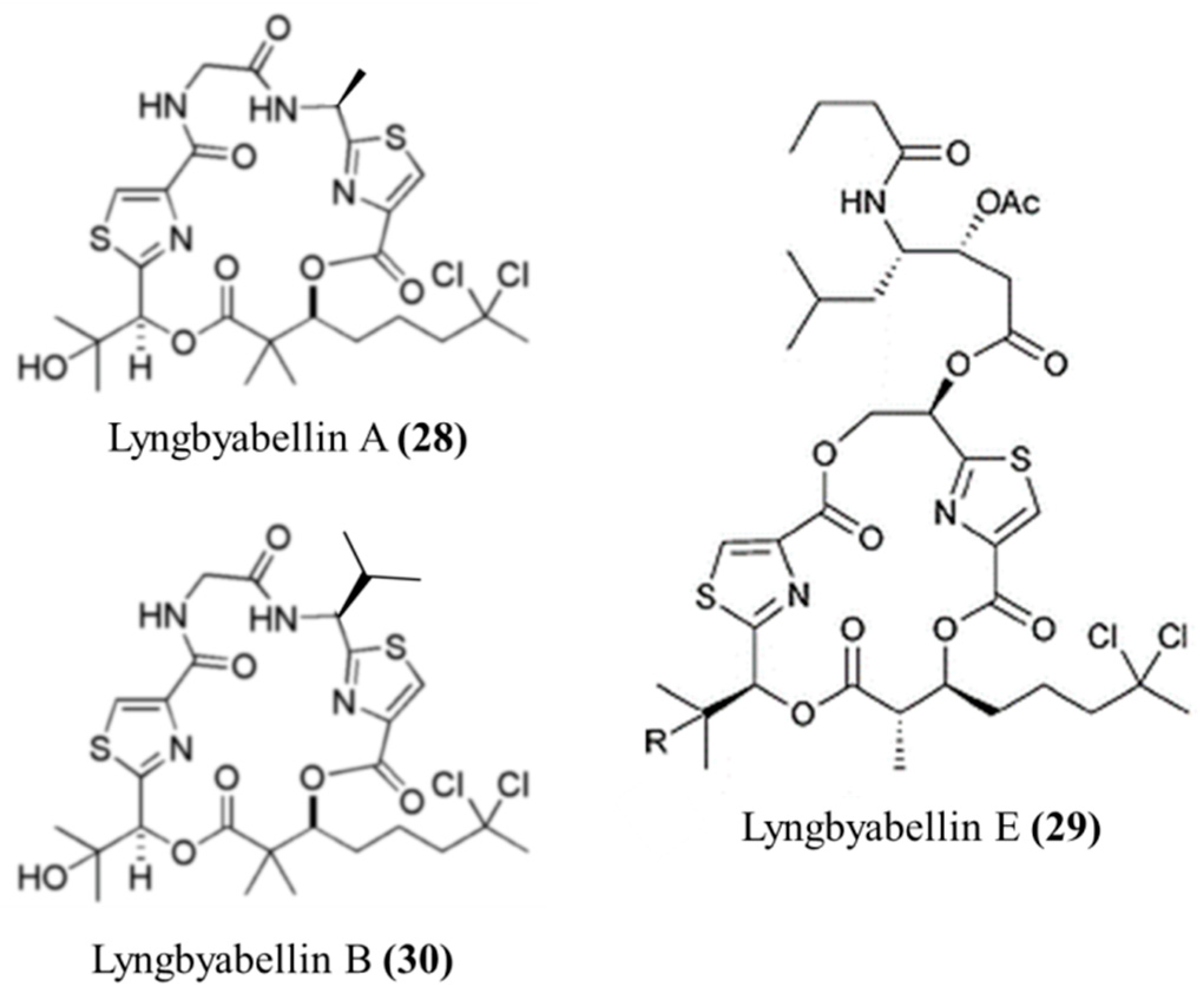
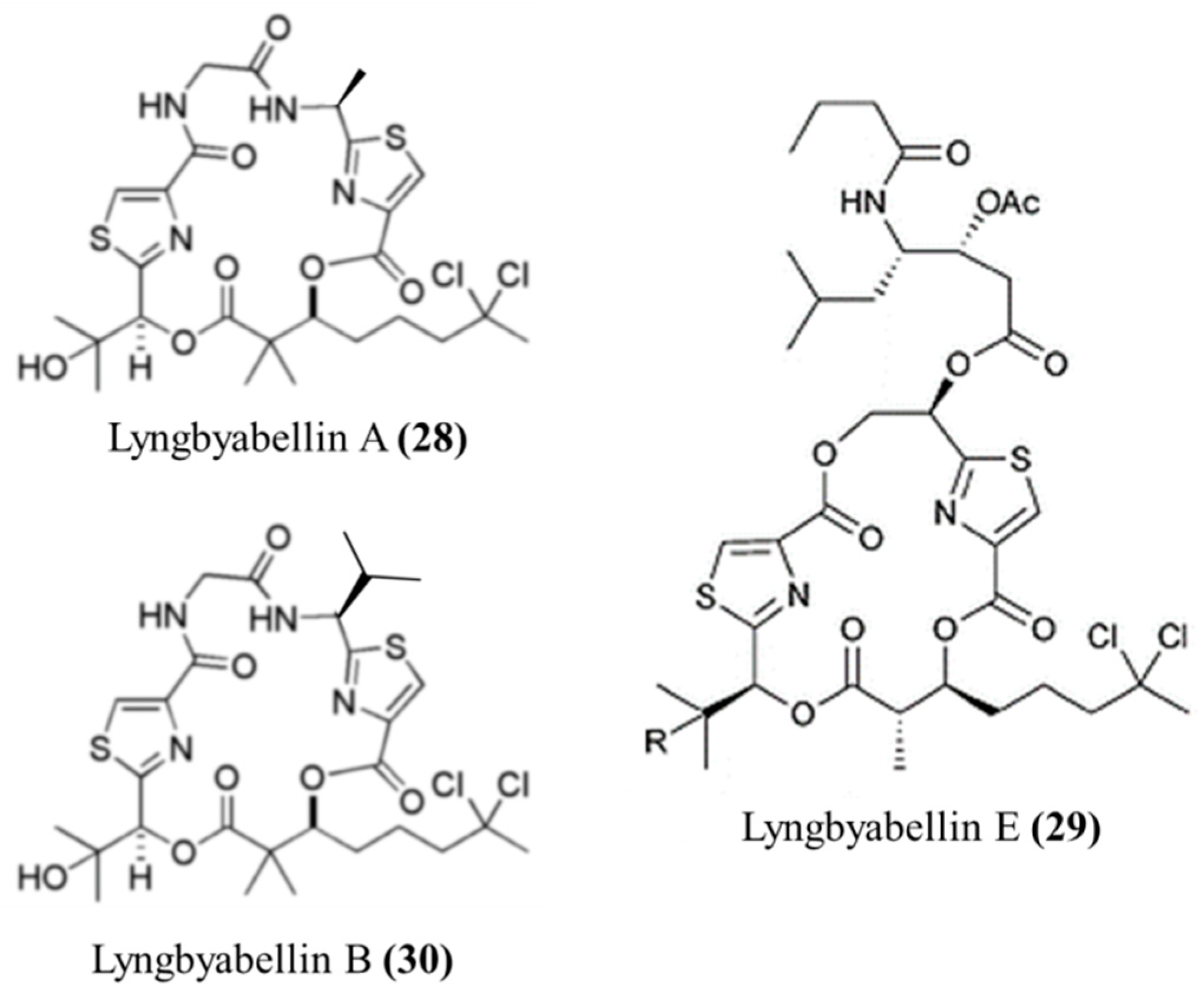
3.1.15. Lyngbyabellin A, E, and B
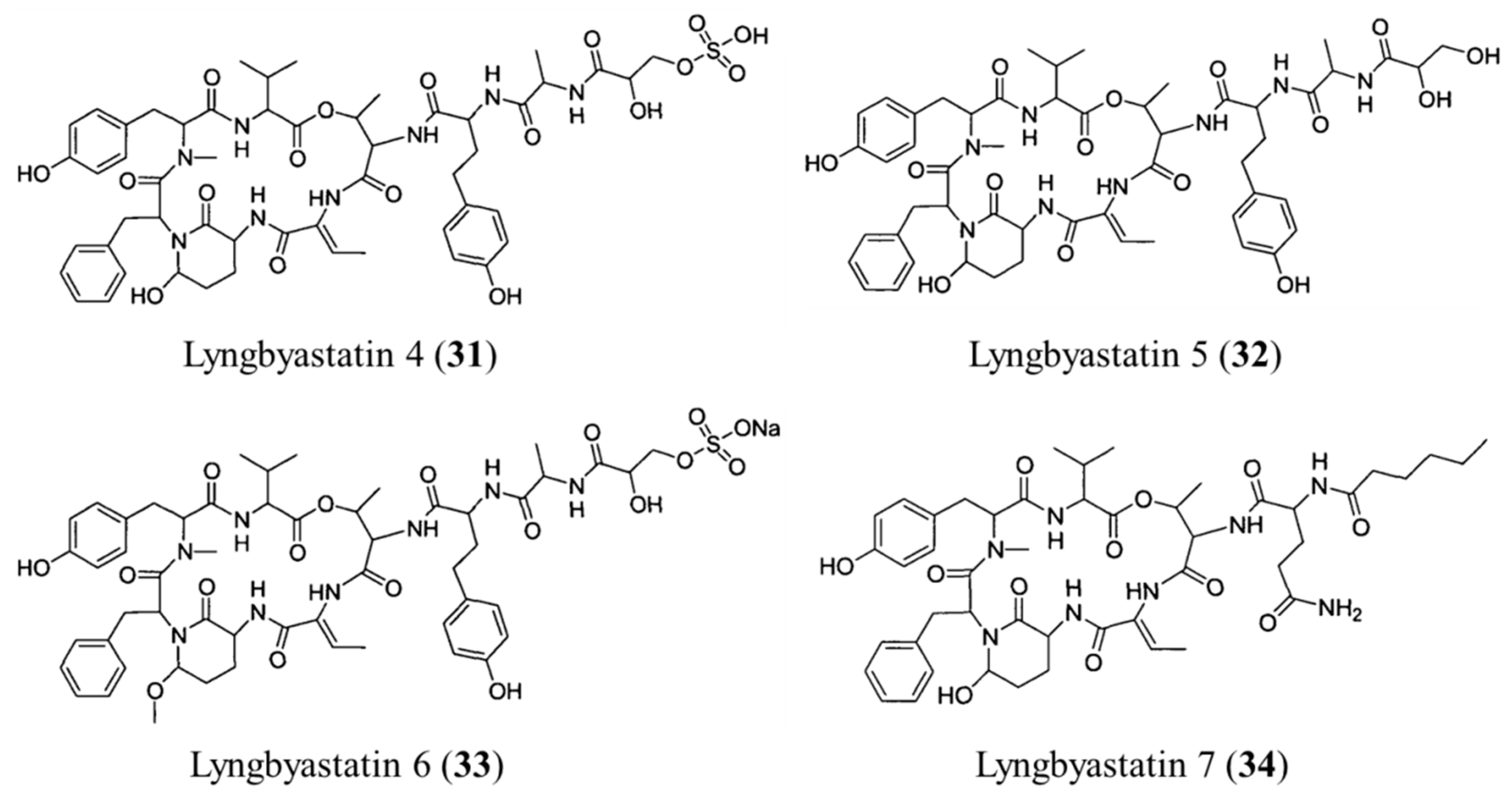
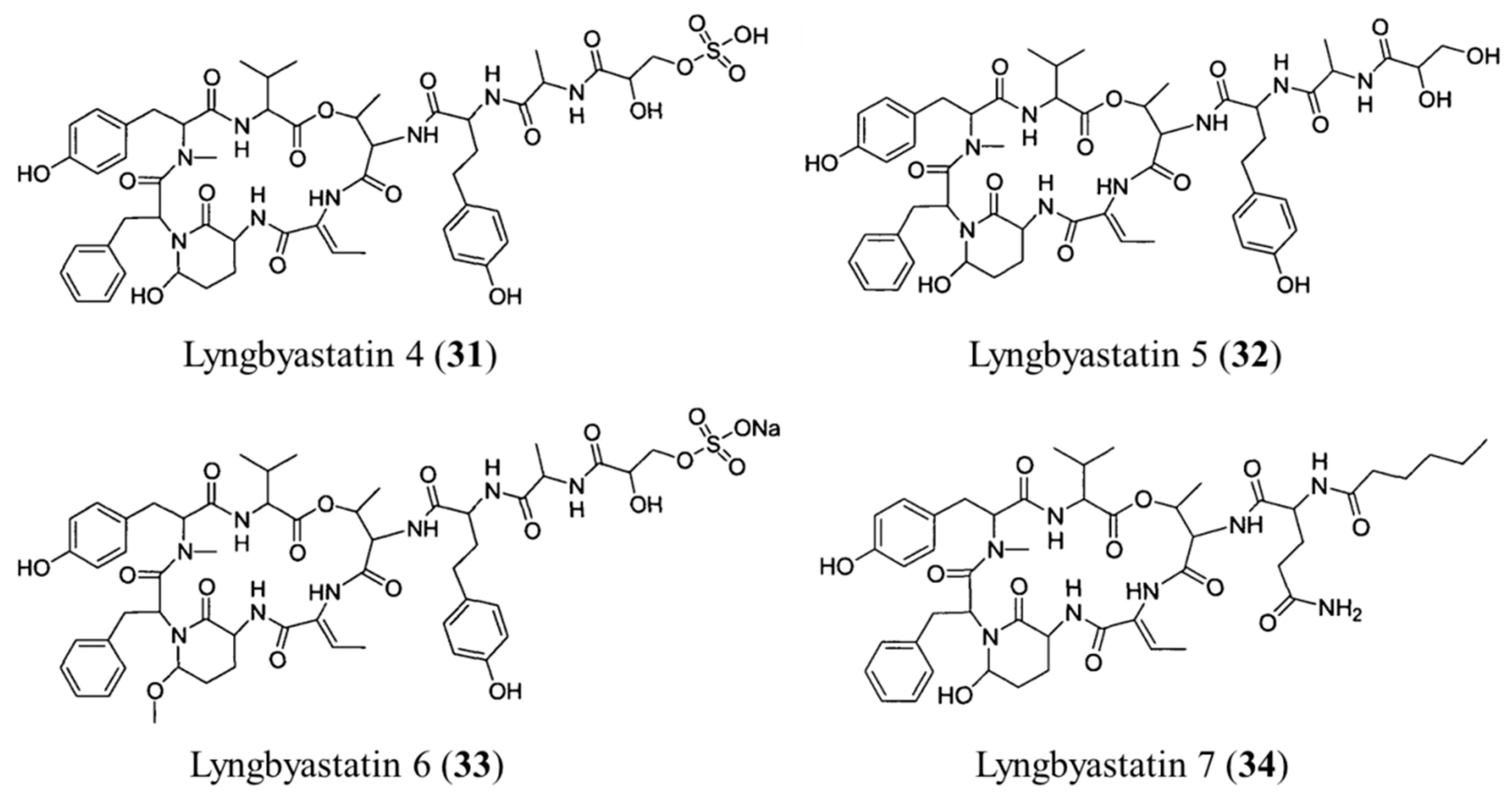
3.1.16. Lyngbyastatin 4–7
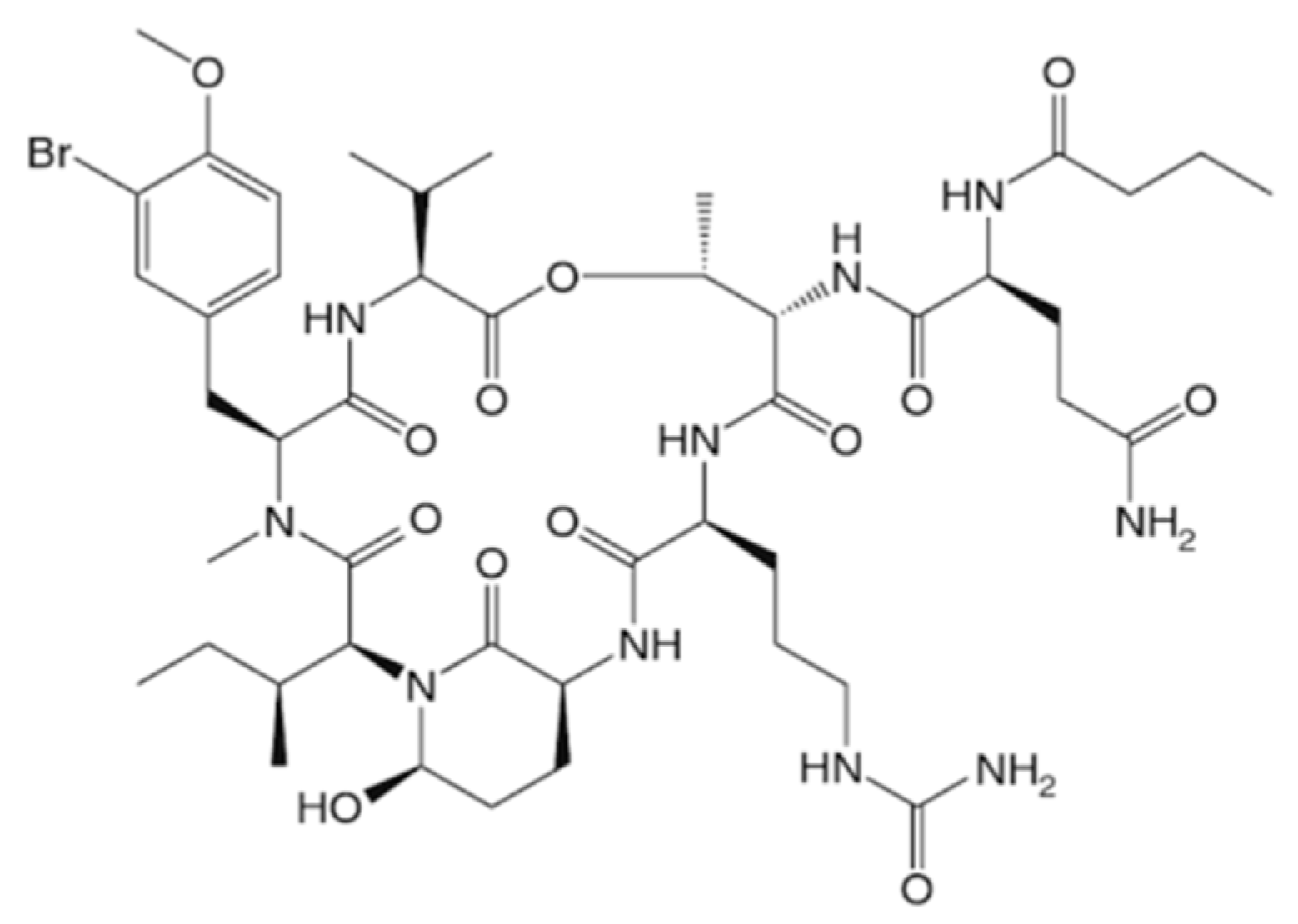
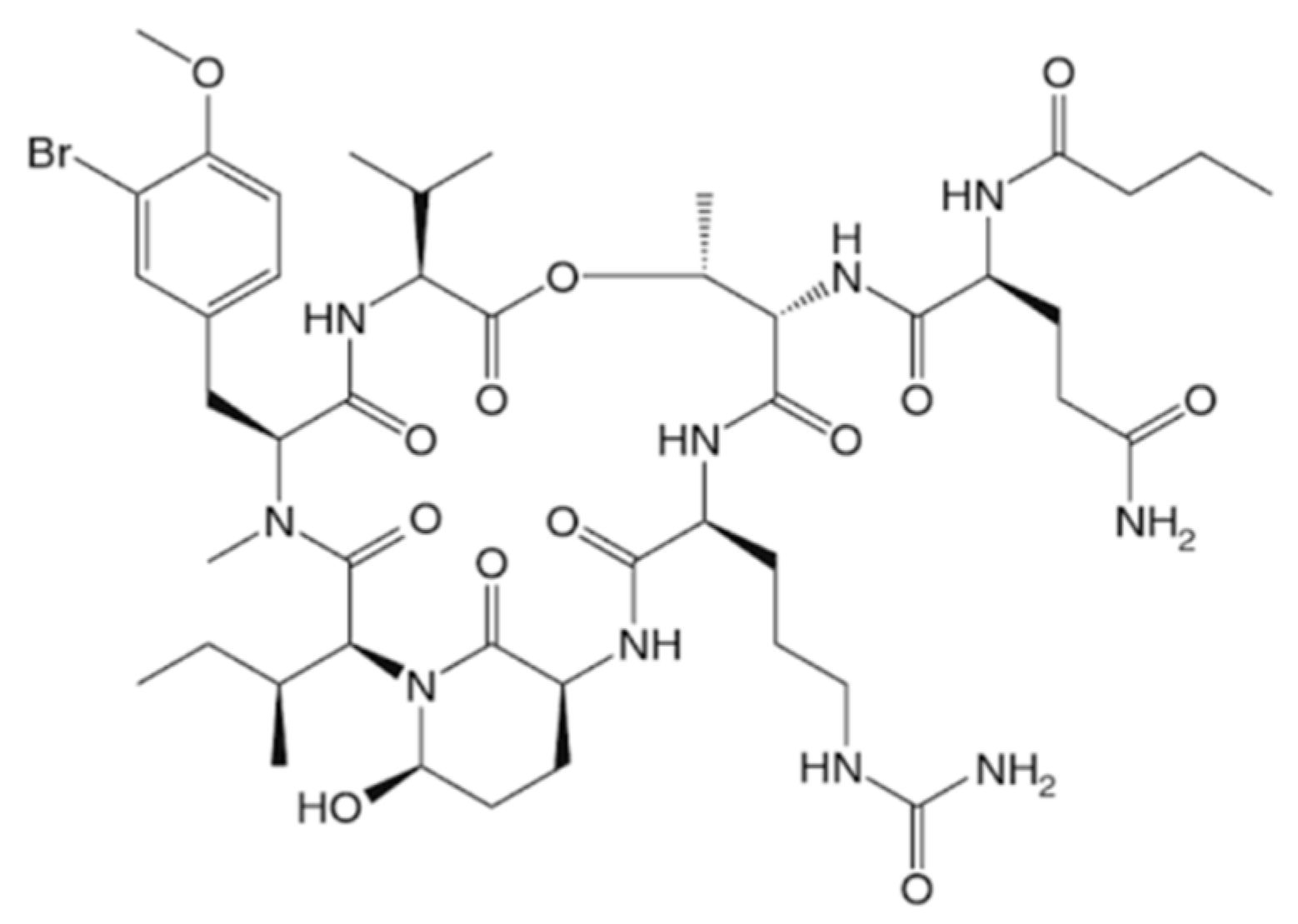
3.1.17. Symplocamide A
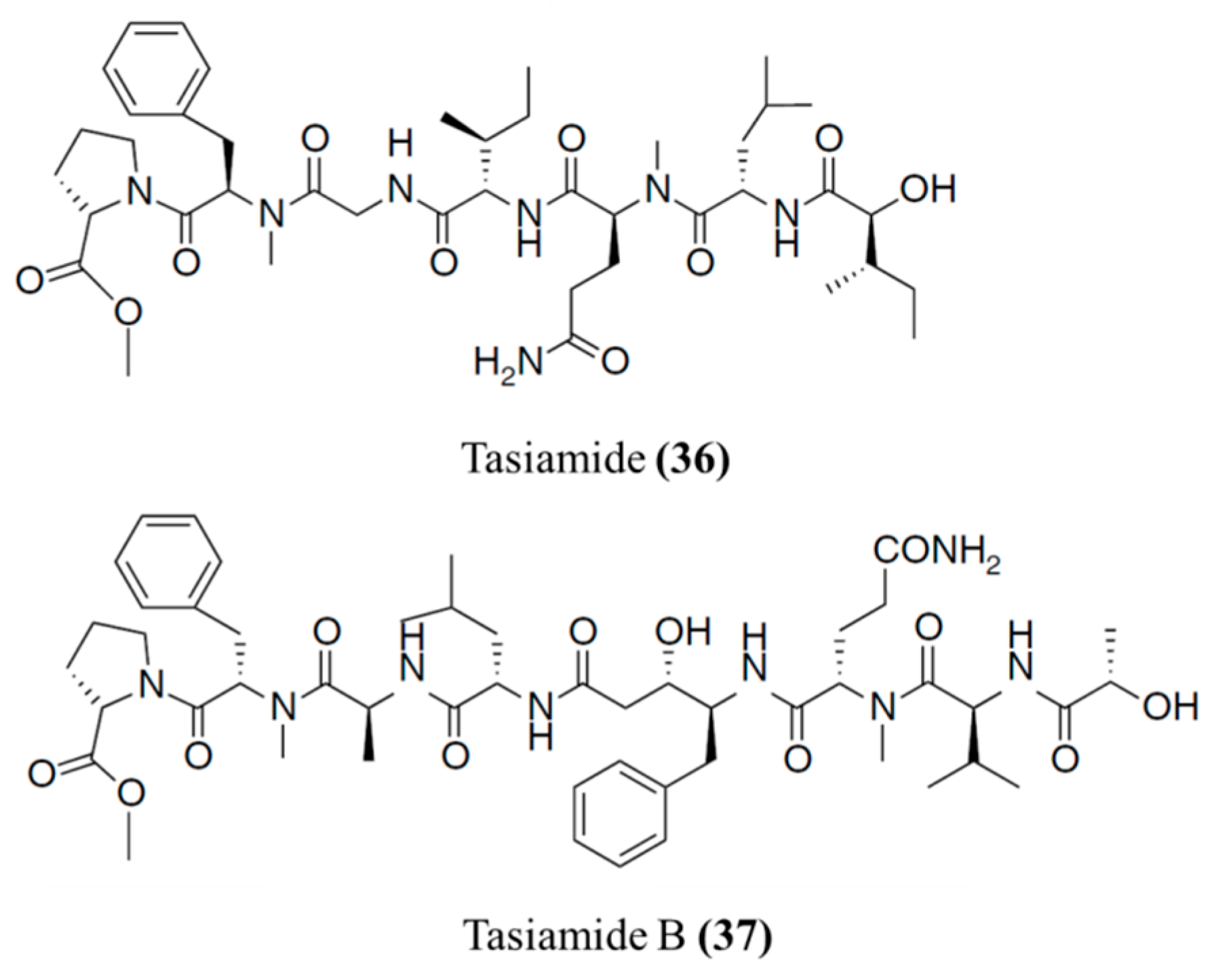
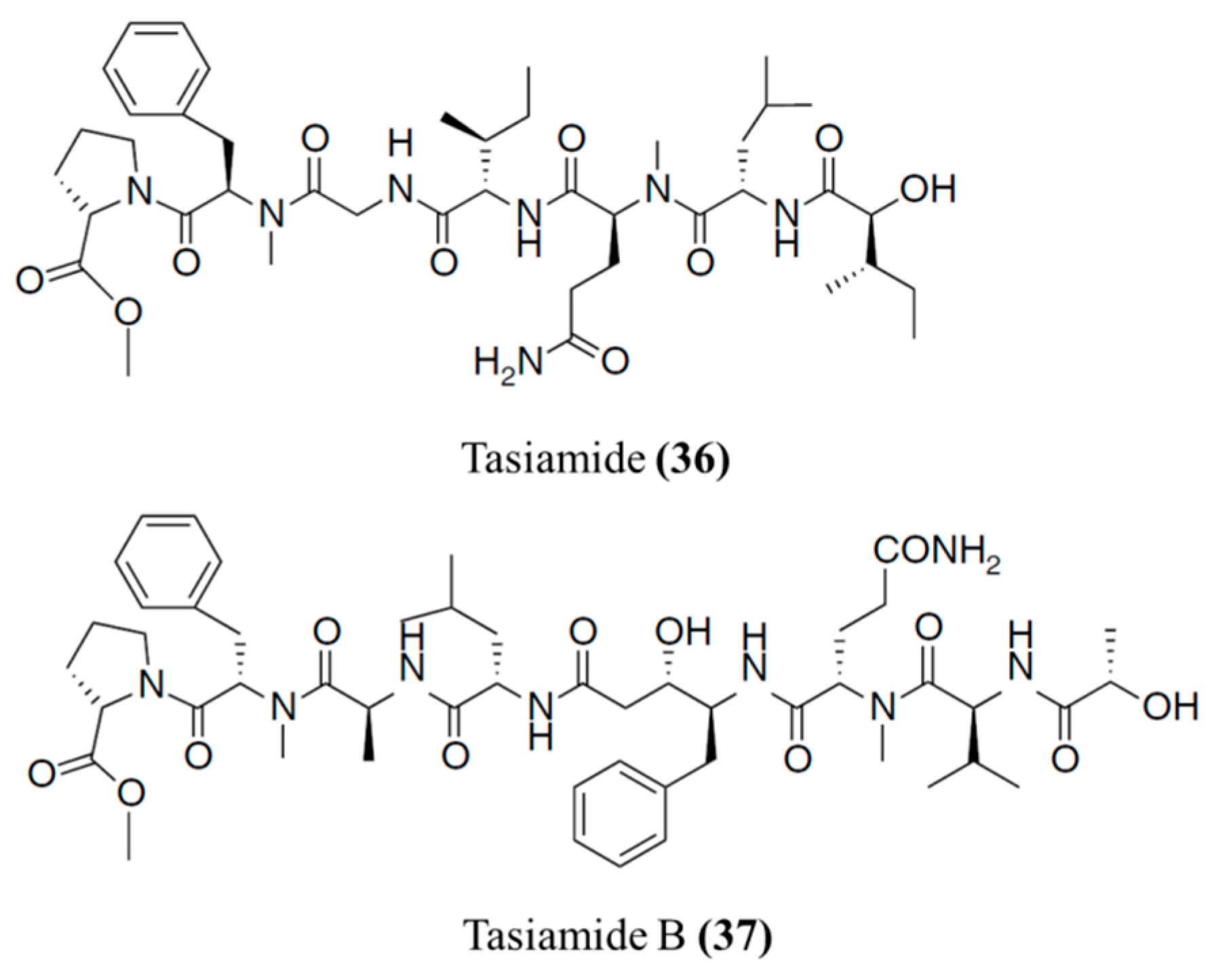
3.1.18. Tasiamides
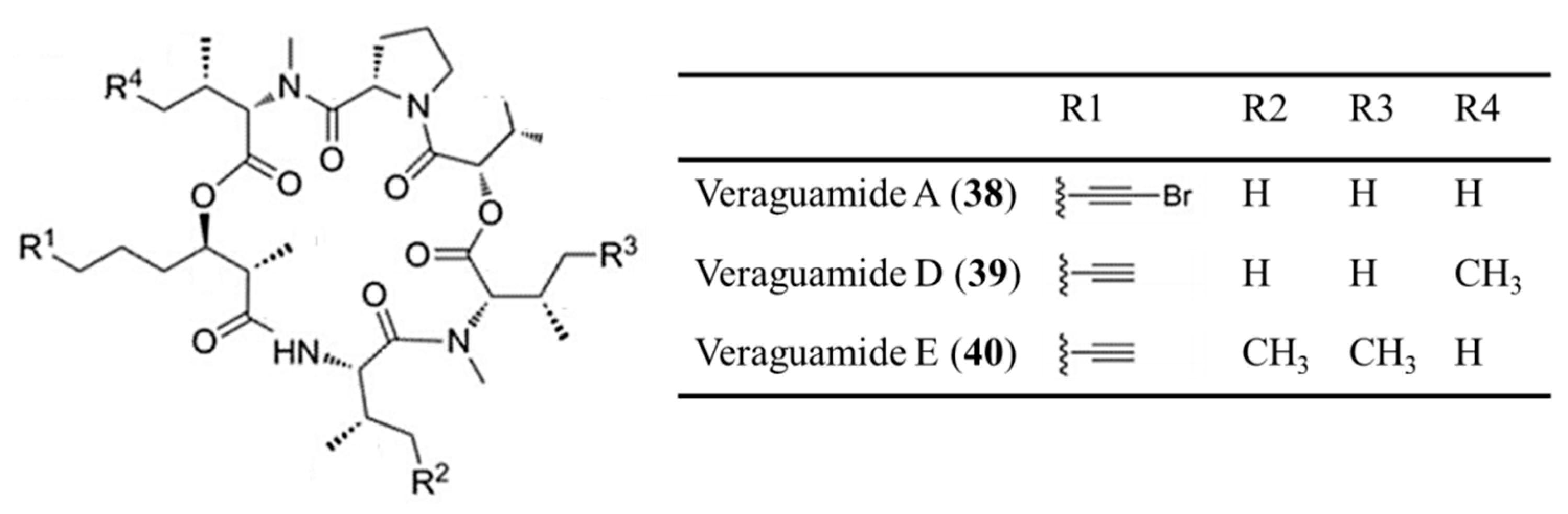
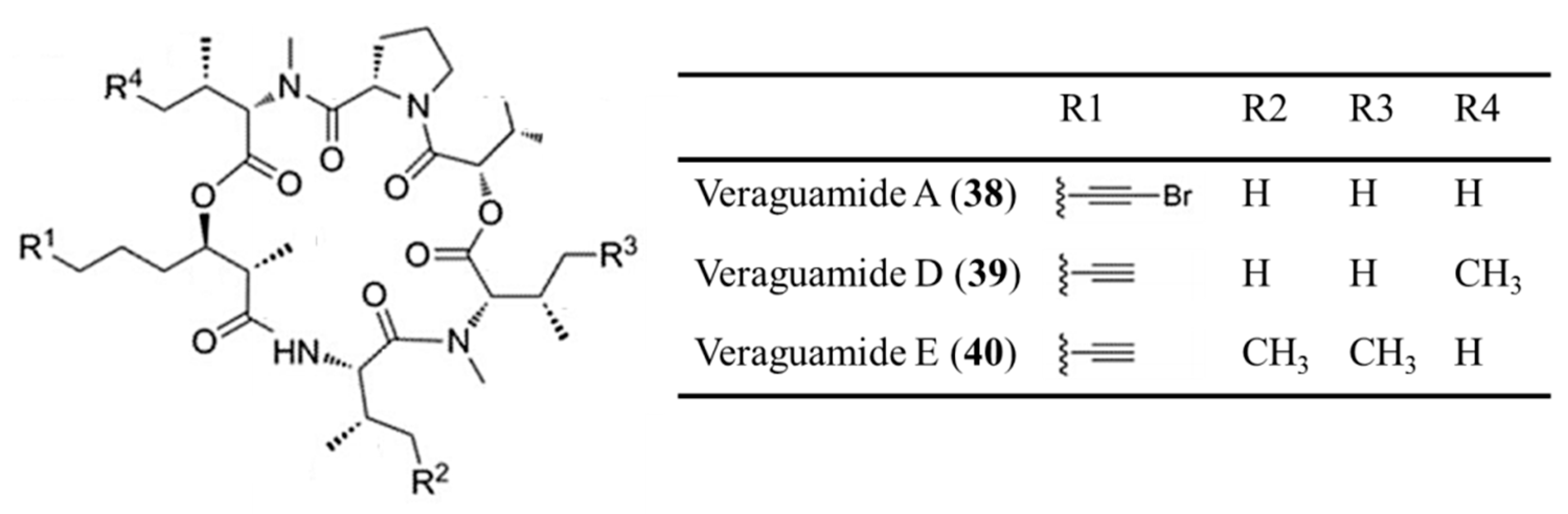
3.1.19. Veraguamide A, D, and E
3.2. Fungi-Derived Peptides
3.2.1. Azonazine
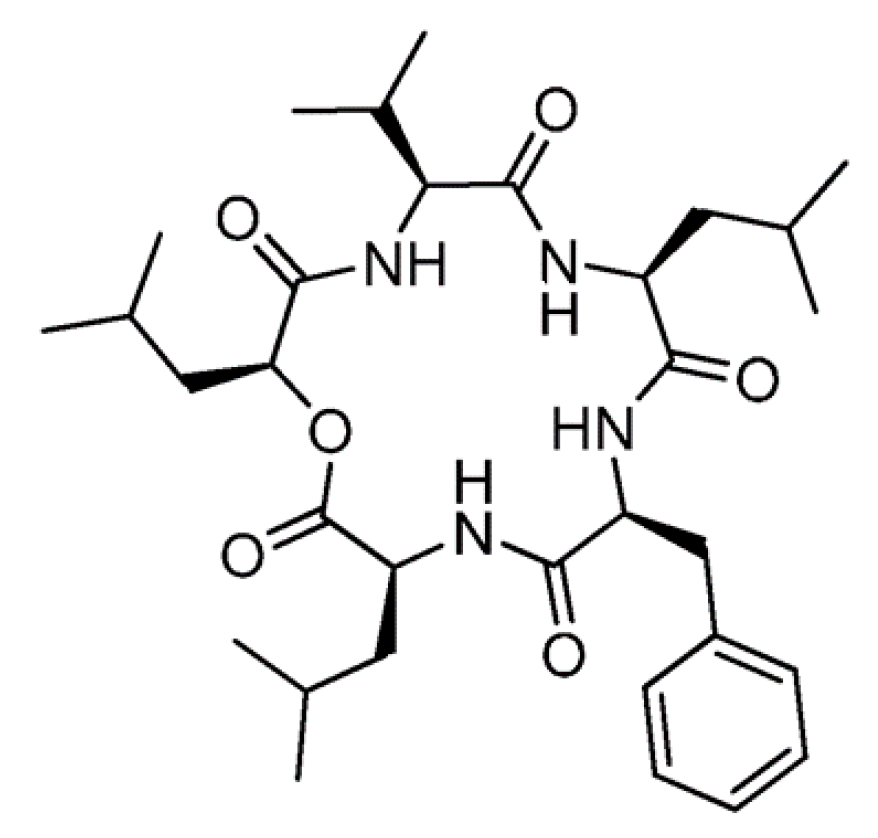
3.2.2. Sansalvamide A
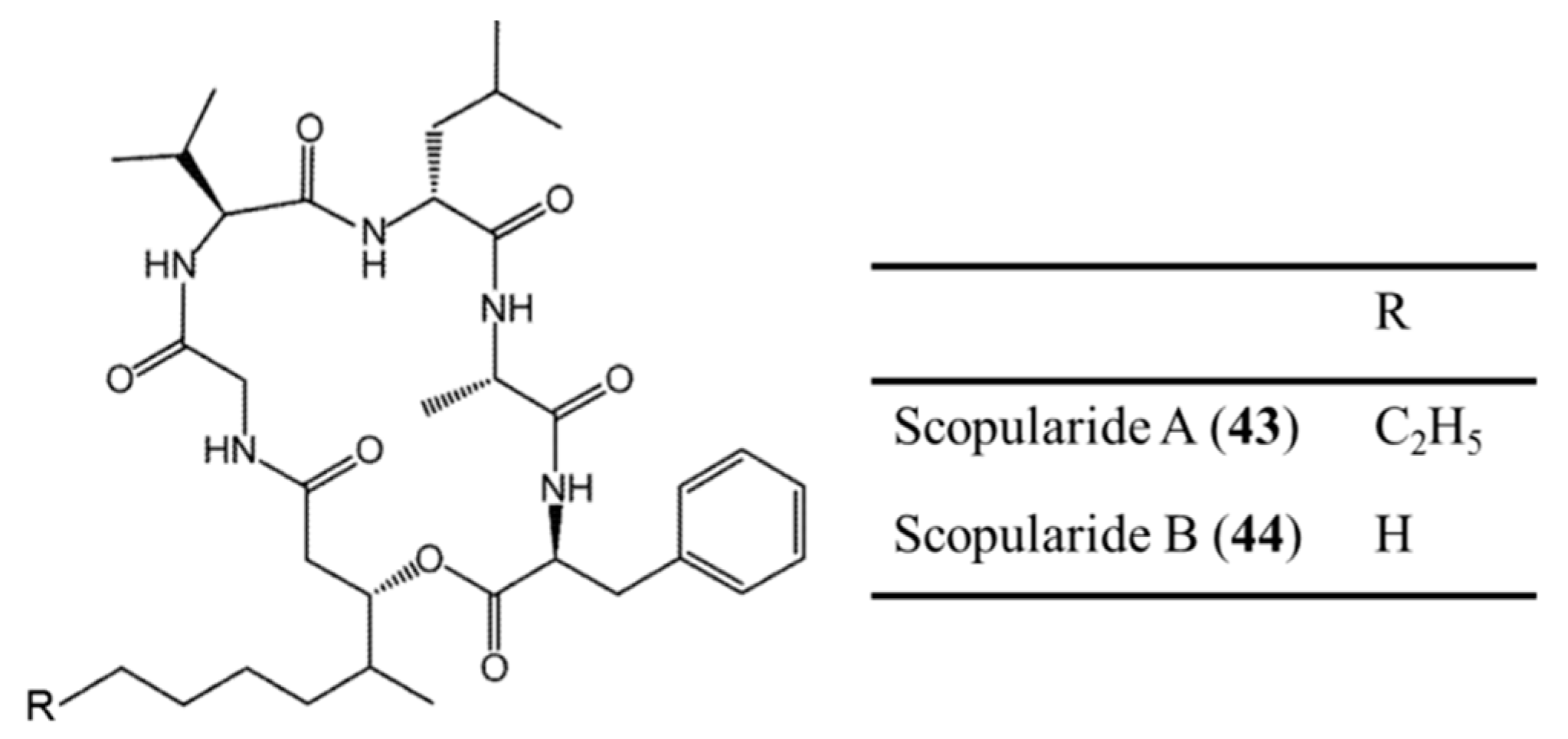
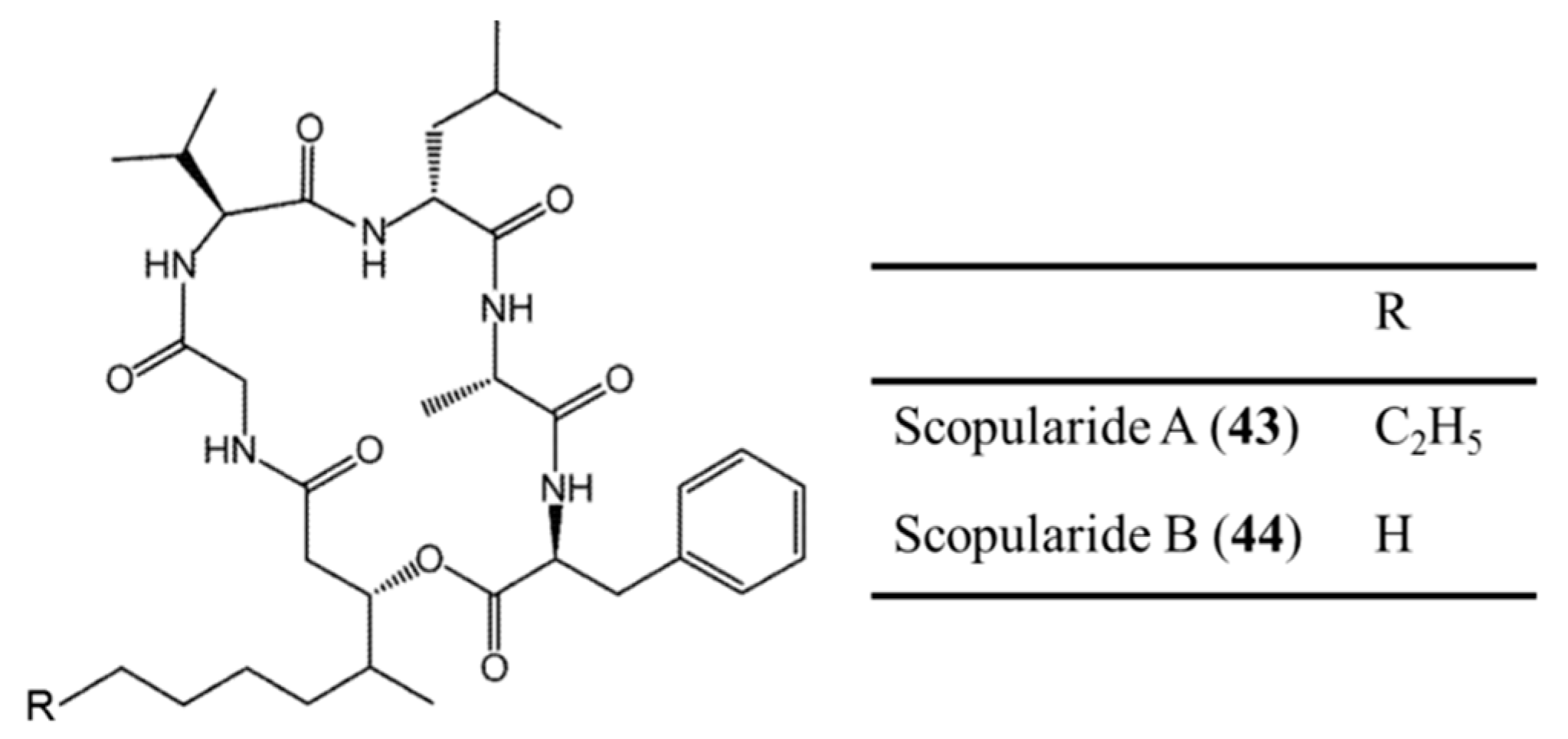
3.2.3. Scopularide A and B
3.3. Sponge-Derived Peptides
3.3.1. Arenastatin A
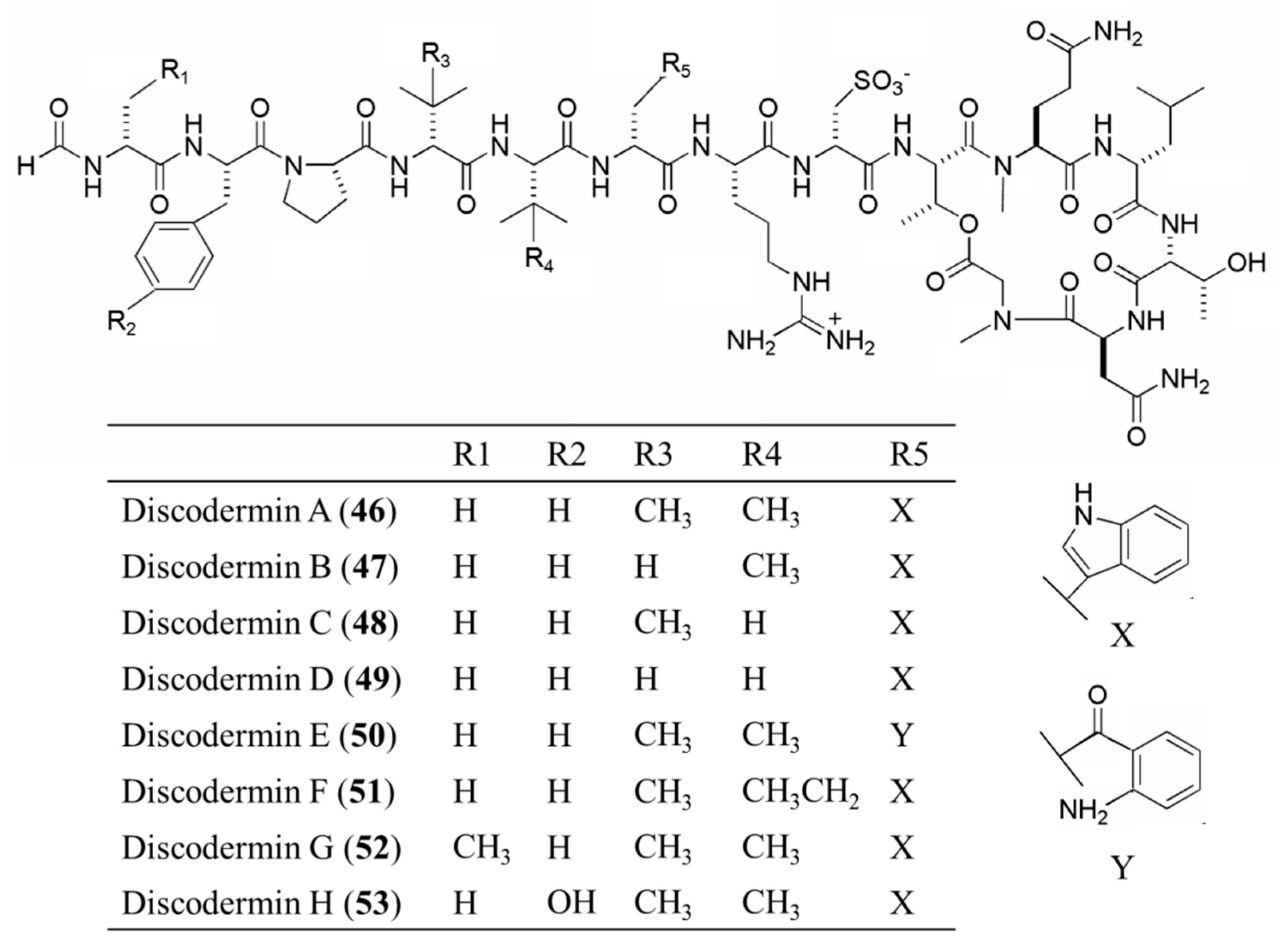
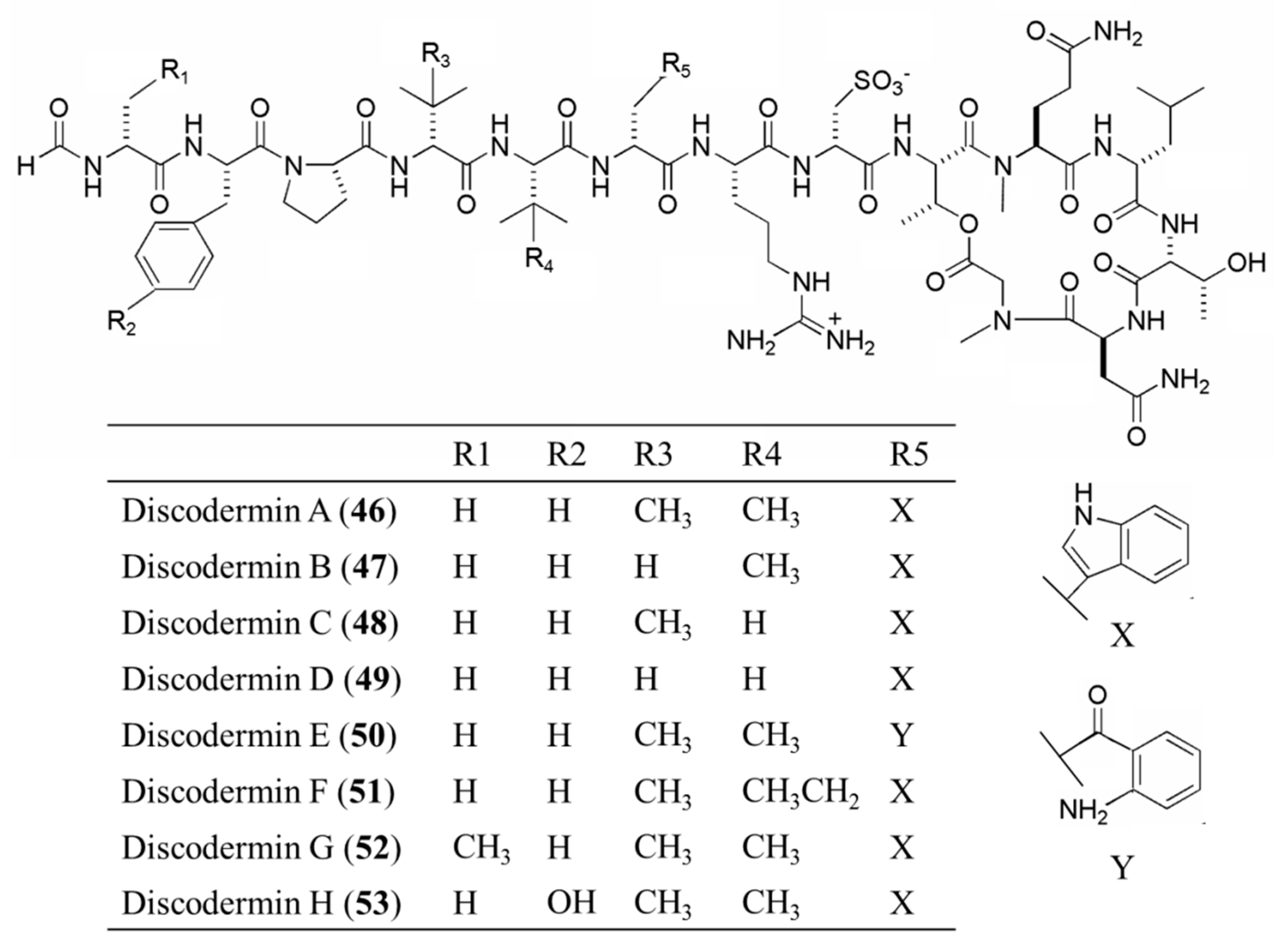
3.3.2. Discodermin A–H
3.3.3. Geodiamolide H
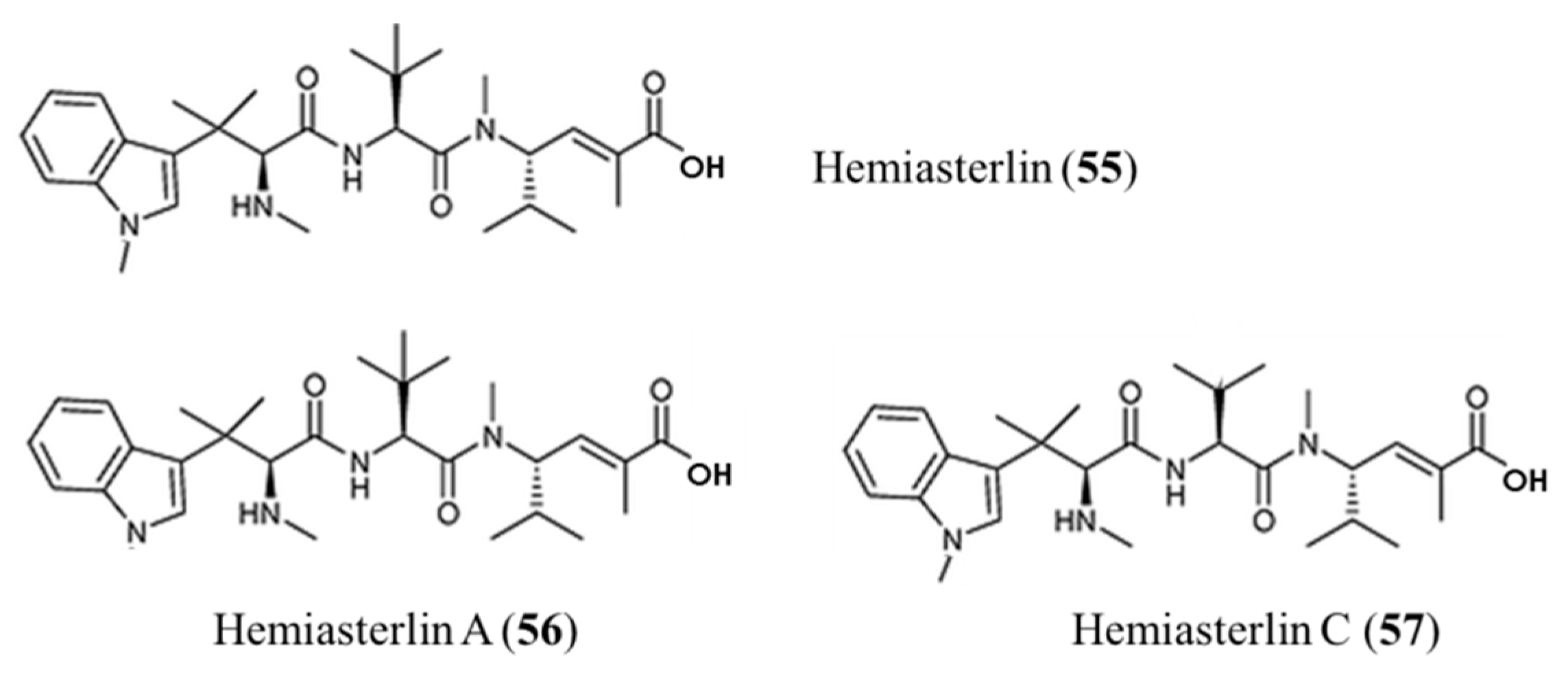
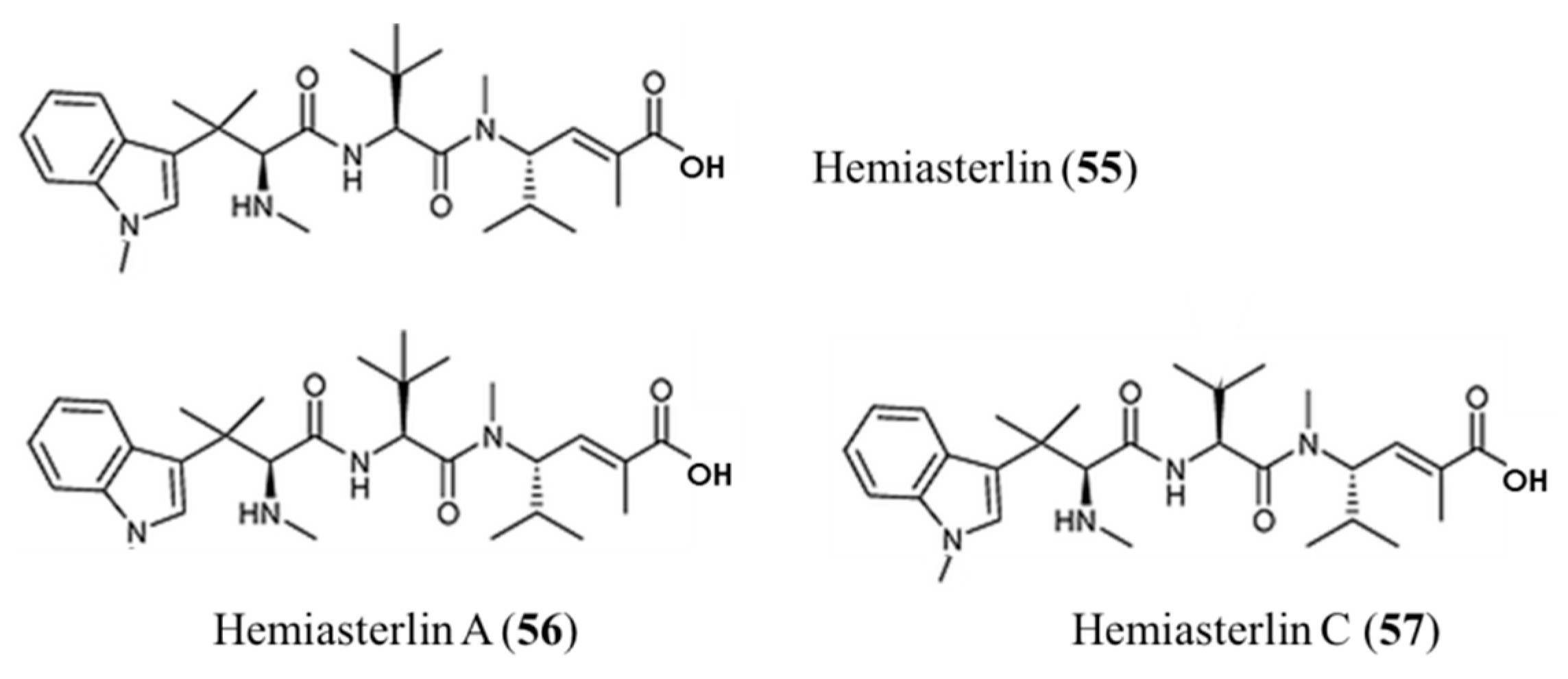
3.3.4. Hemiasterlin
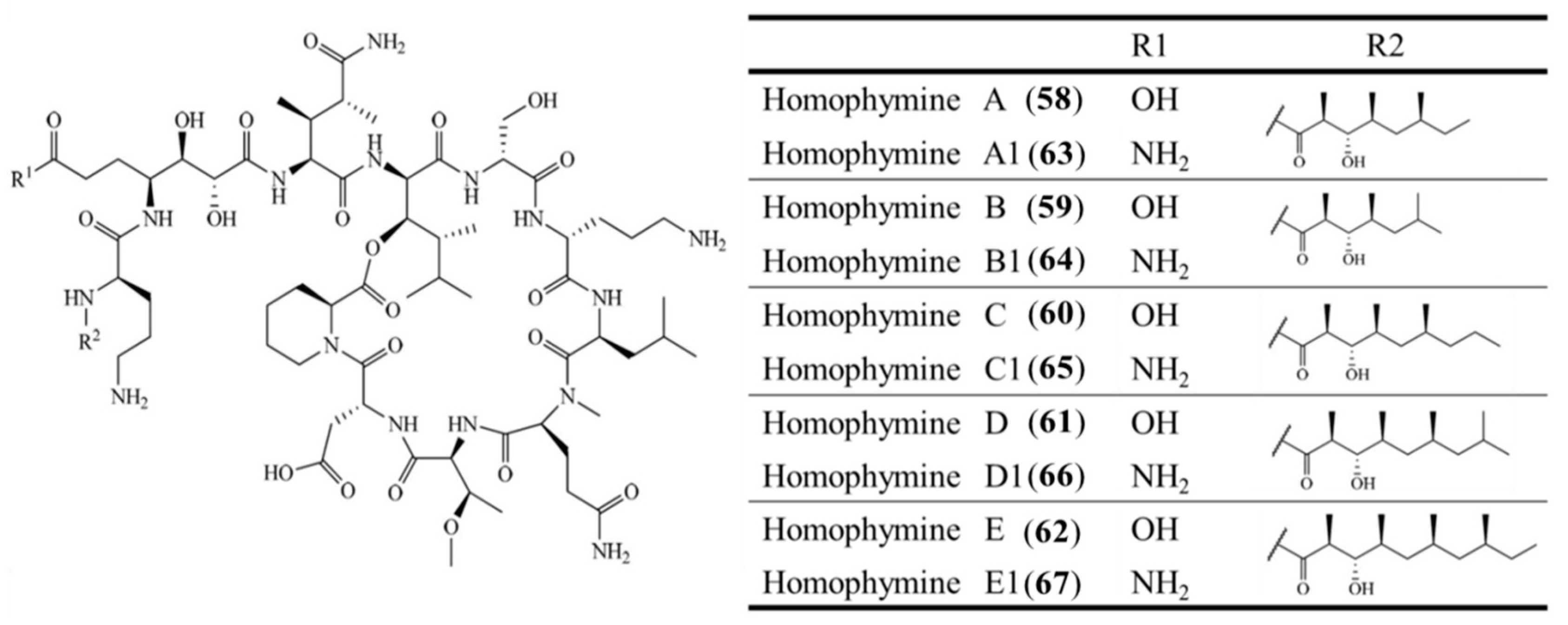
3.3.5. Homophymine A–E and A1–E1
3.3.6. Jaspamide
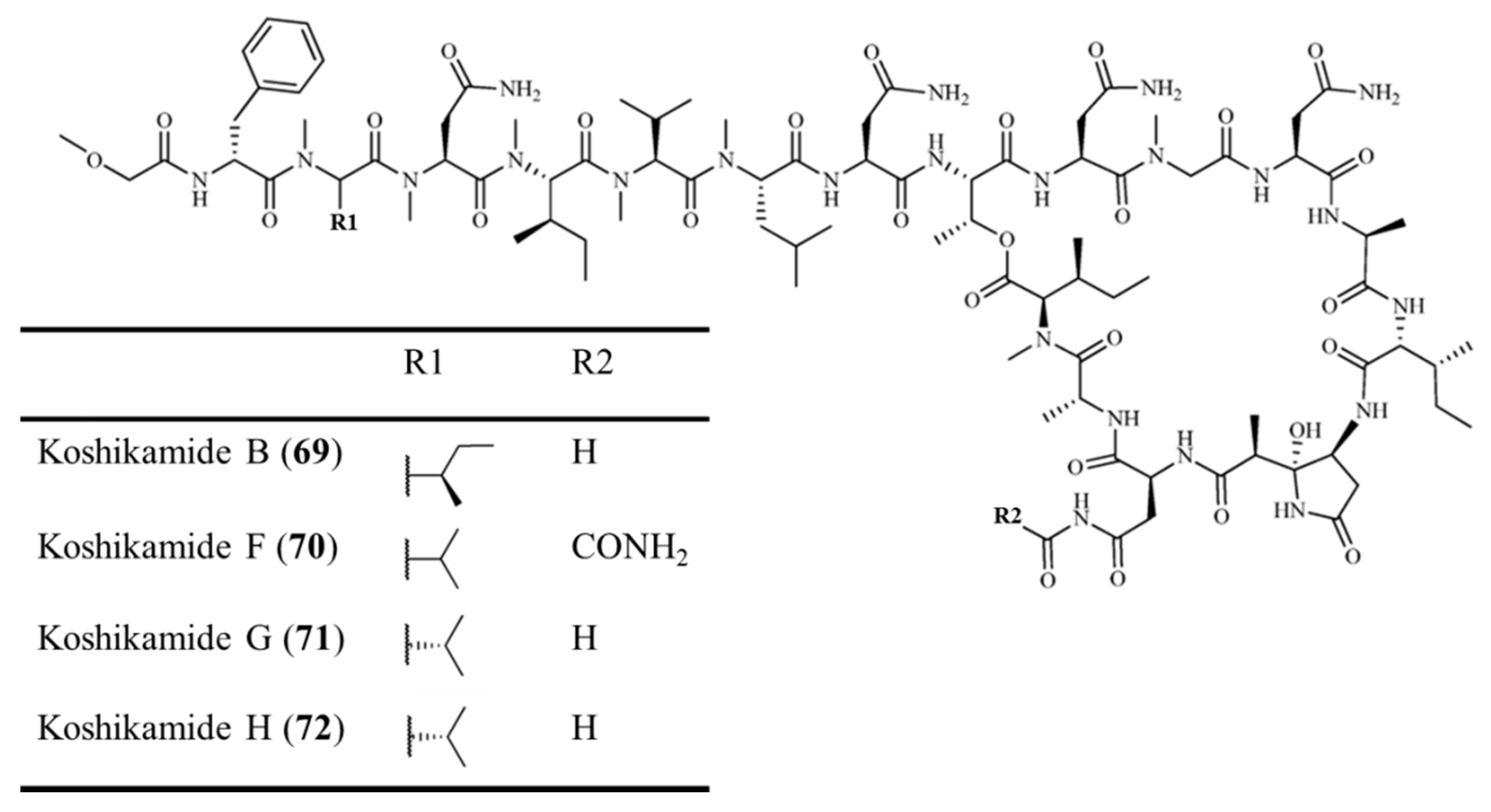
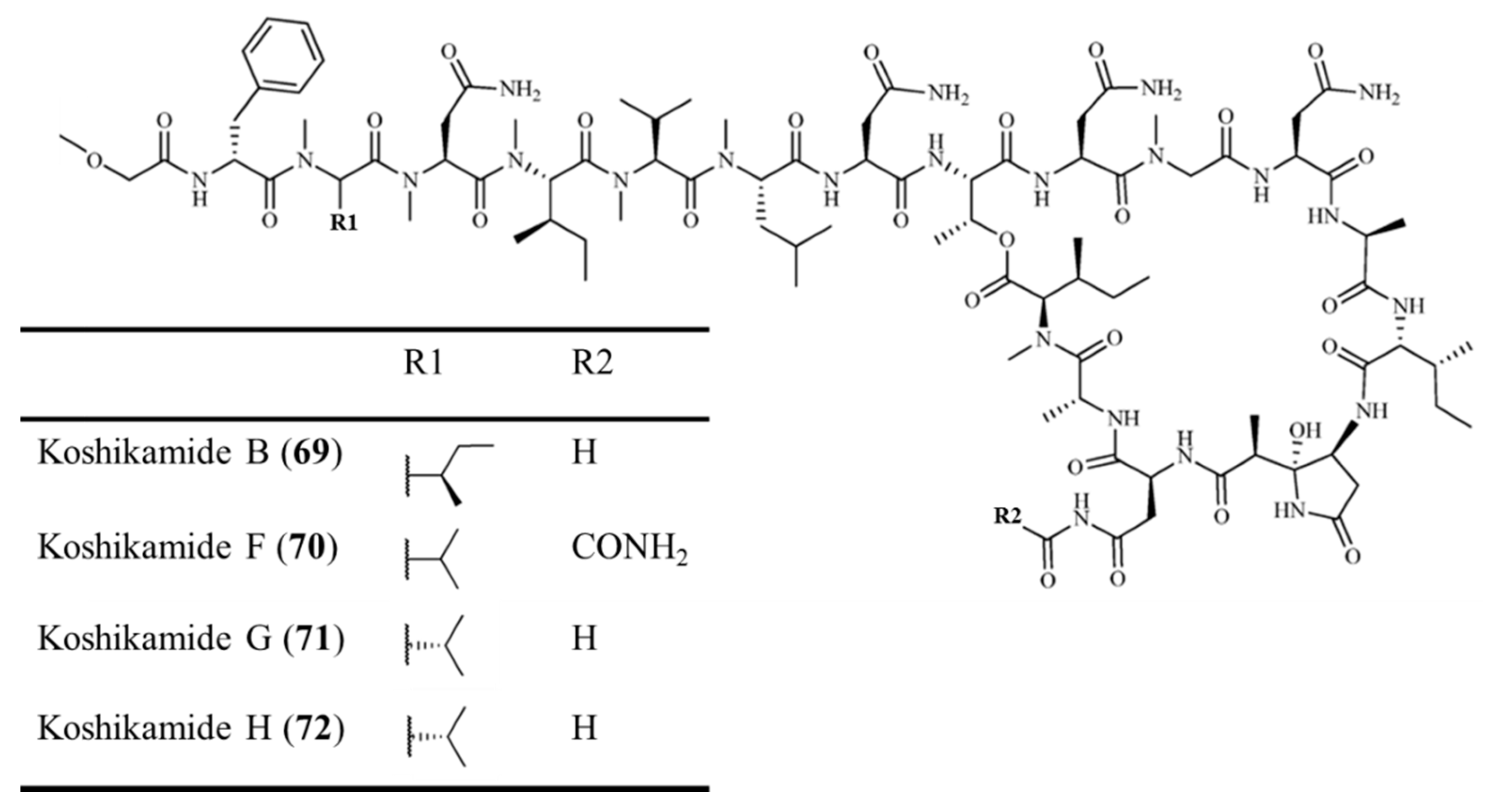
3.3.7. Koshikamide B and F–H
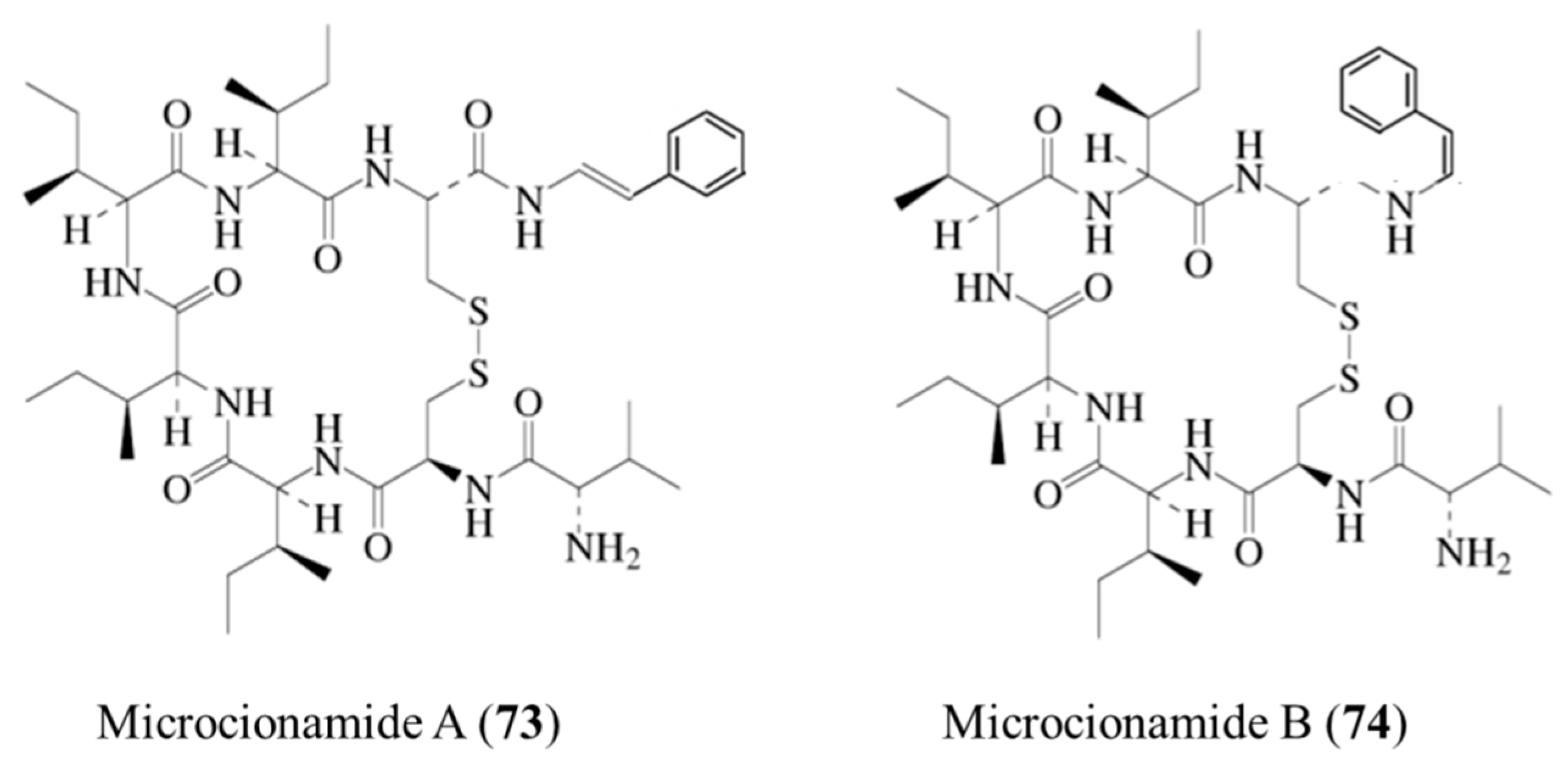
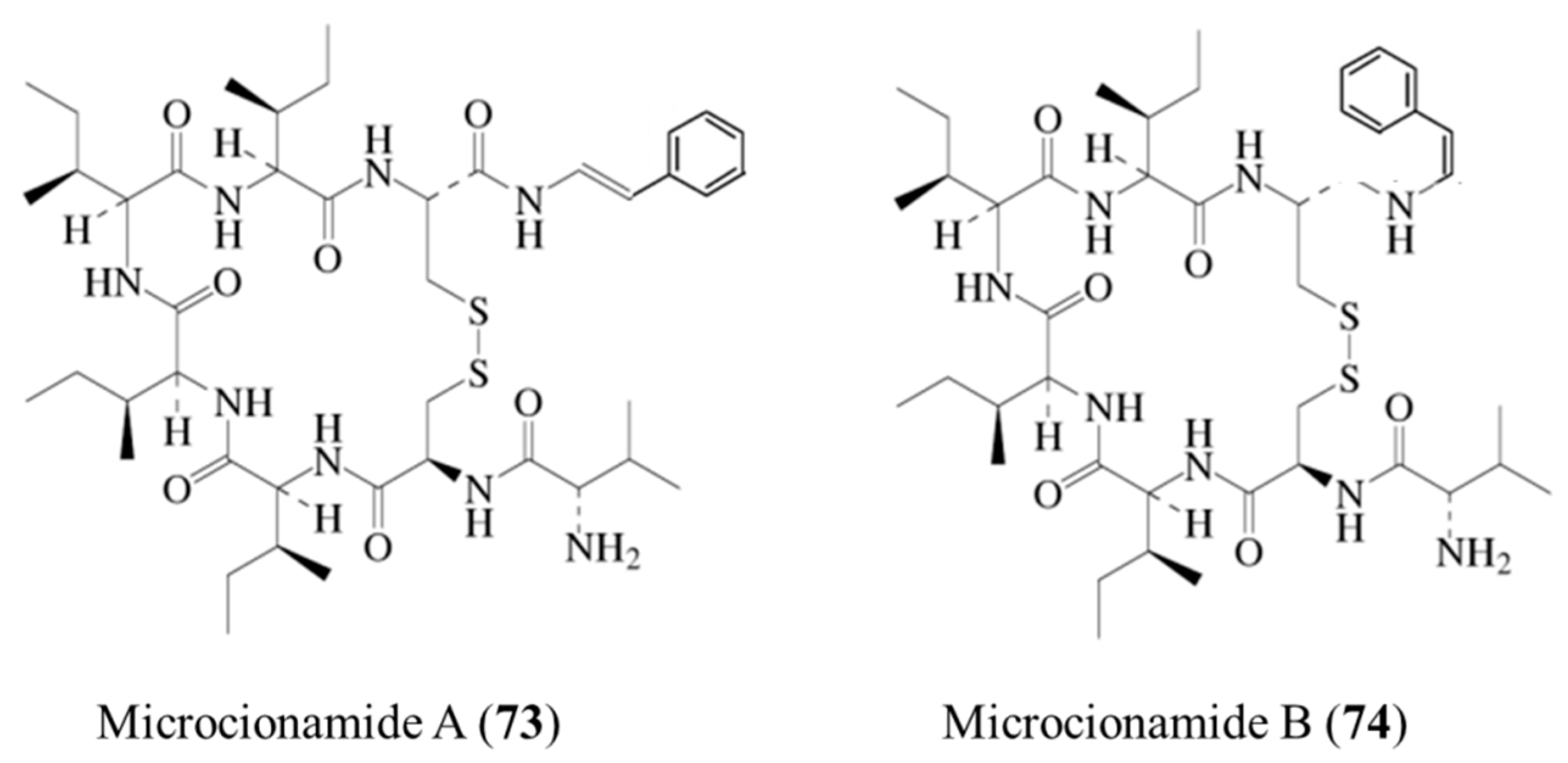
3.3.8. Microcionamide A and B
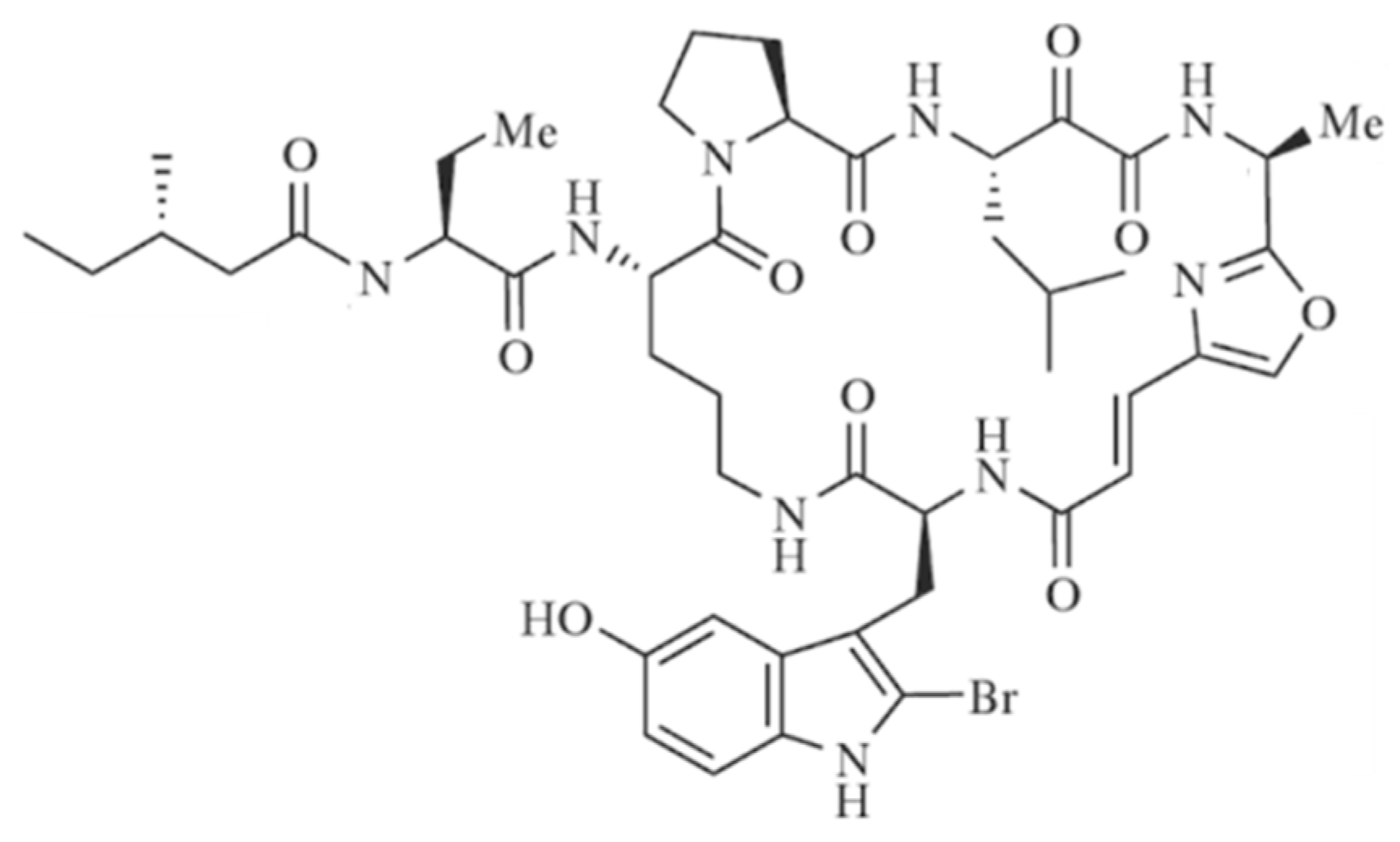
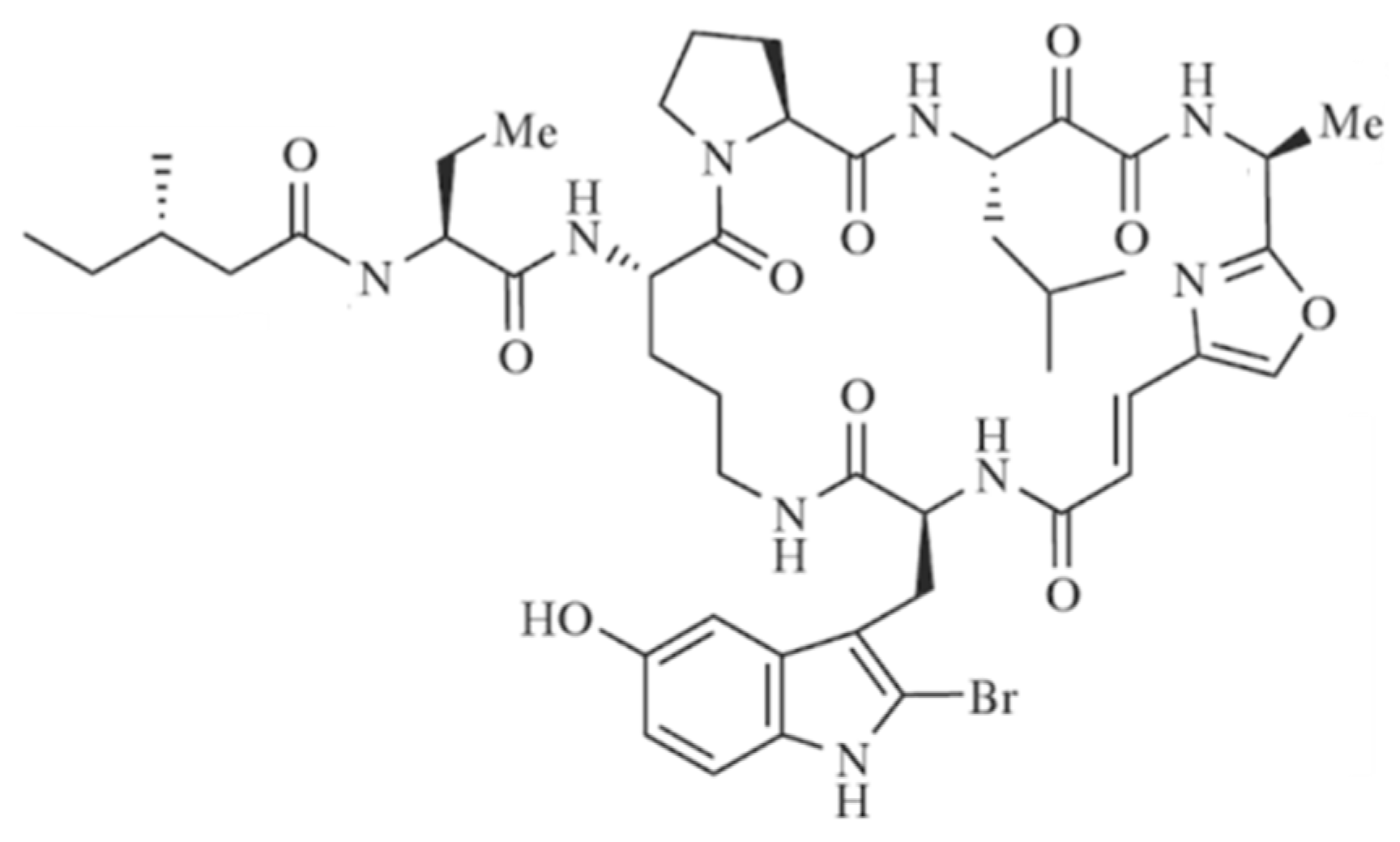
3.3.9. Orbiculamide A
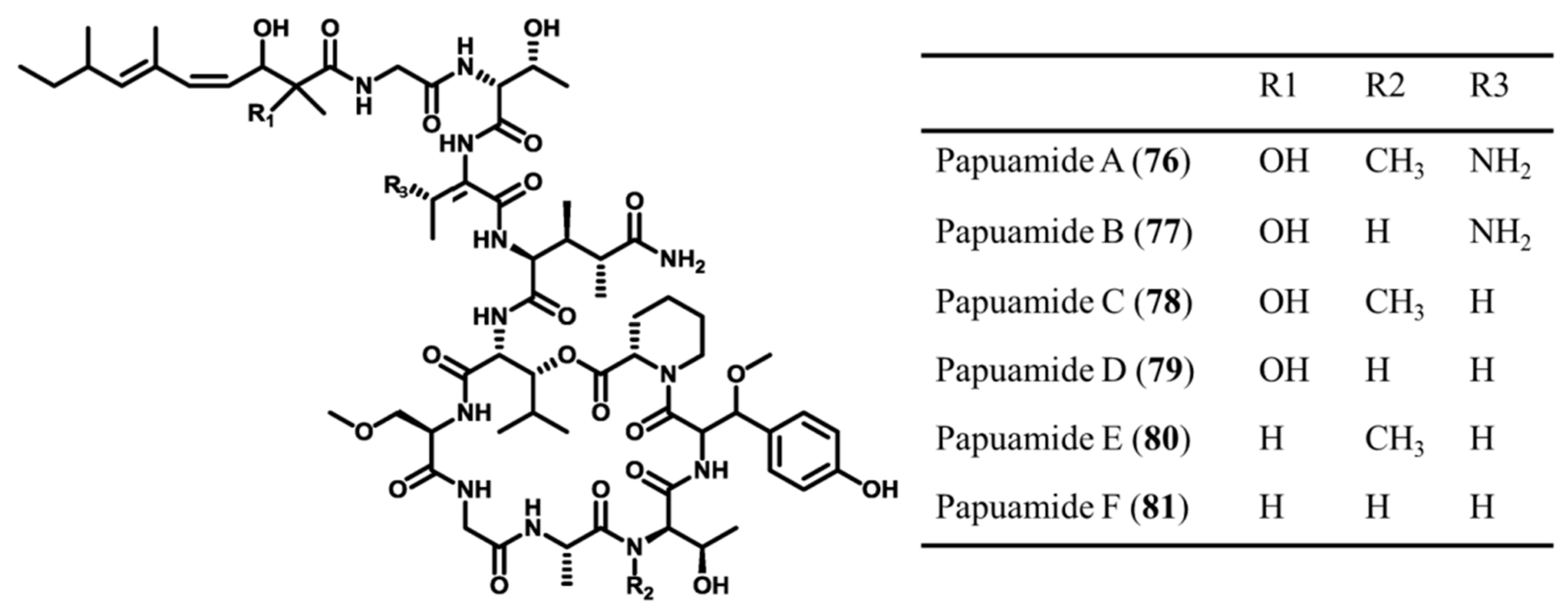
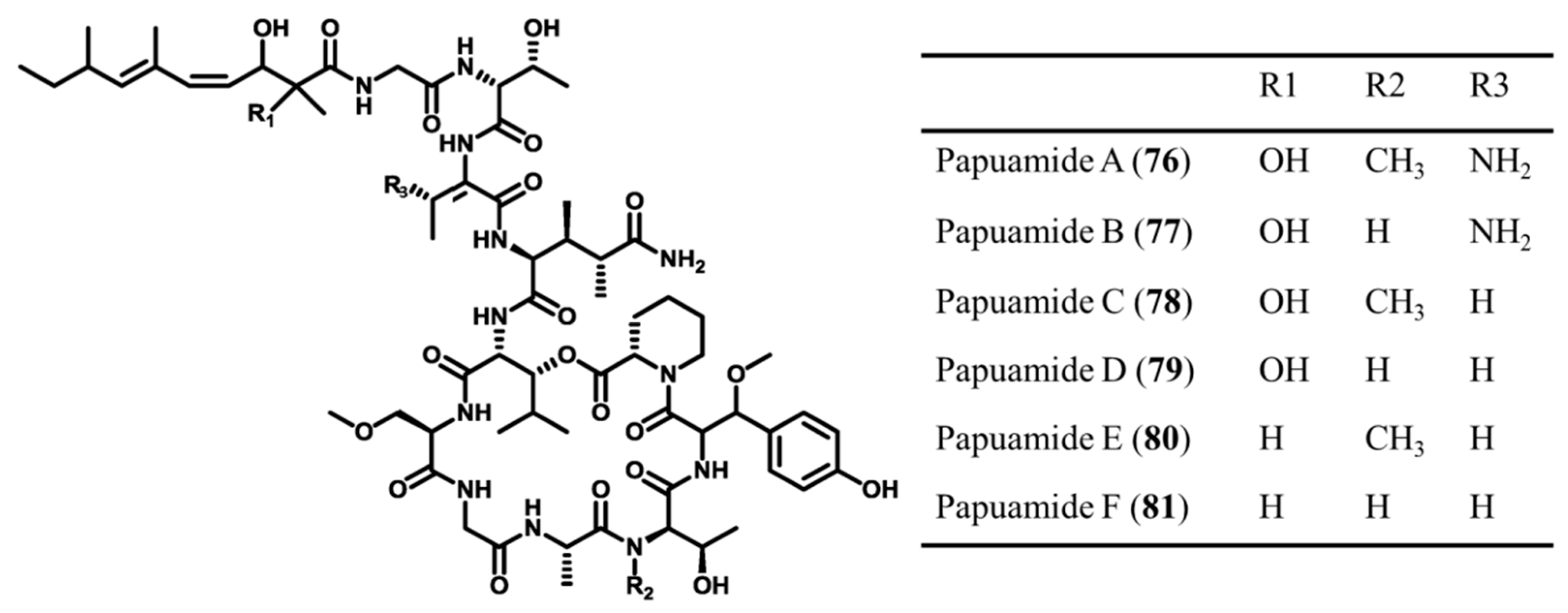
3.3.10. Papuamides
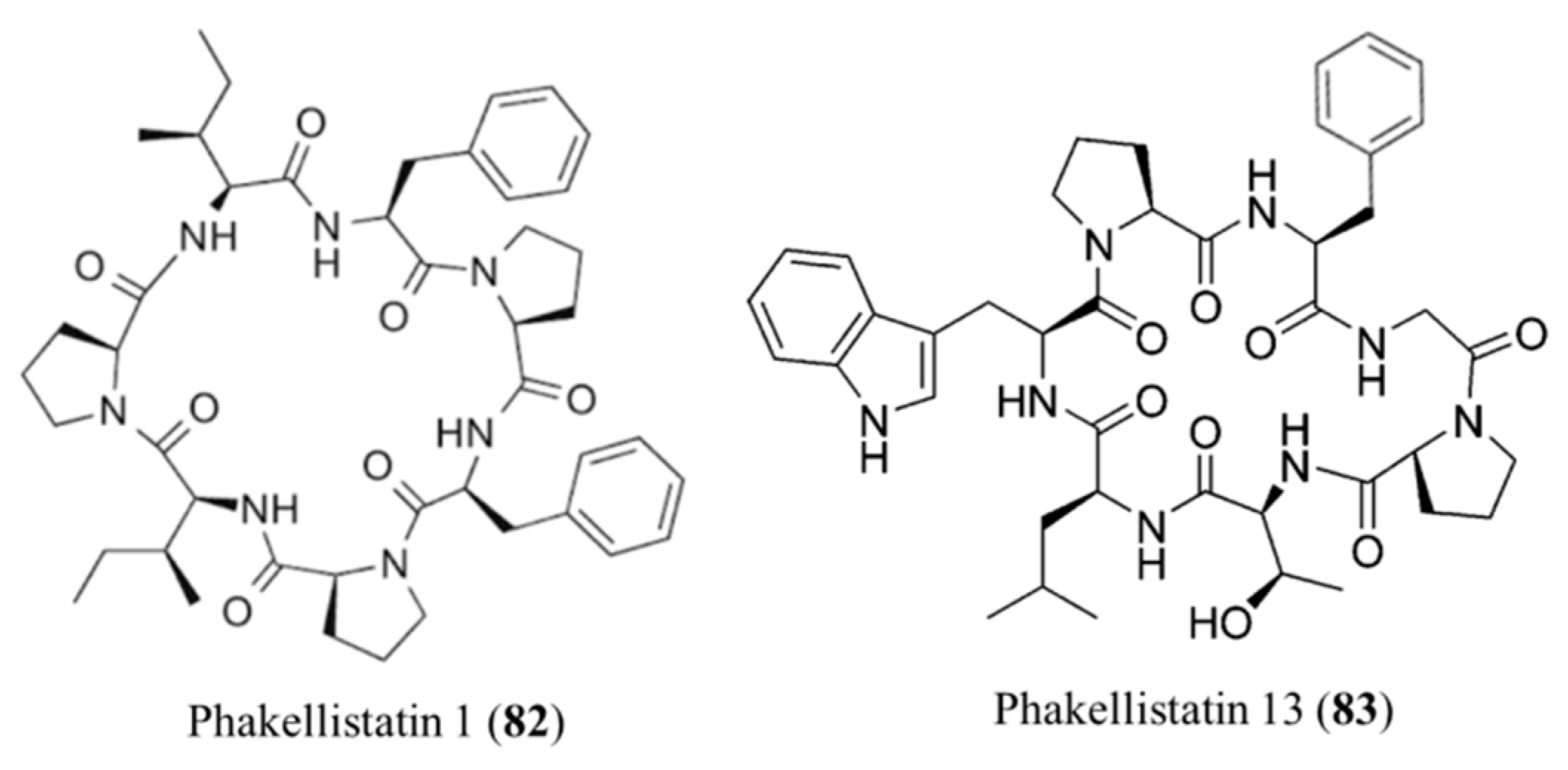
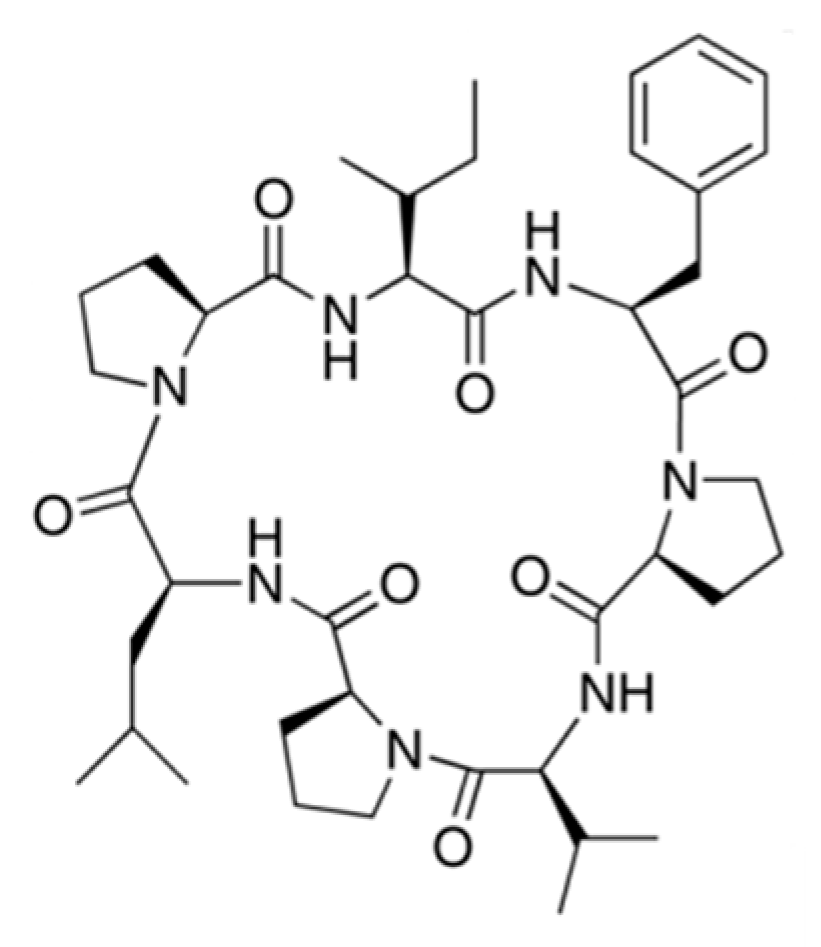
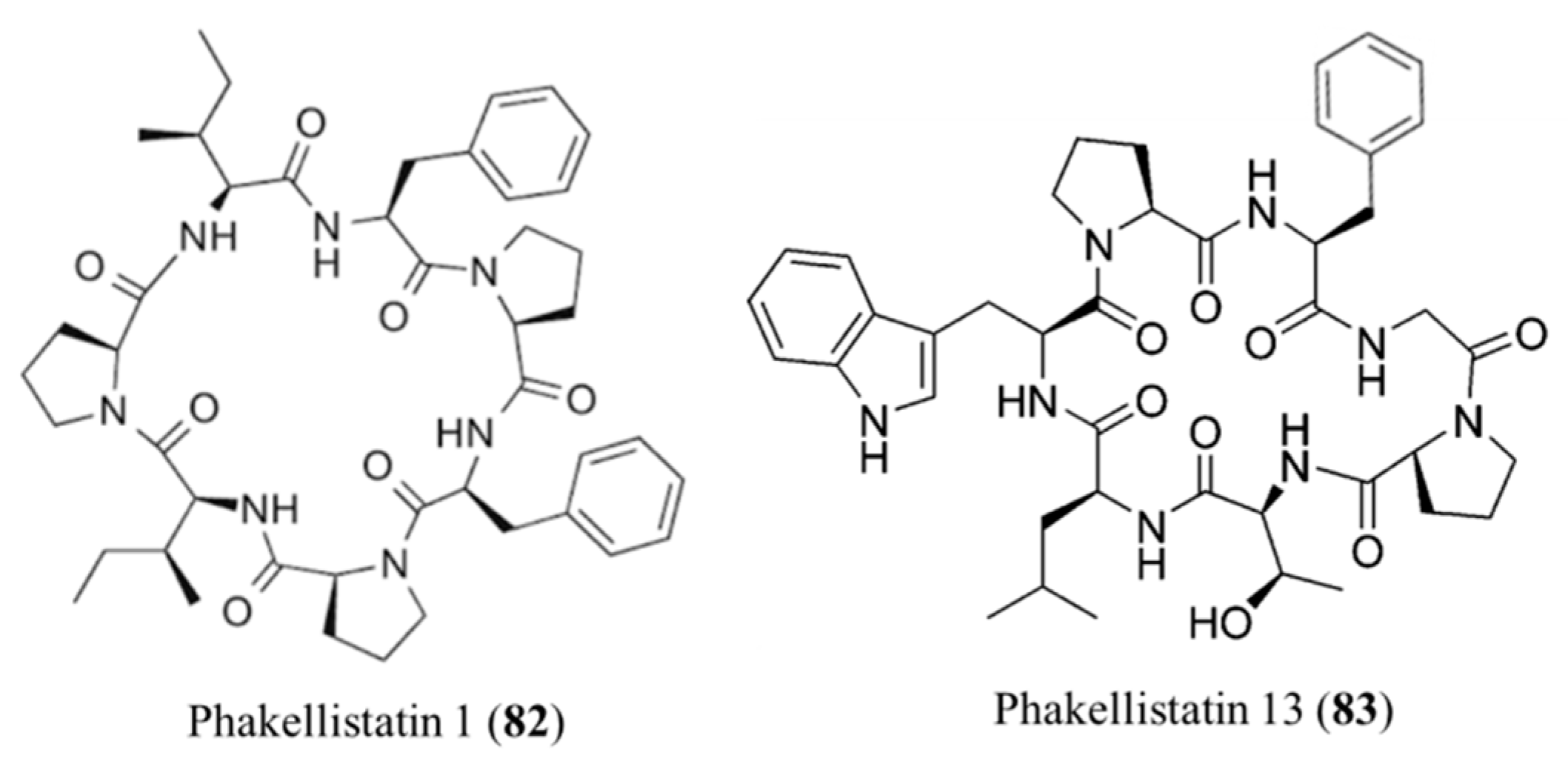
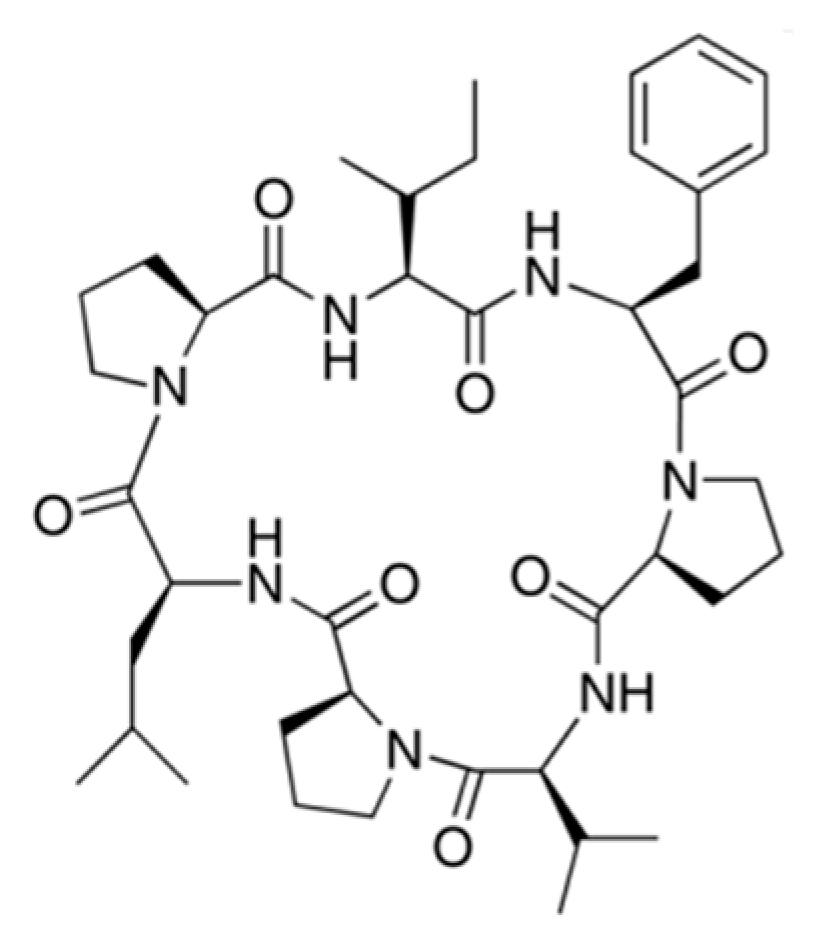
3.3.11. Phakellistatins
3.3.12. Rolloamide A and B
3.3.13. Scleritodermin A
3.4. Tunicate and Ascidia-Derived Peptides
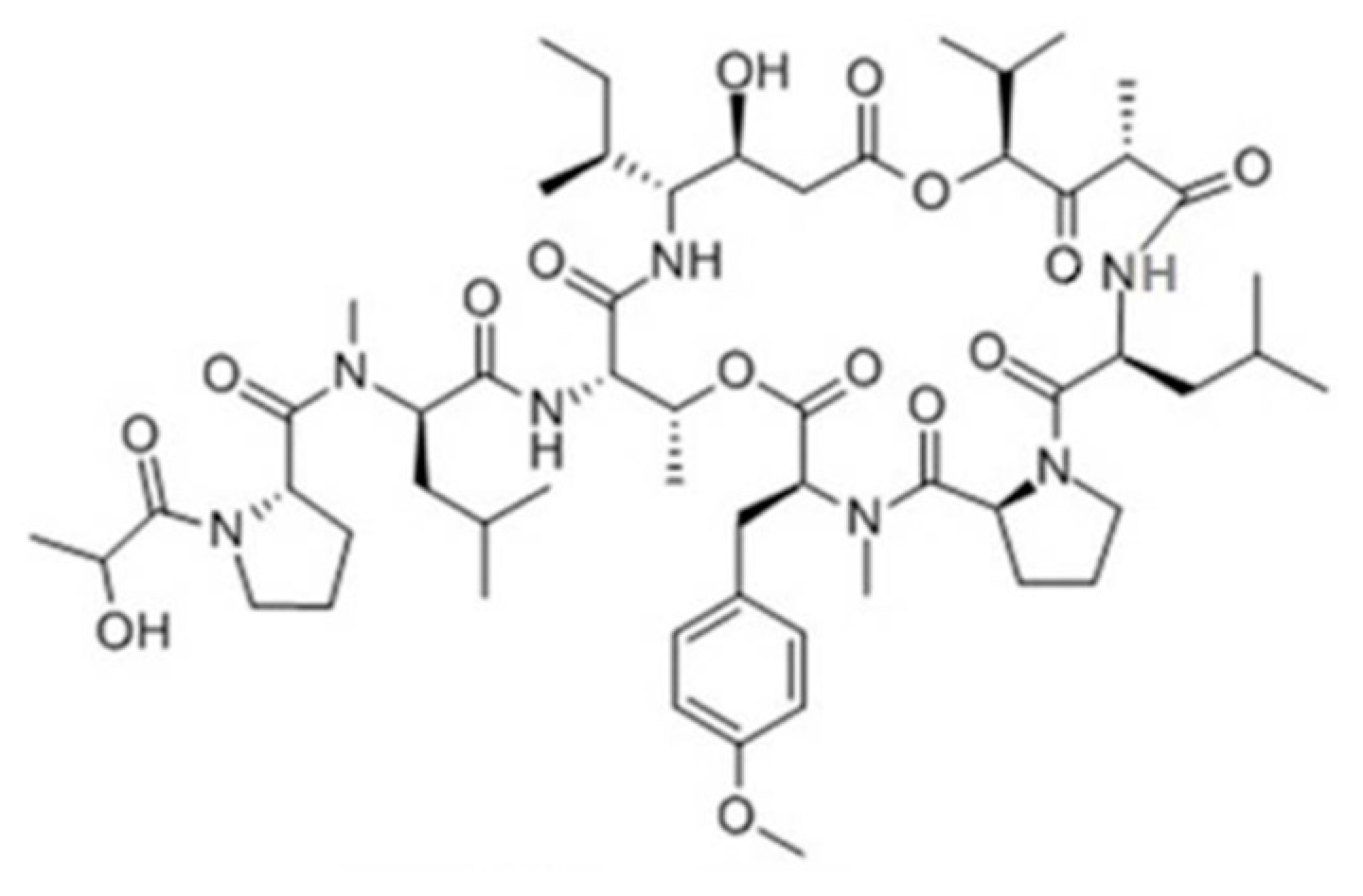
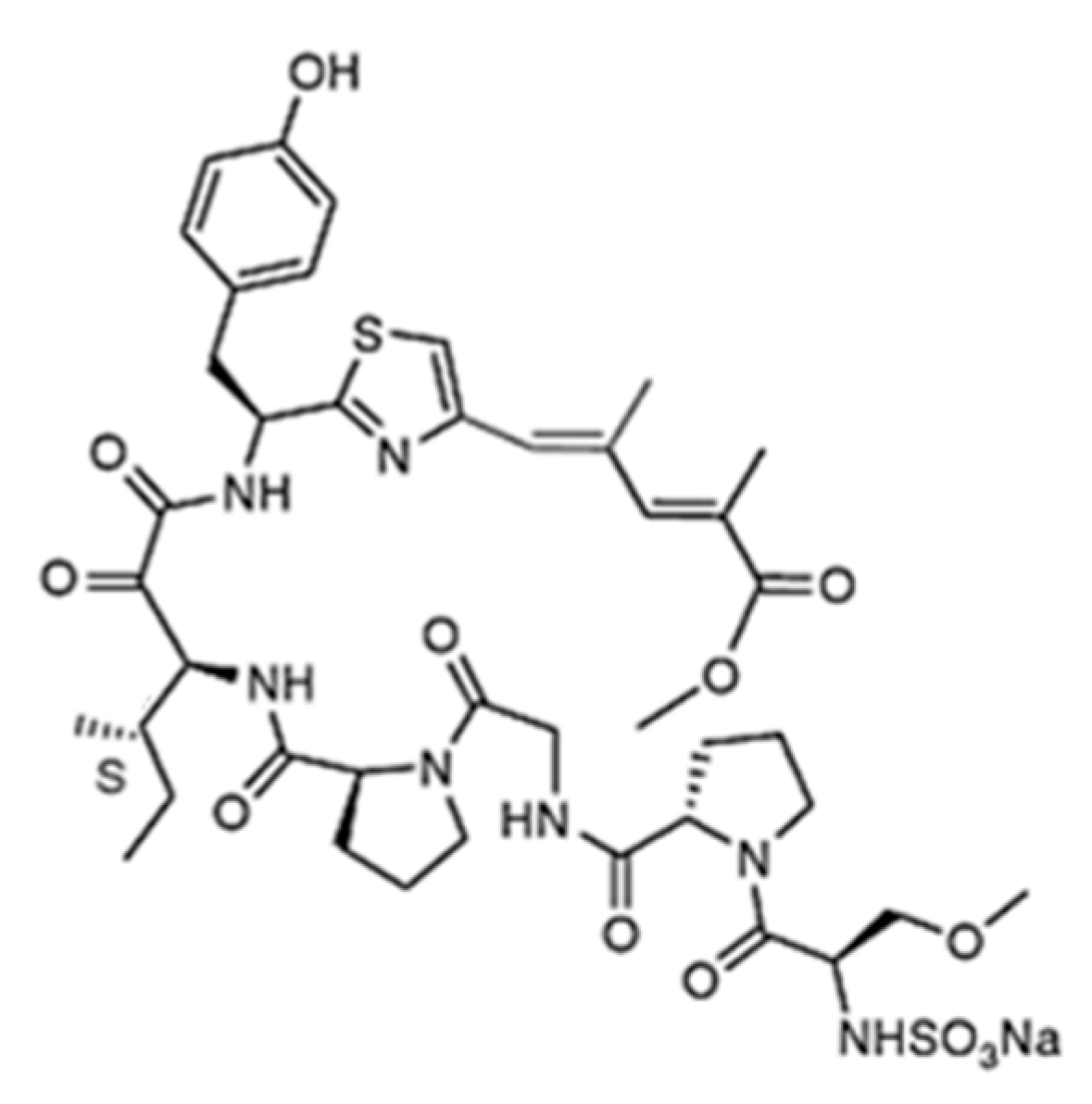
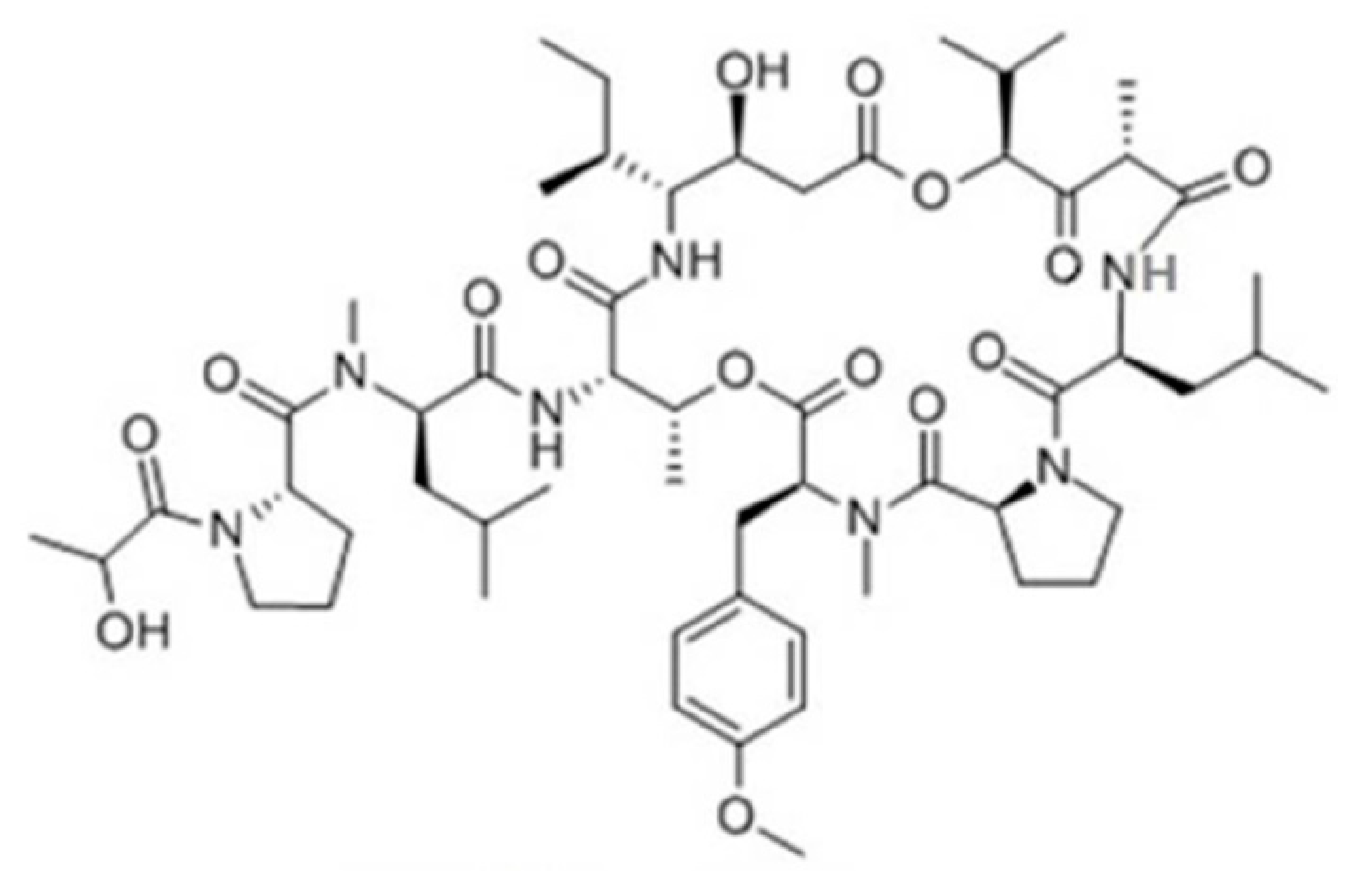
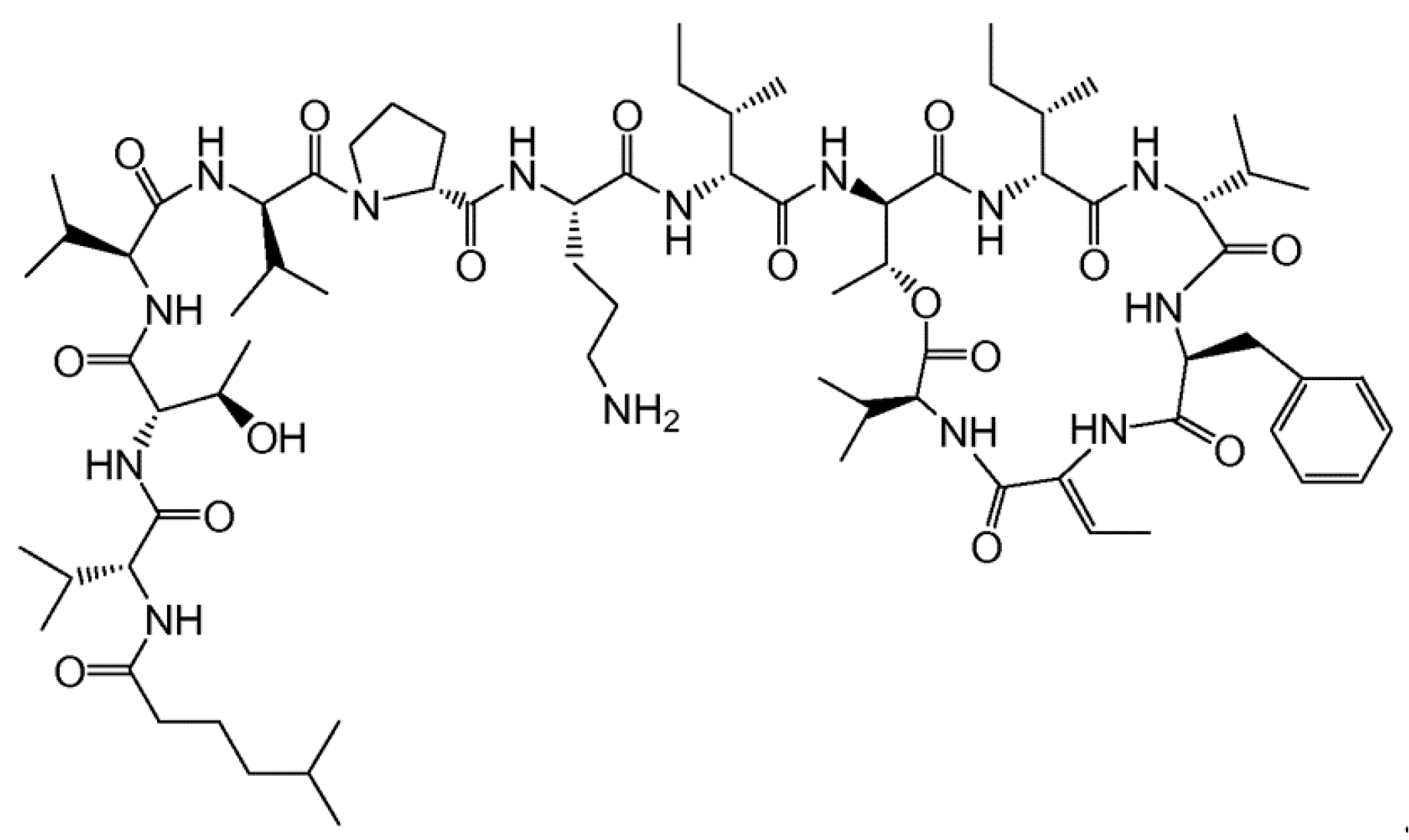
3.4.1. Aplidin
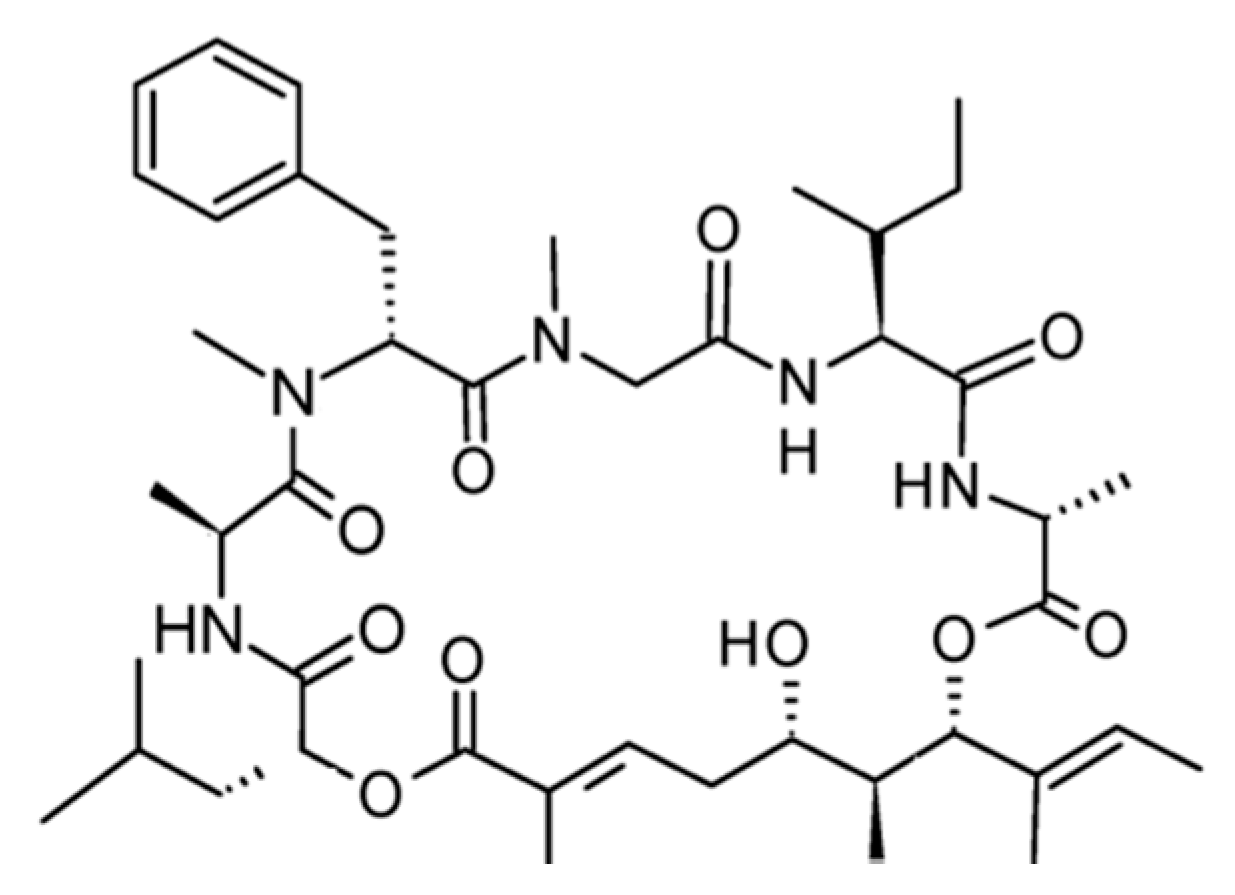
3.4.2. Didemnin B
3.4.3. Cycloxazoline
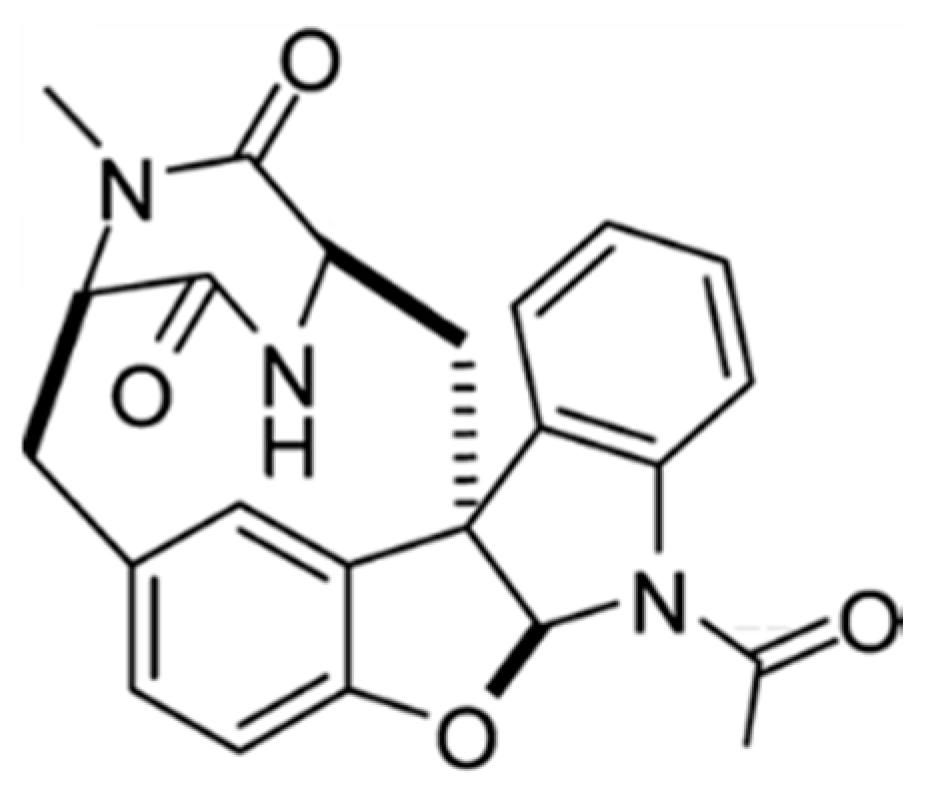
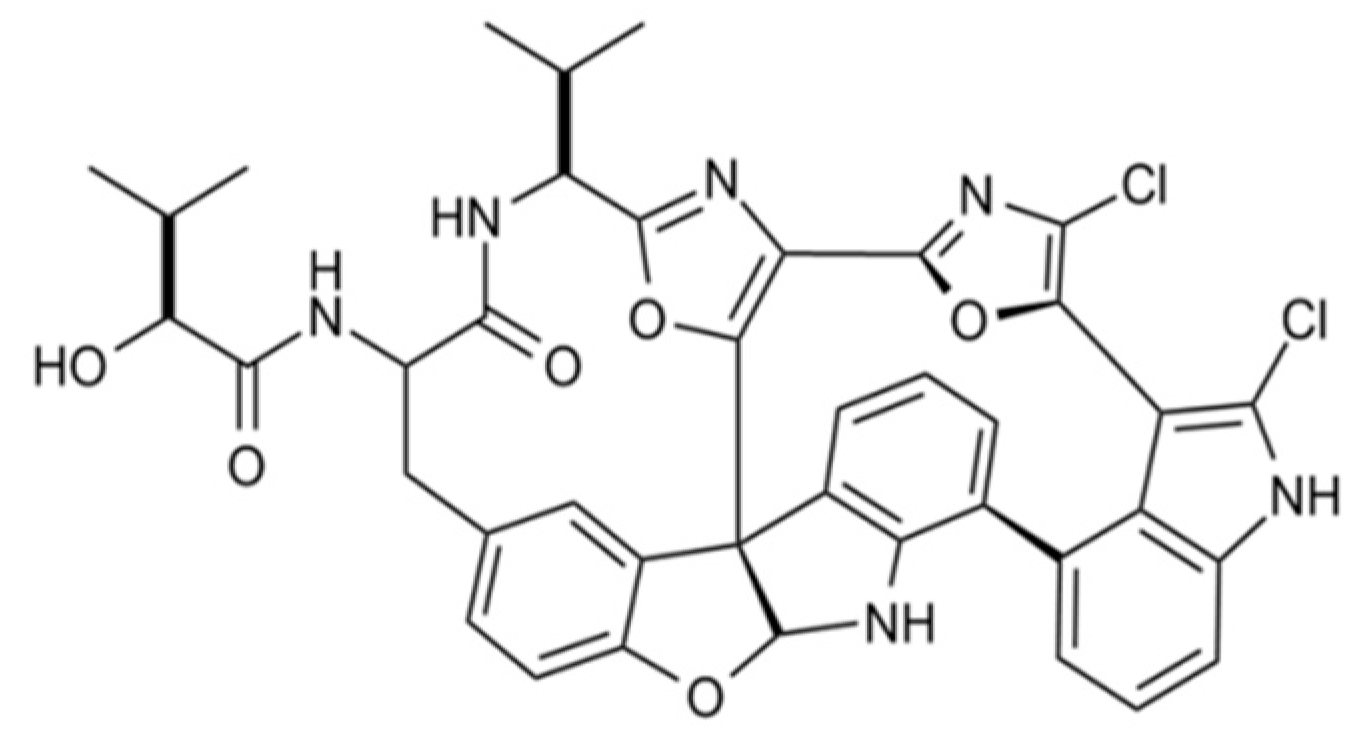
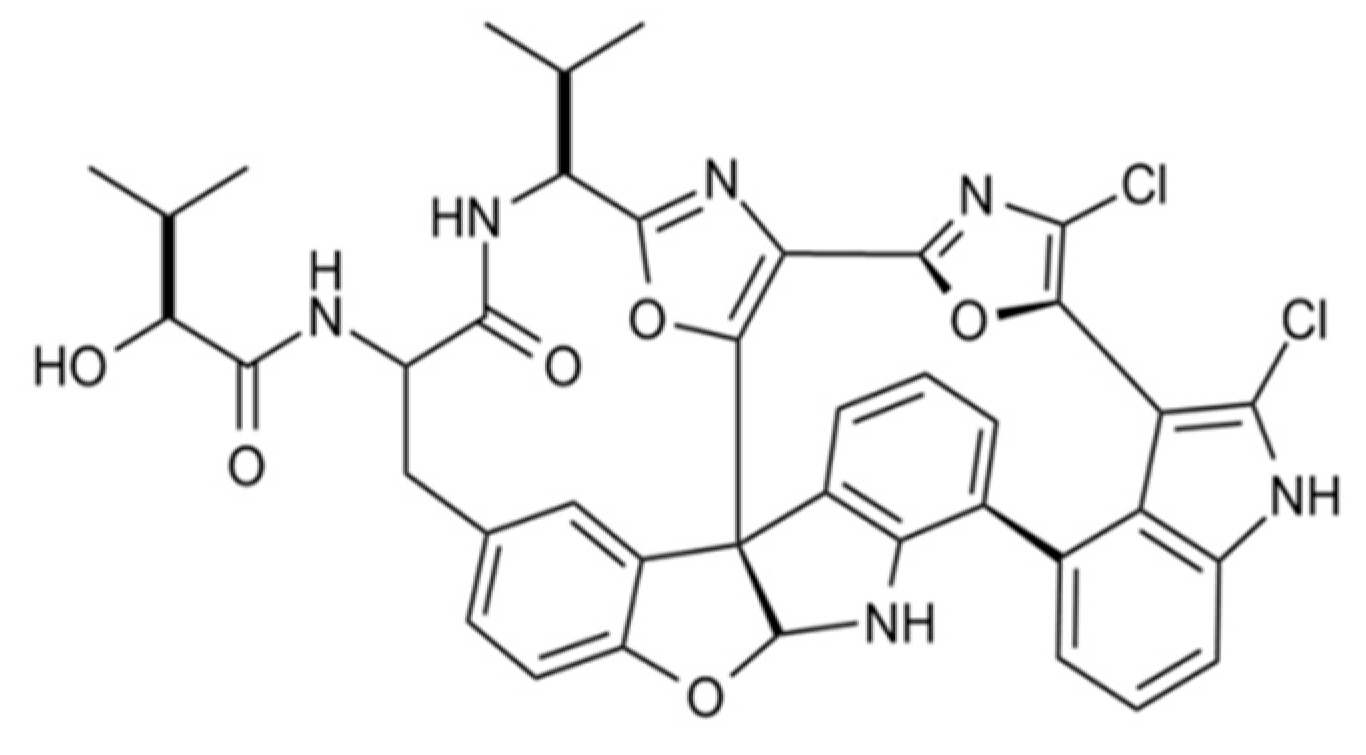
3.4.4. Diazonamide A
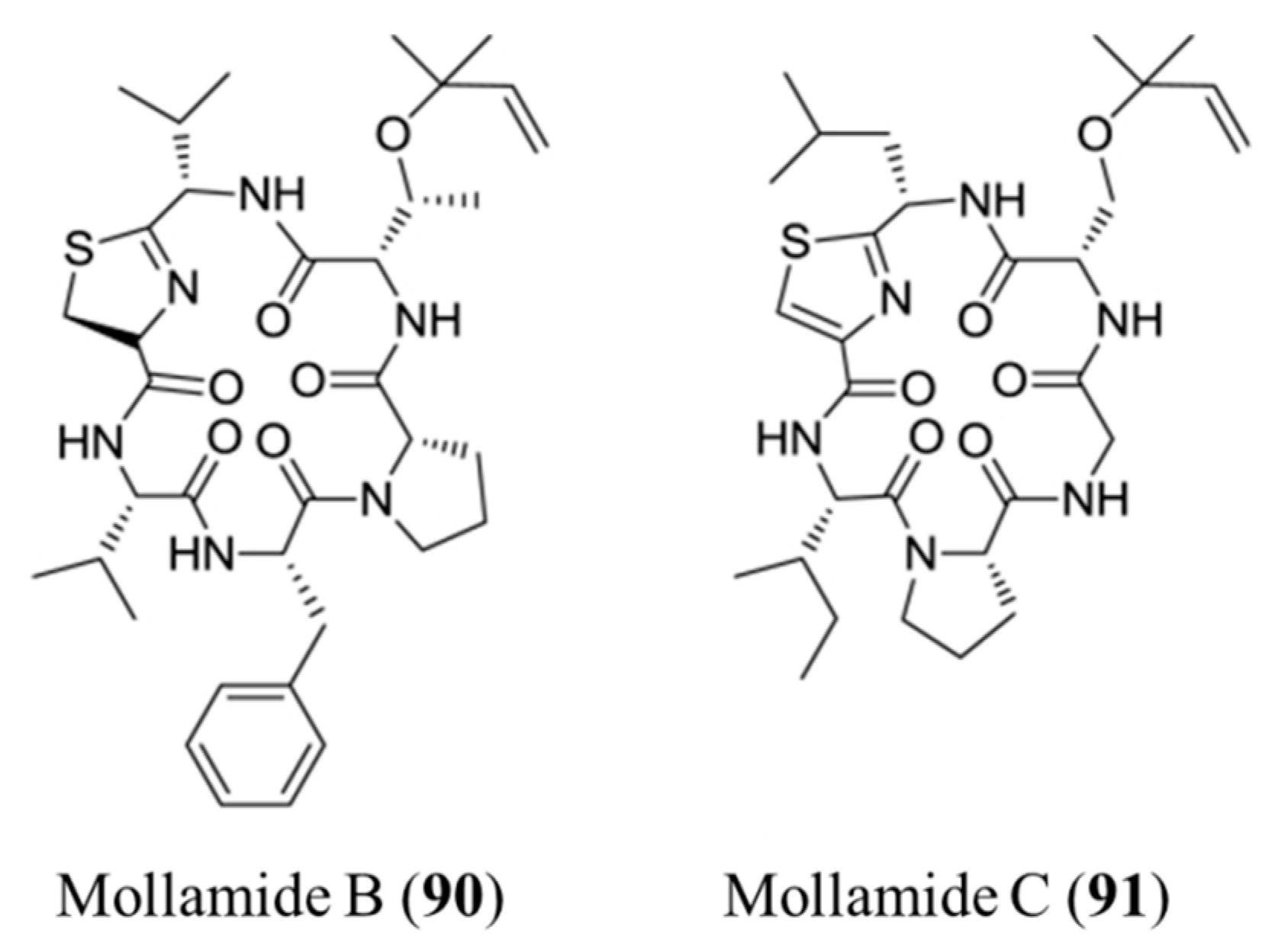
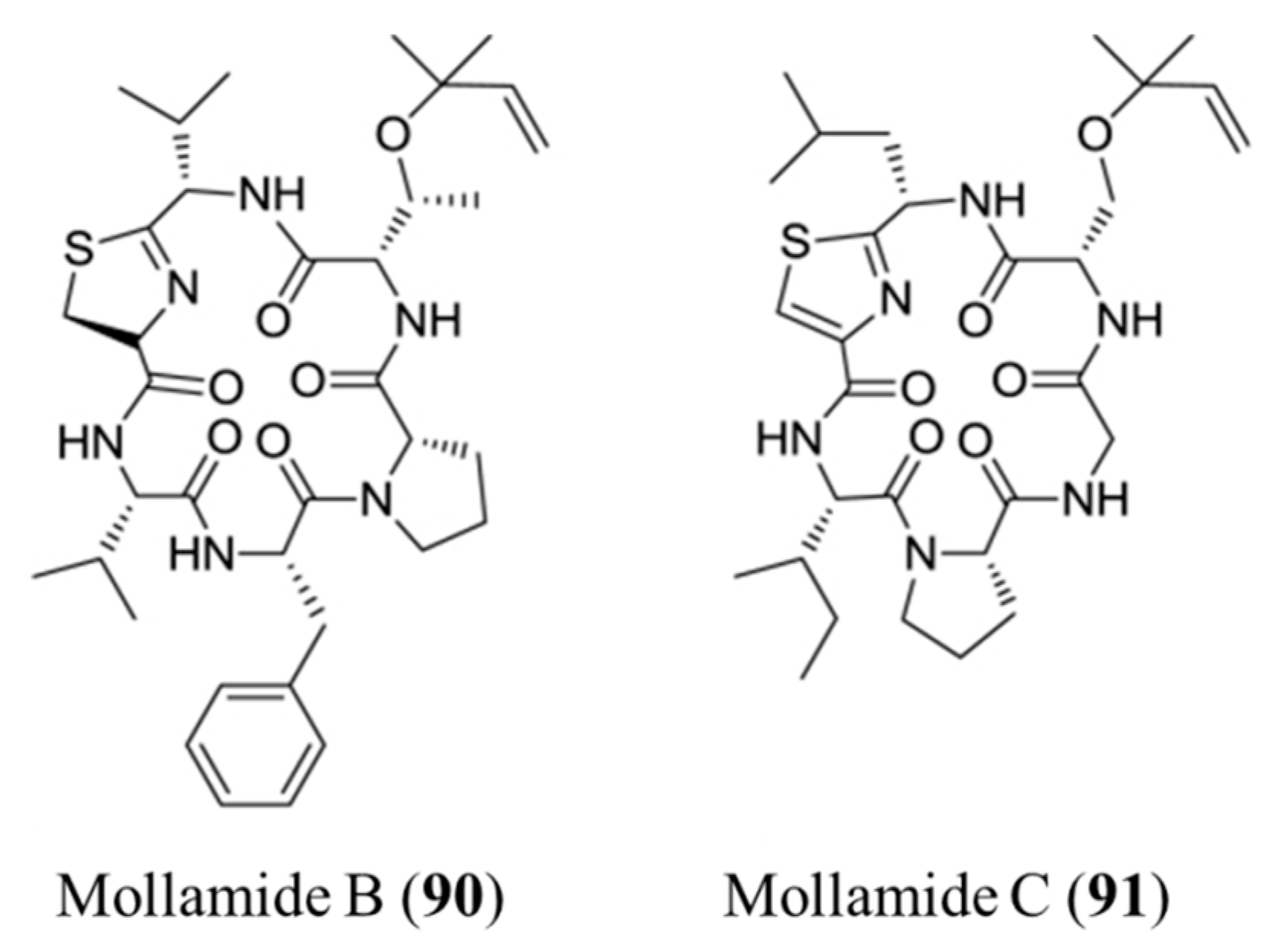
3.4.5. Mollamide B and C
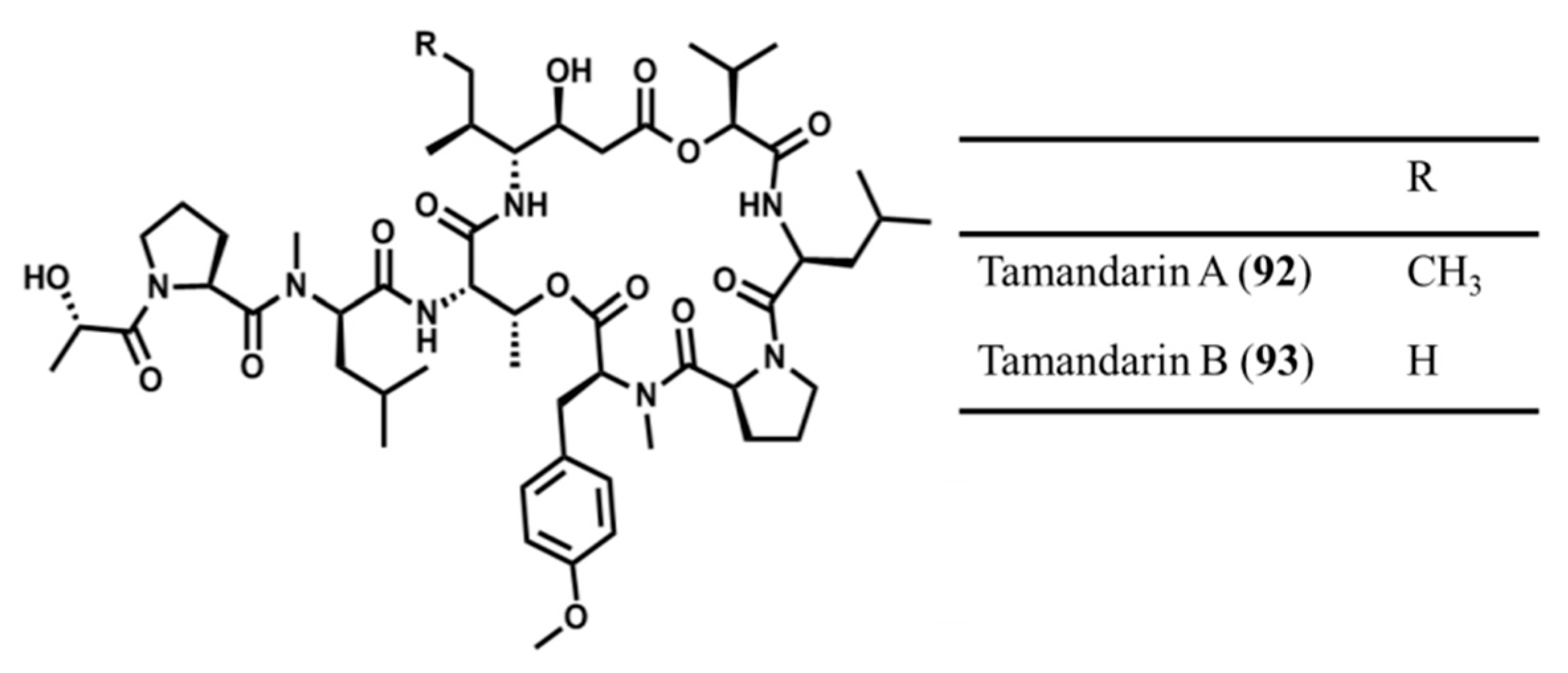
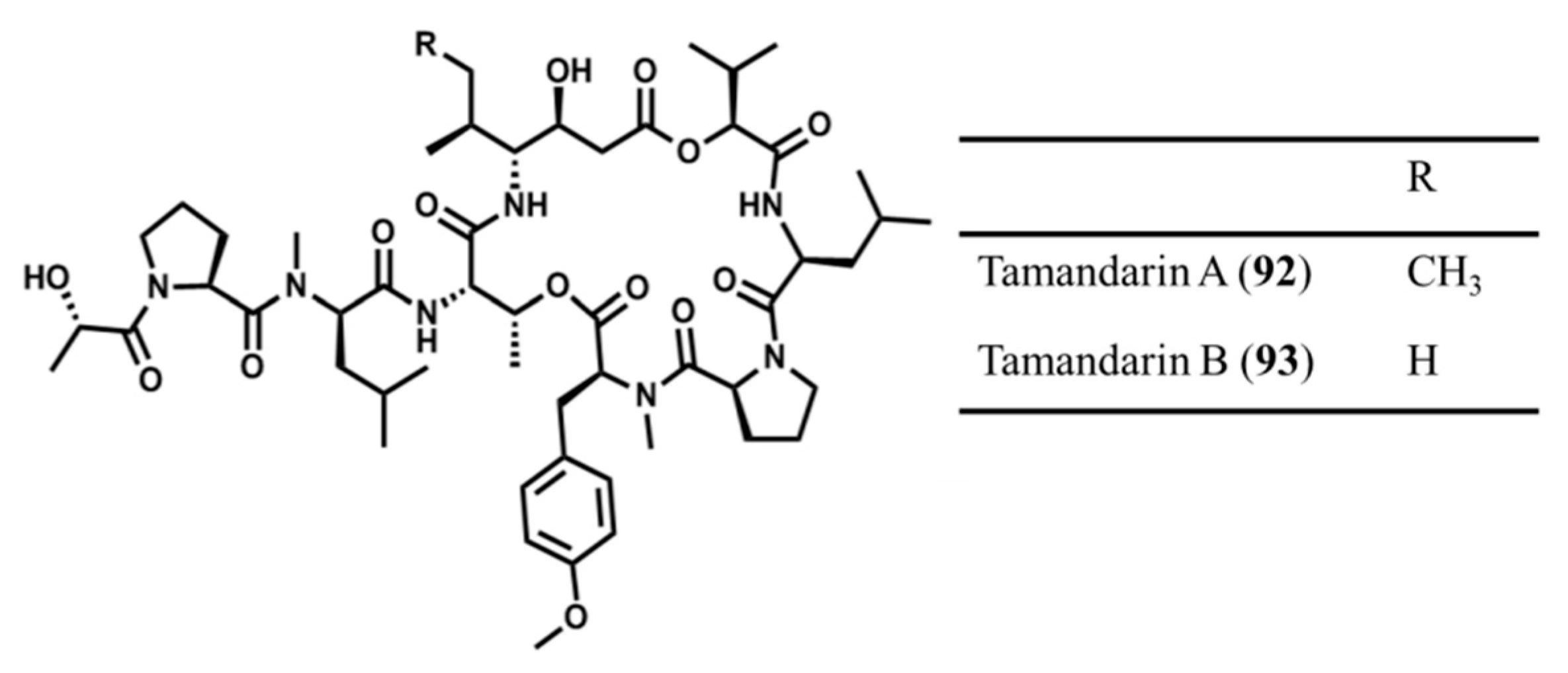
3.4.6. Tamandarin A and B
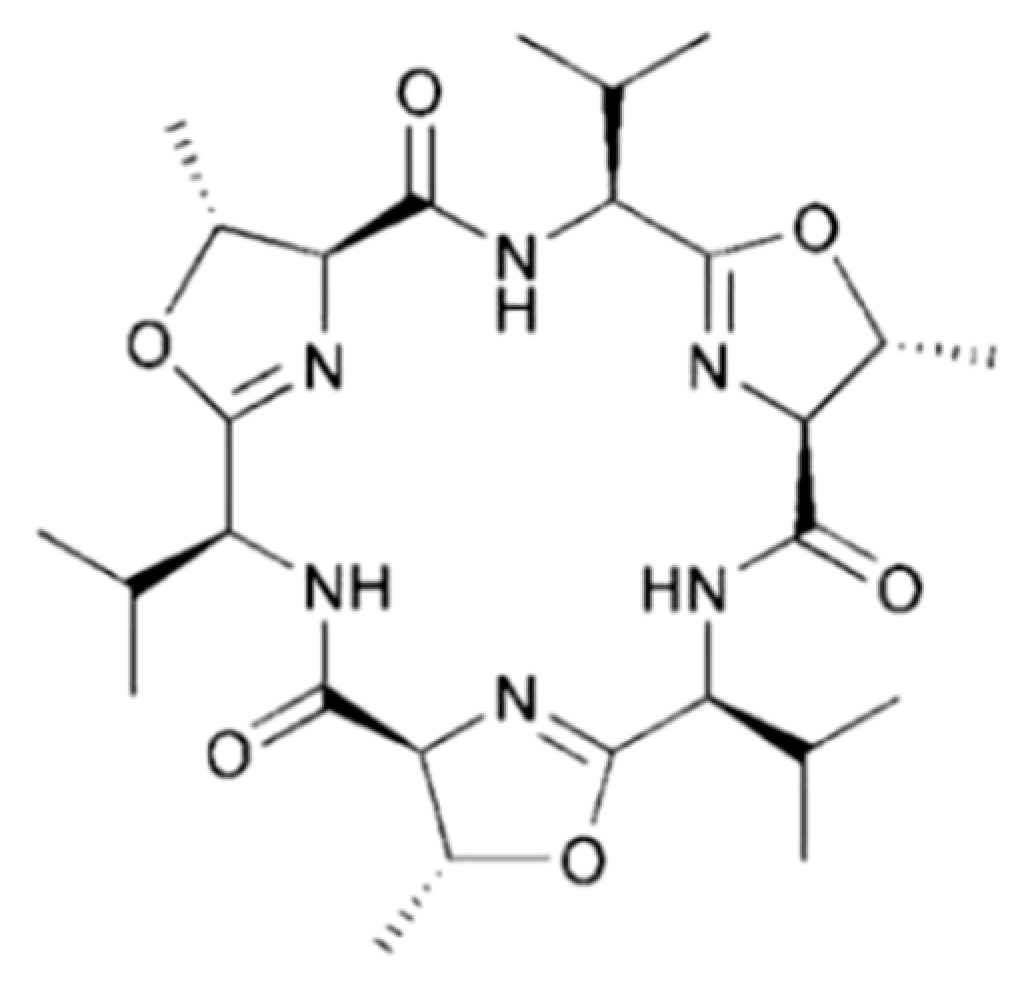
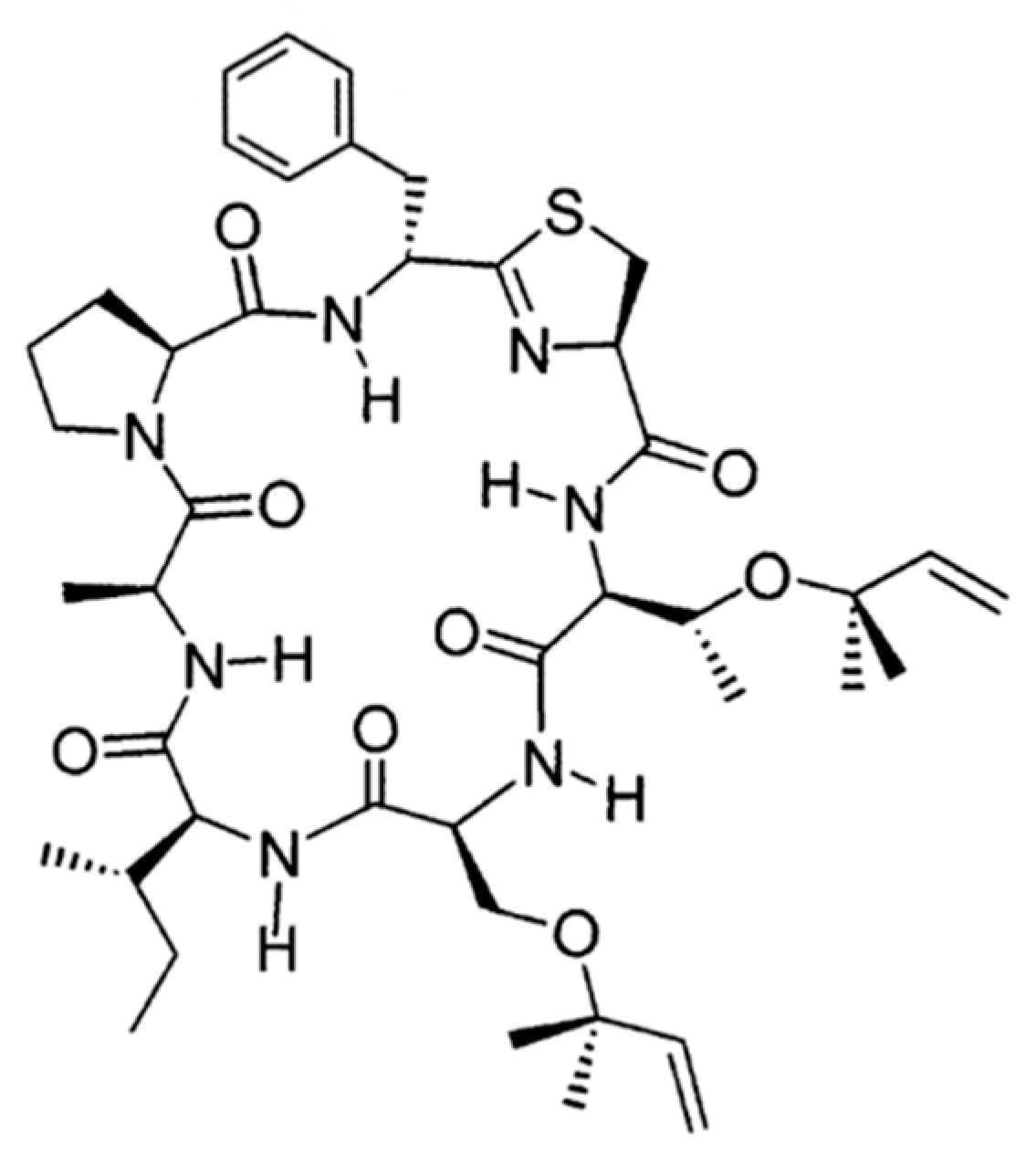
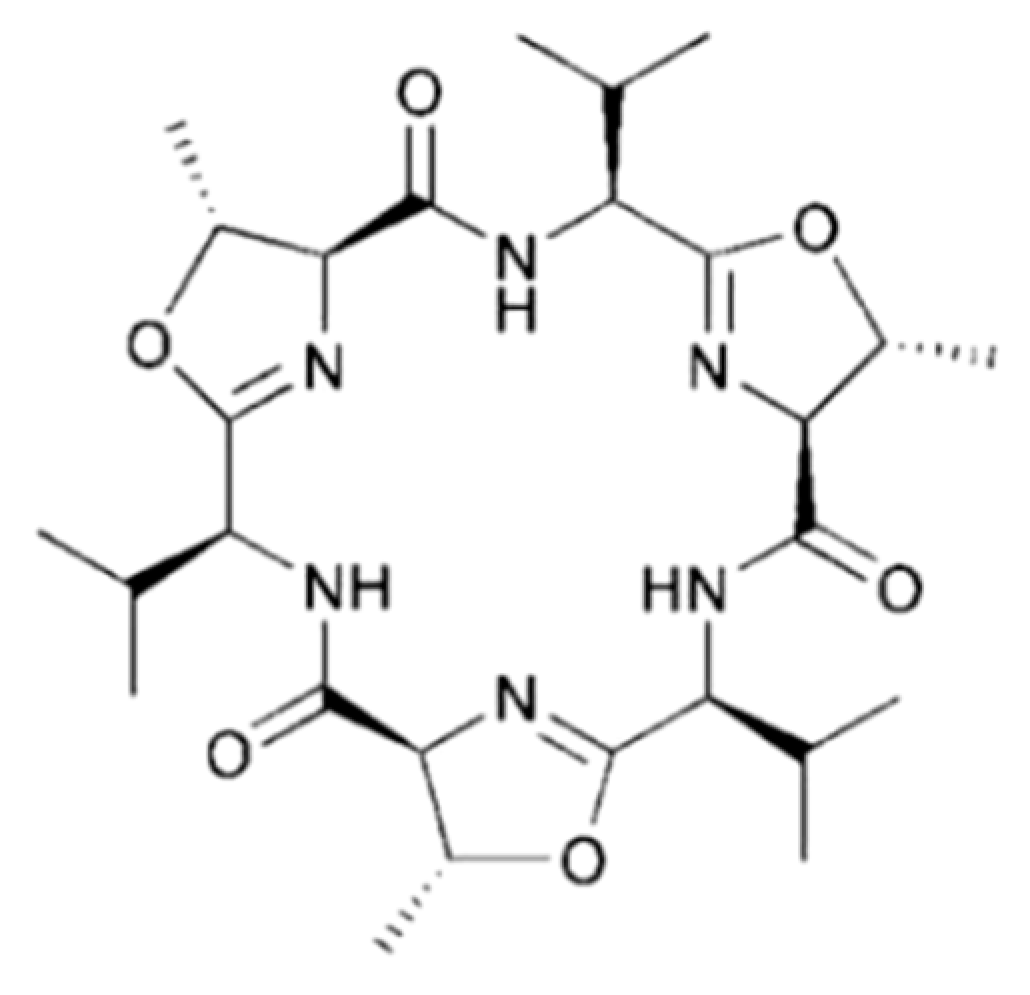
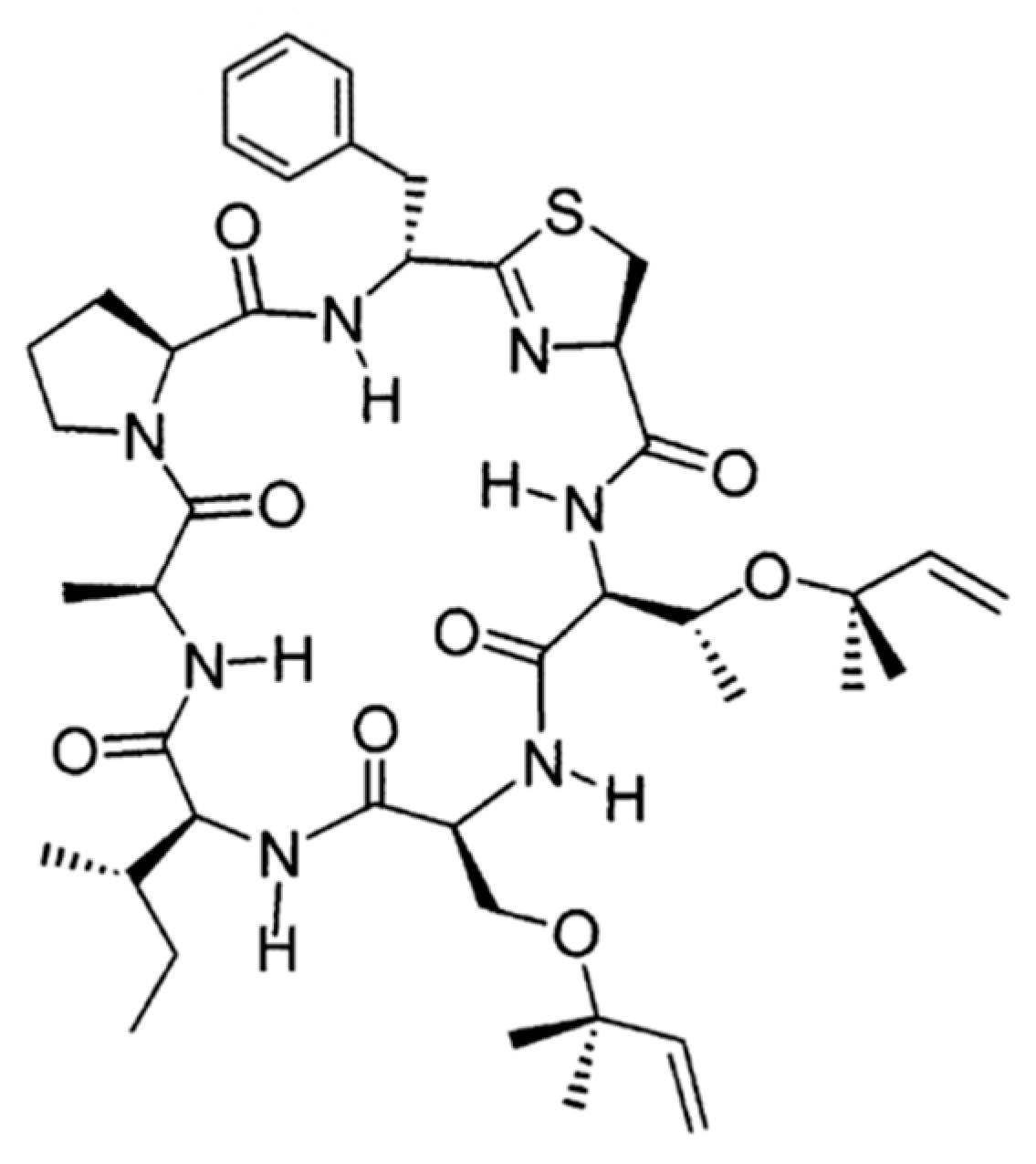
3.4.7. Trunkamide A
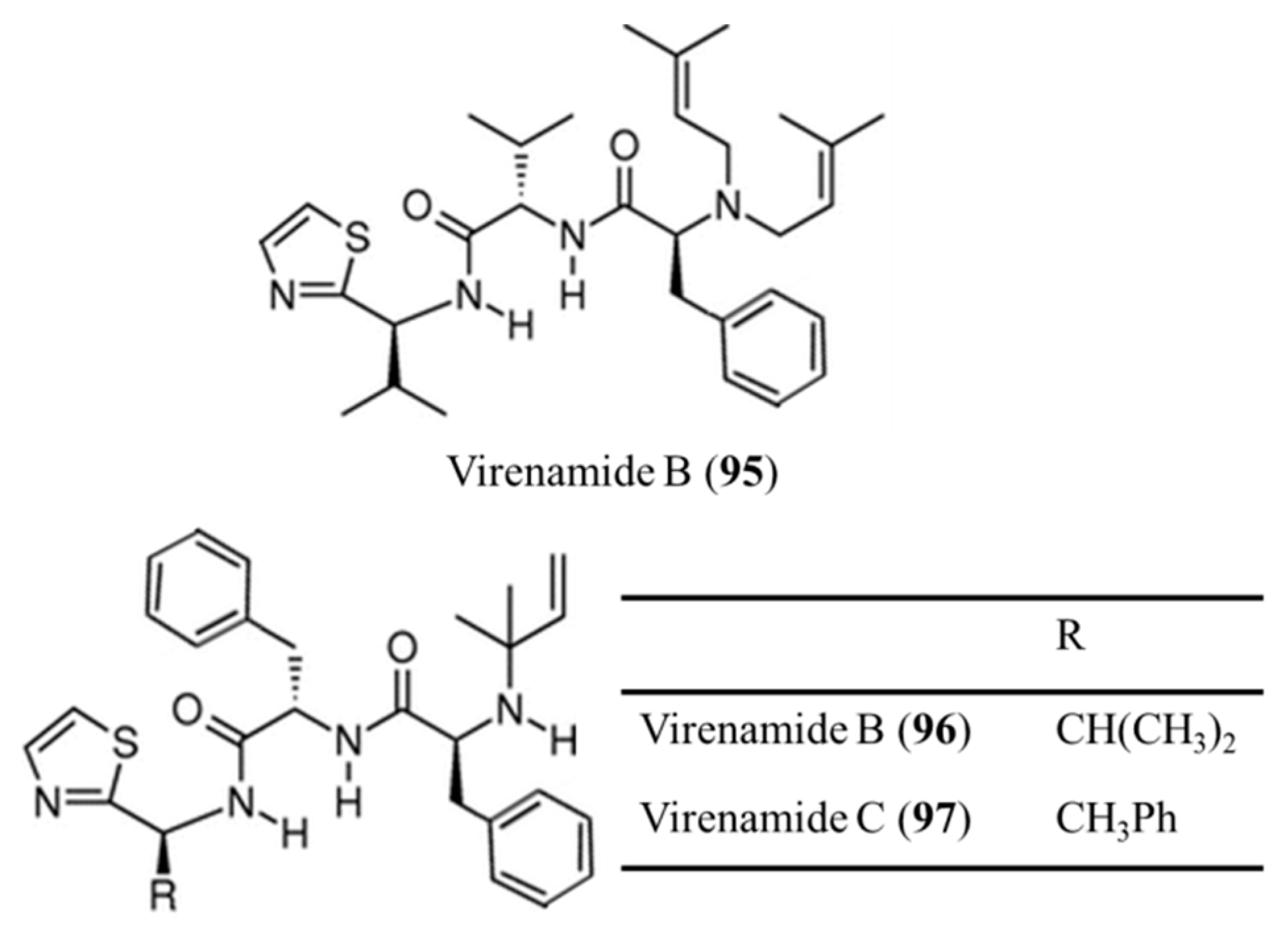
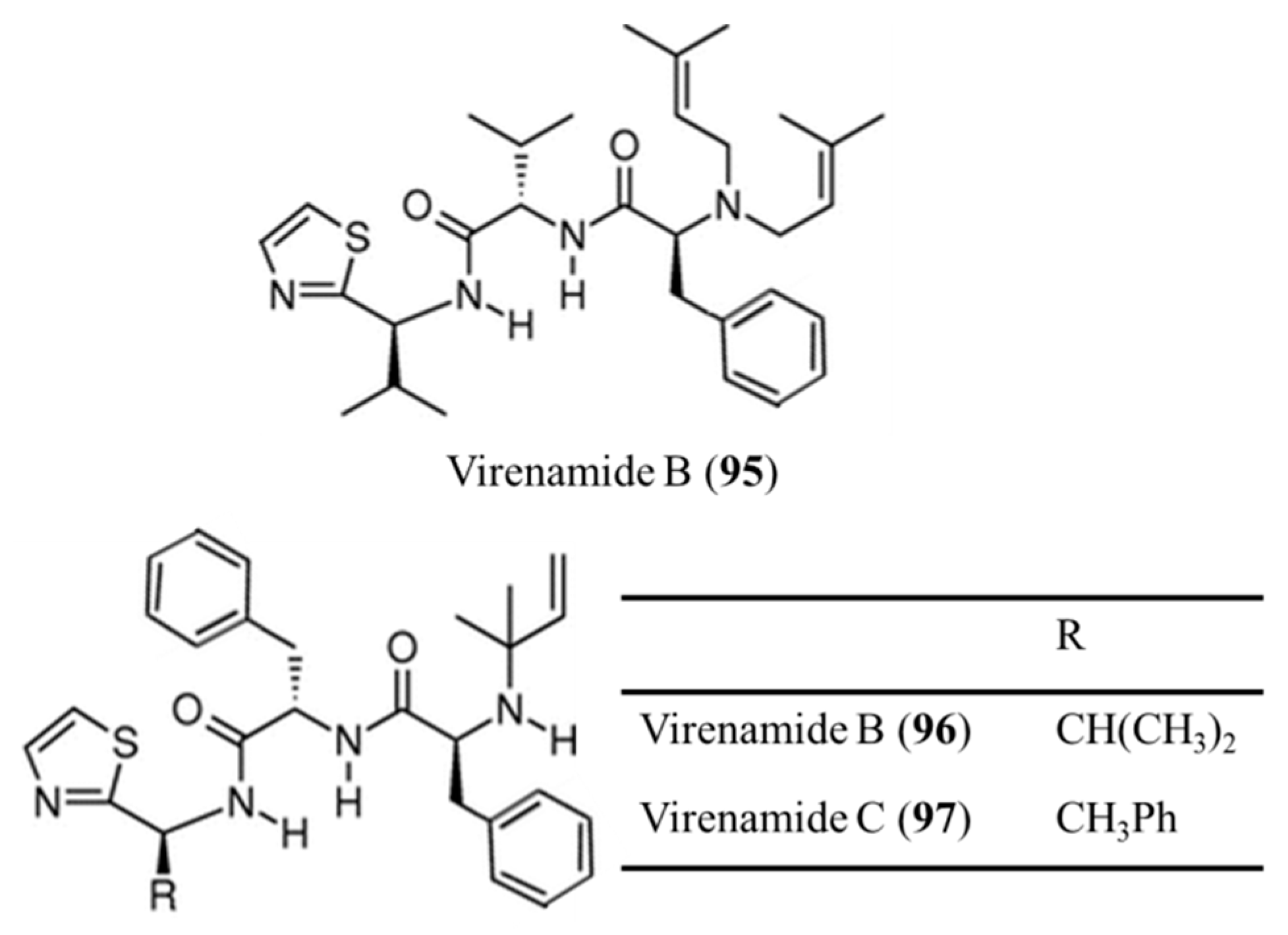
3.4.8. Virenamide A–C
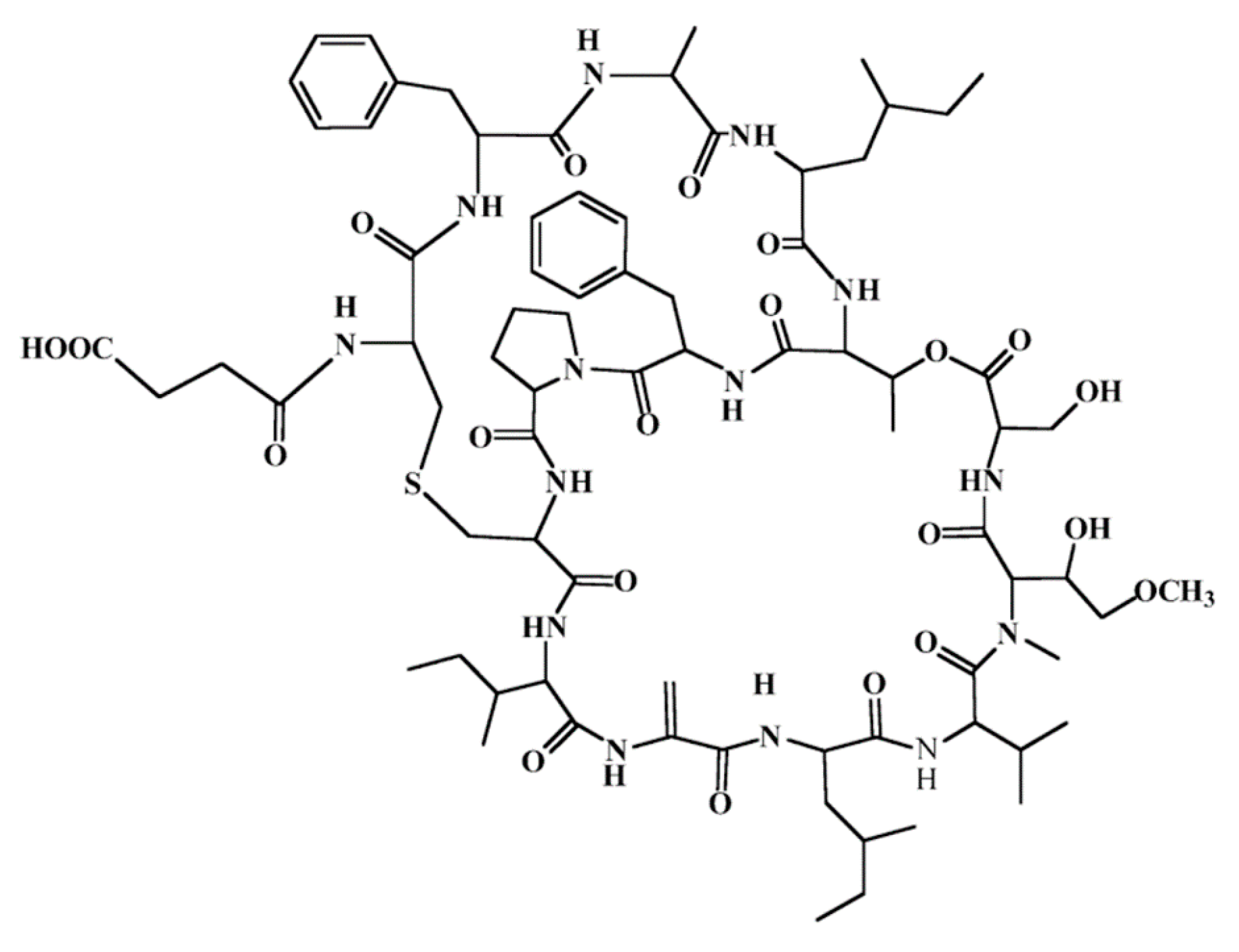
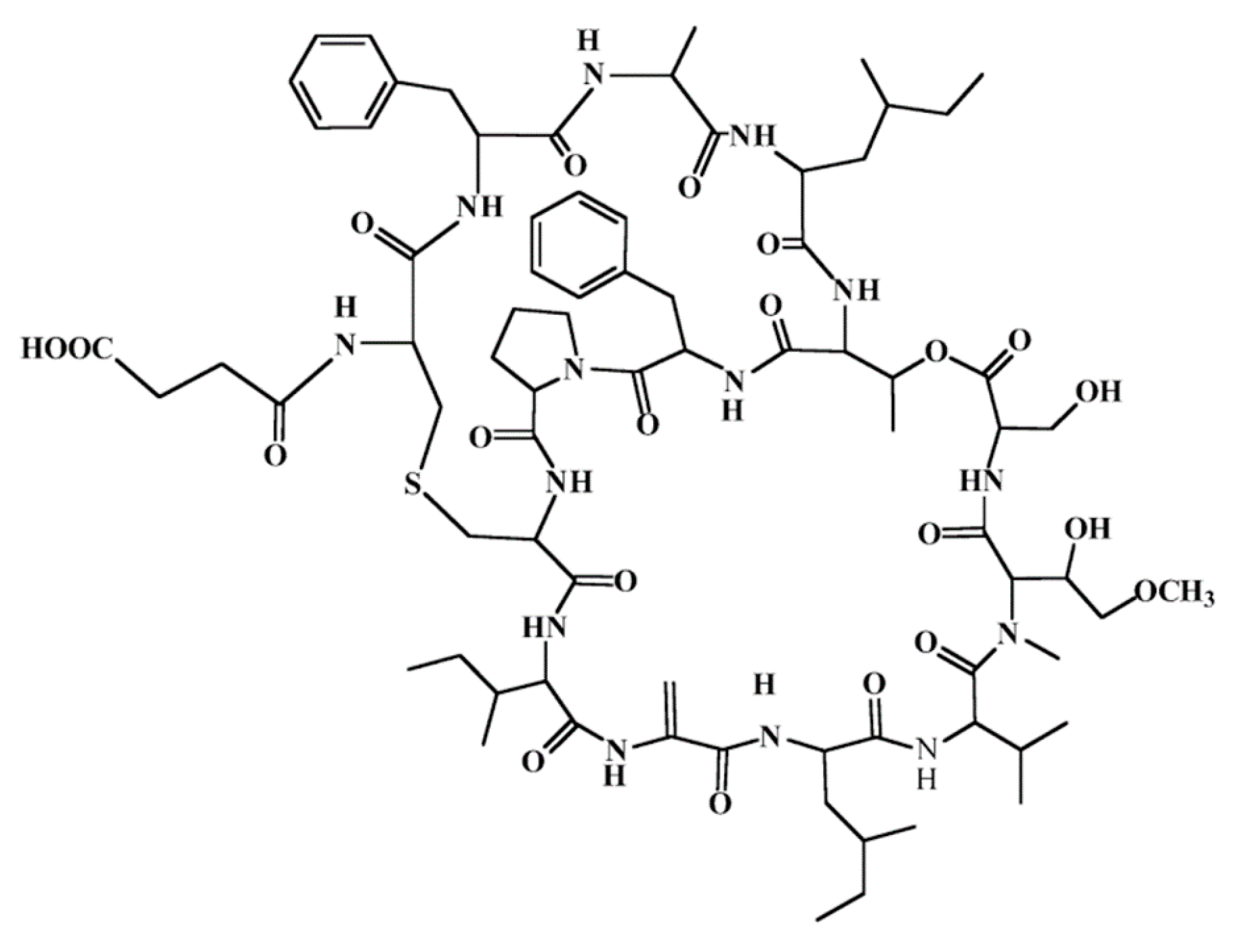
3.4.9. Vitilevuamide
3.5. Mollusk and Fish-Derived Anticancer Peptides
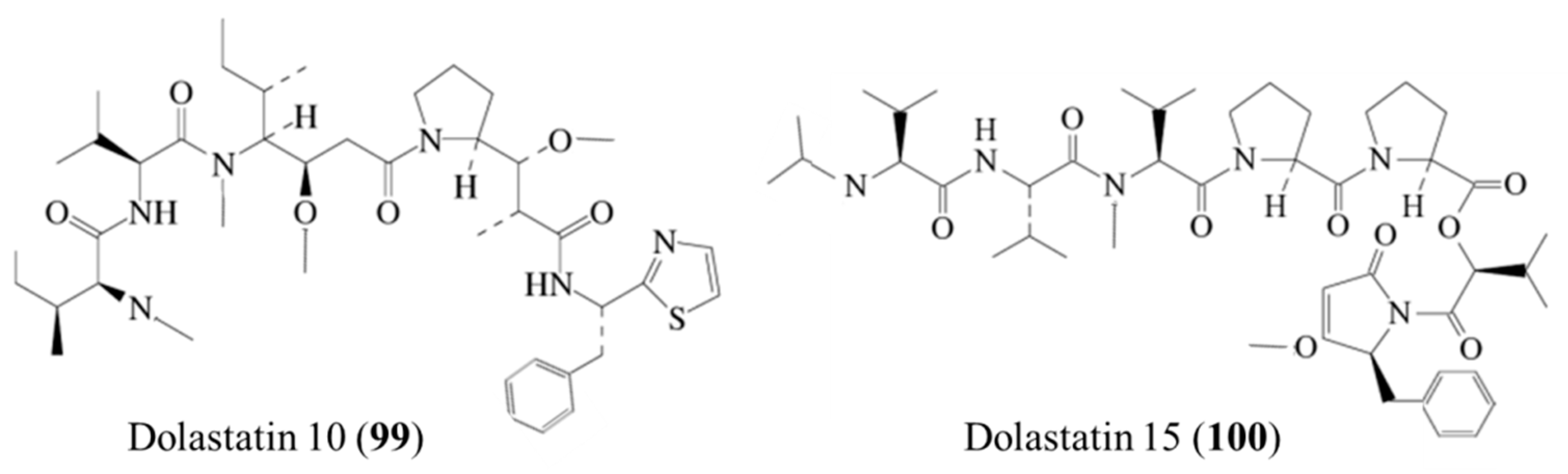
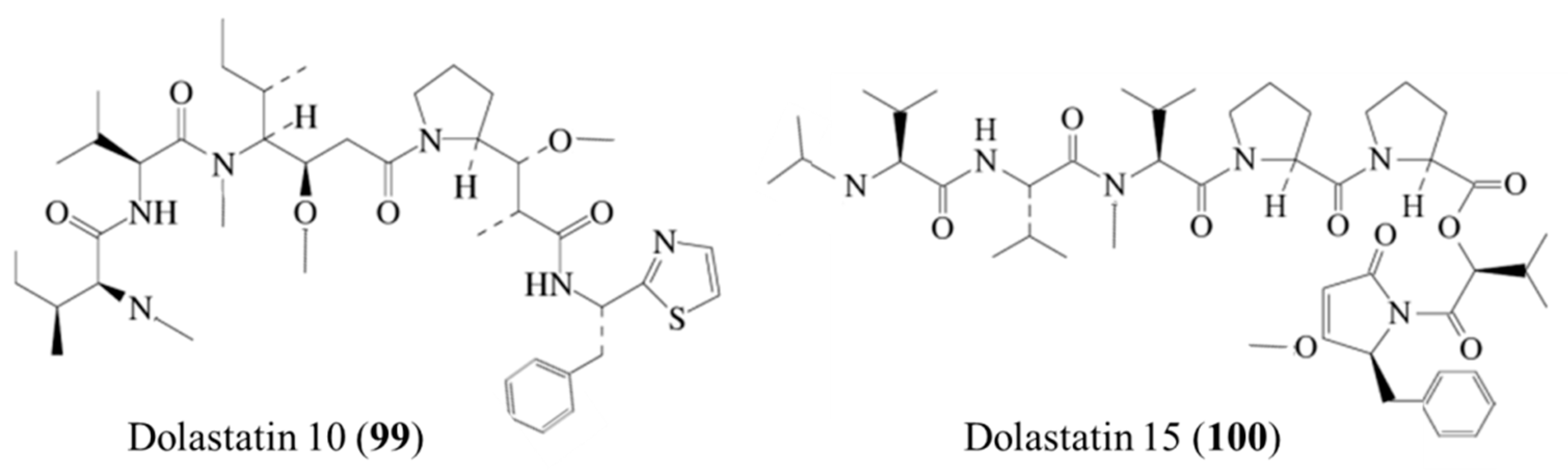
3.5.1. Dolastatins
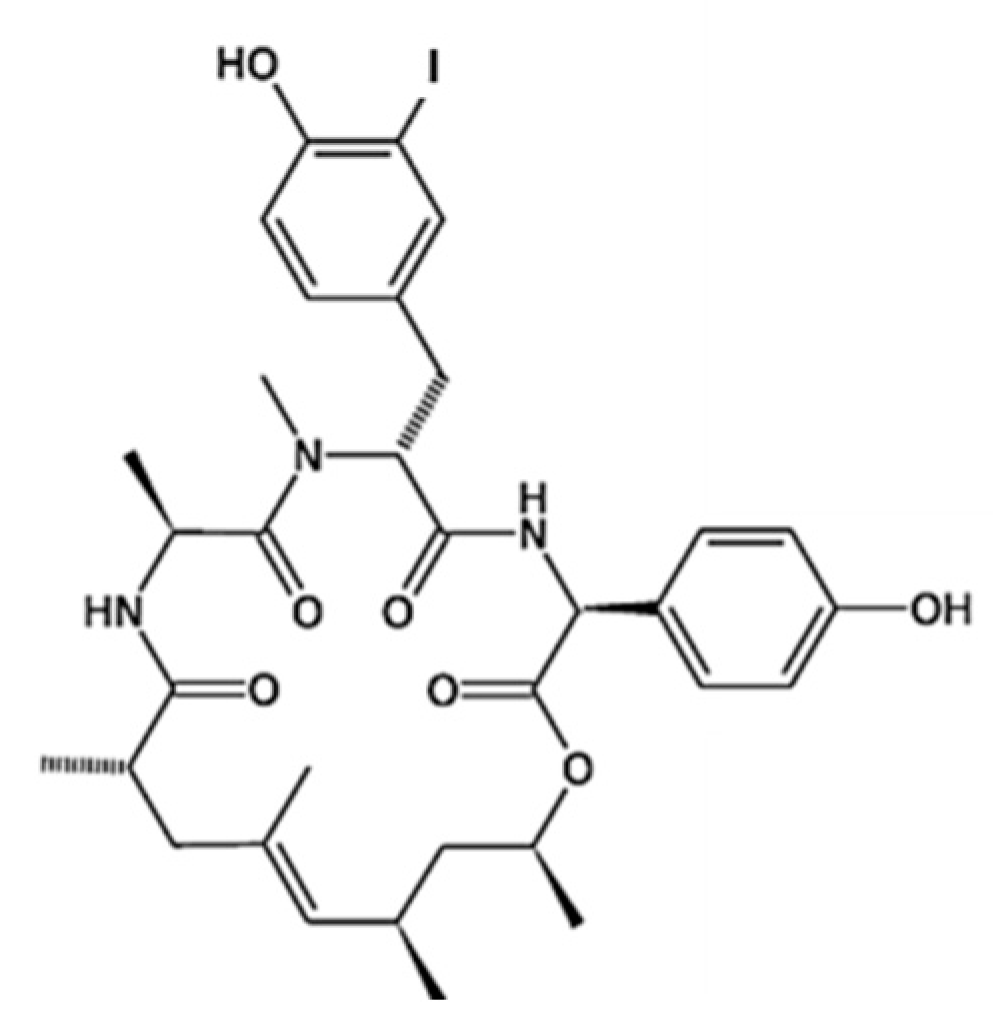
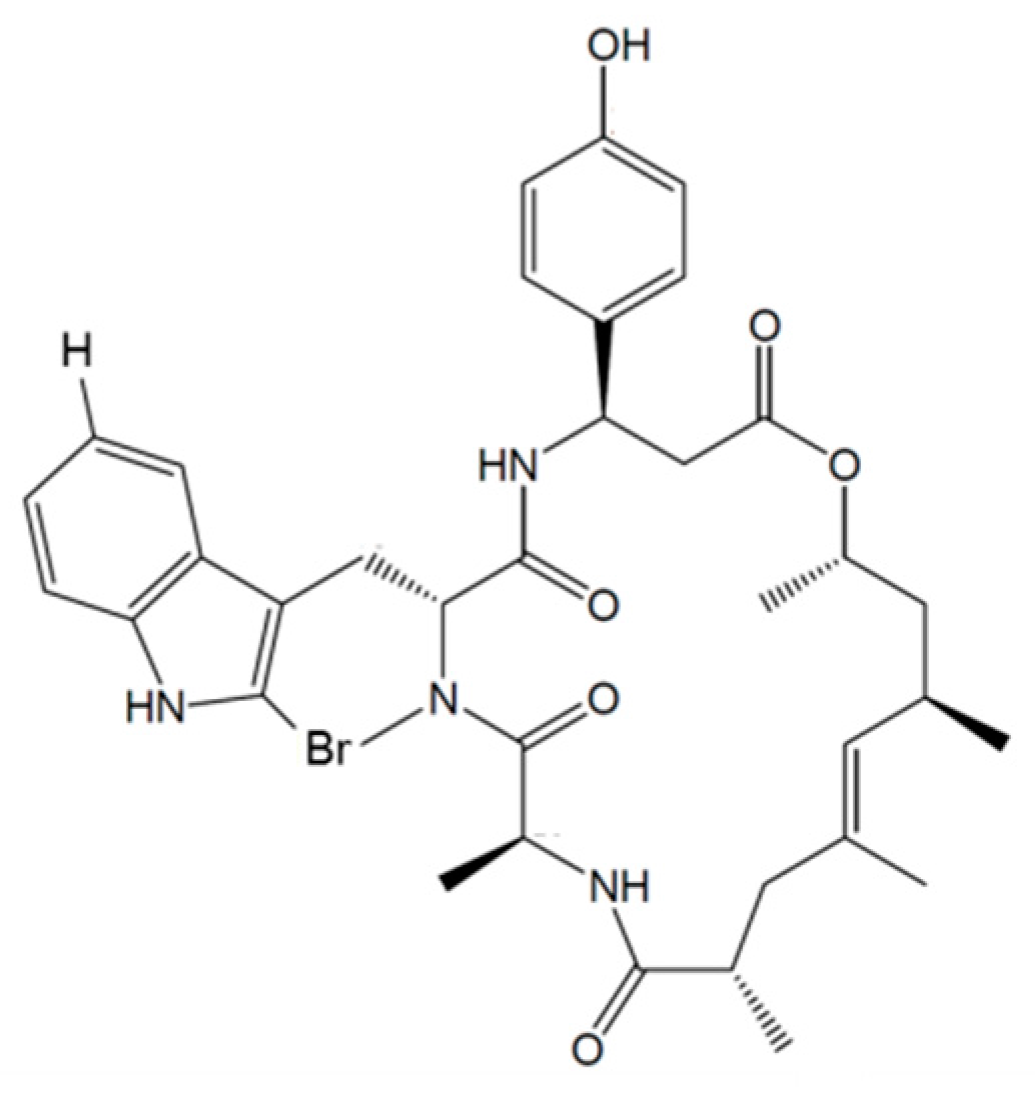
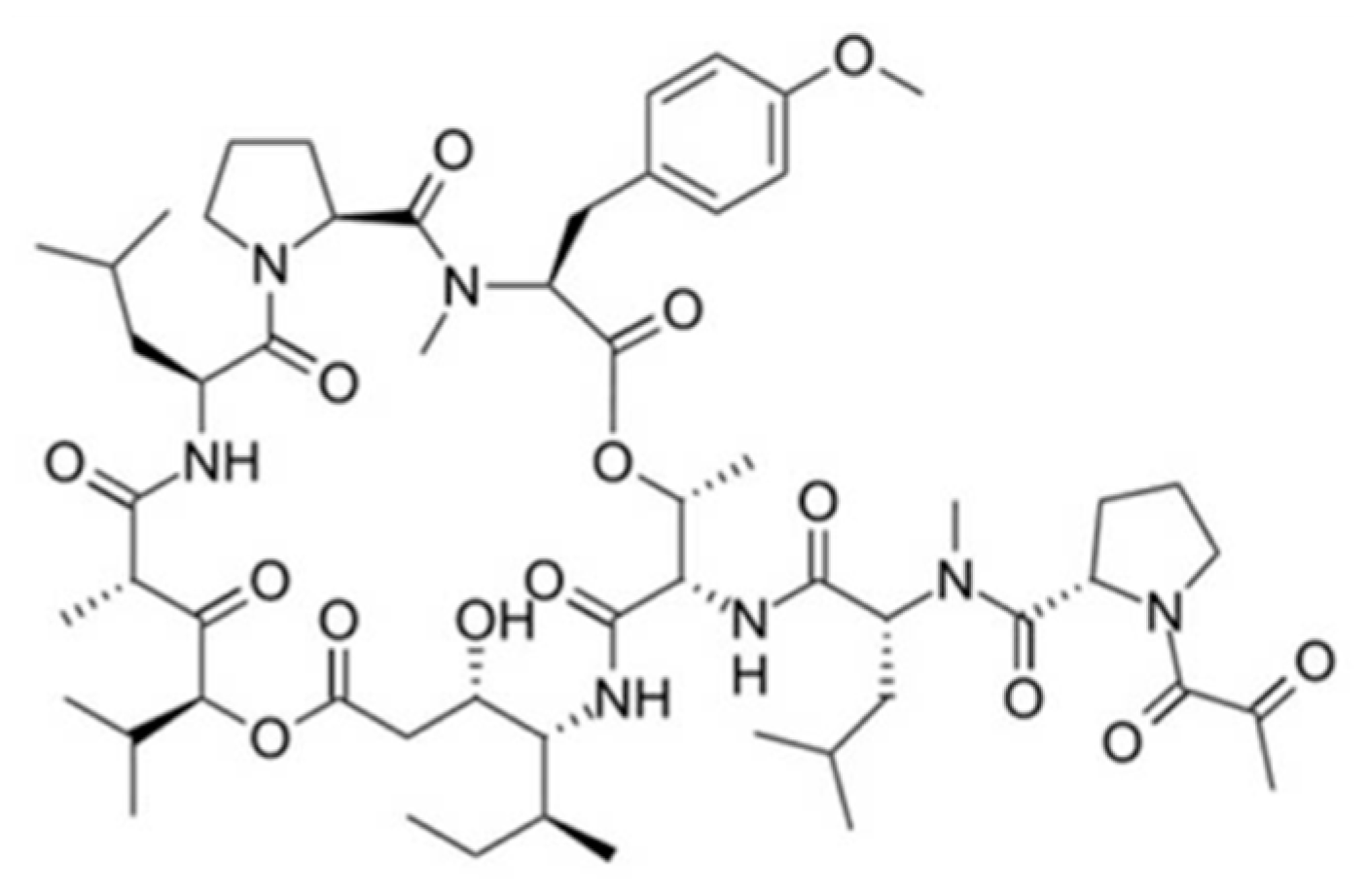
3.5.2. Kahalalides
3.5.3. Keenamide A
3.5.4. Kulokekahilide-2
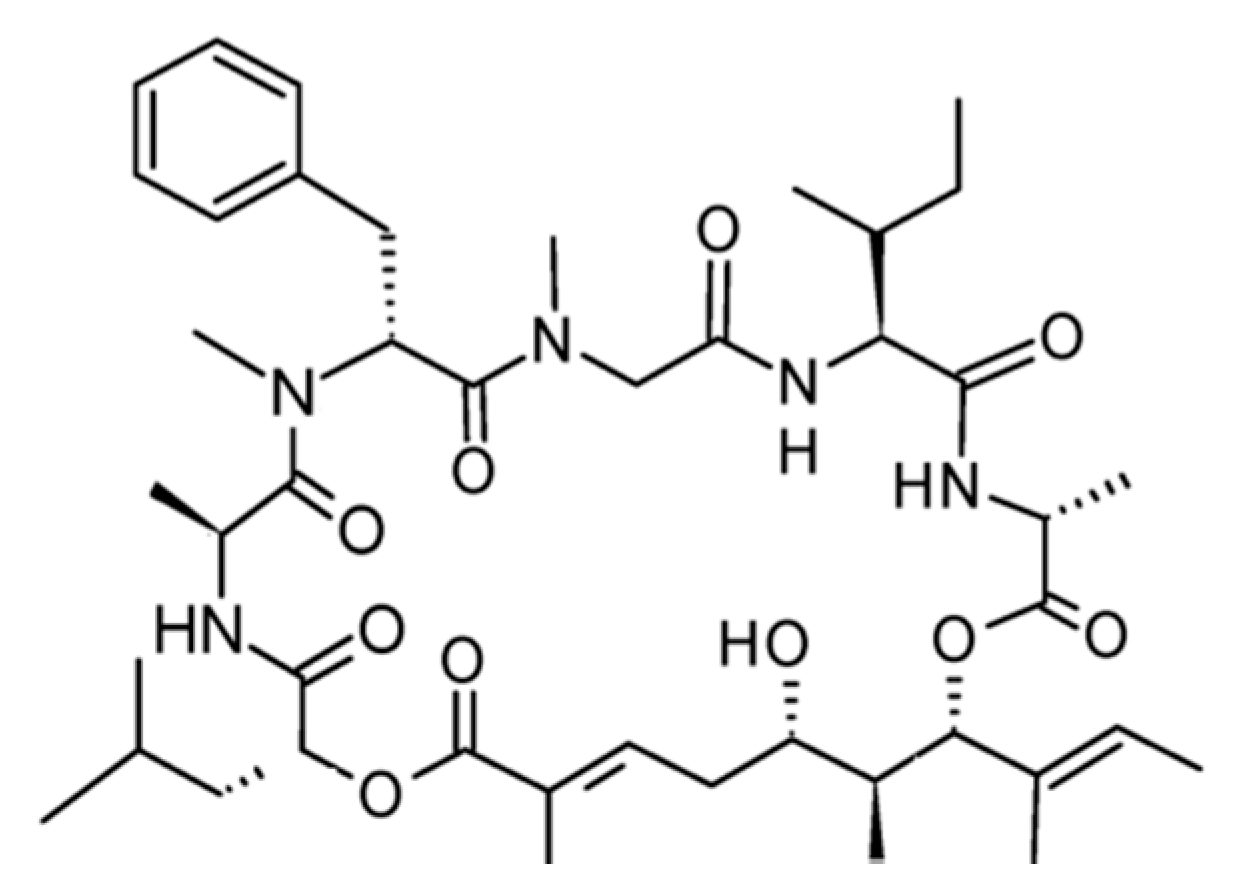
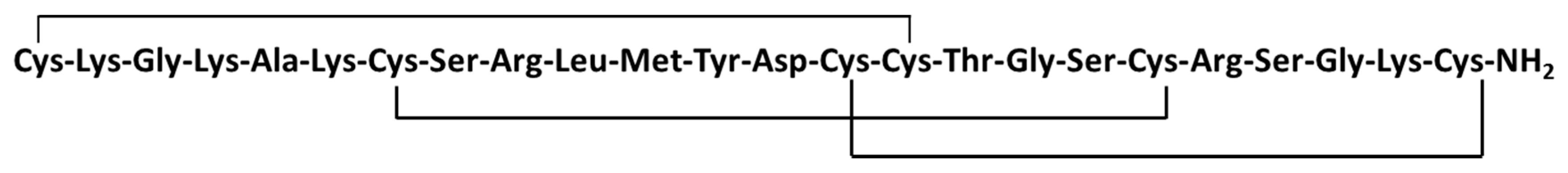
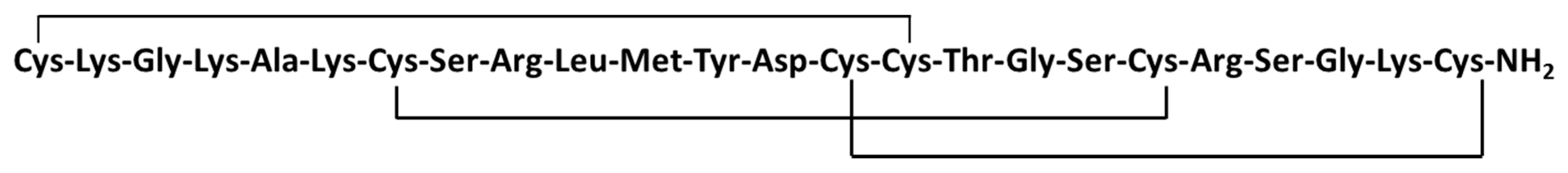
3.5.5. Ziconotide
3.5.6. Pardaxin
3.5.7. YALRAH
4. Anticancer Peptide-Based Drug Therapeutics Developed from Marine Organisms and Future Prospects
Author Contributions
Conflicts of Interest
References
- Burgess, J.G. New and emerging analytical techniques for marine biotechnology. Curr. Opin. Biotechnol. 2012, 23, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, X. Discovery of antitumor agents with peptides from marine sources. JSM Clin. Oncol. Res. 2014, 2, 1034. [Google Scholar]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Jimenez, G.M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Sithranga Boopathy, N.; Kathiresan, K. Anticancer Drugs from Marine Flora: An Overview. J. Oncol. 2010, 2010, 214186. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Micol, V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Blunden, G. Biologically active compounds from marine organisms. Phytother. Res. 2001, 15, 89–94. [Google Scholar] [CrossRef] [PubMed]
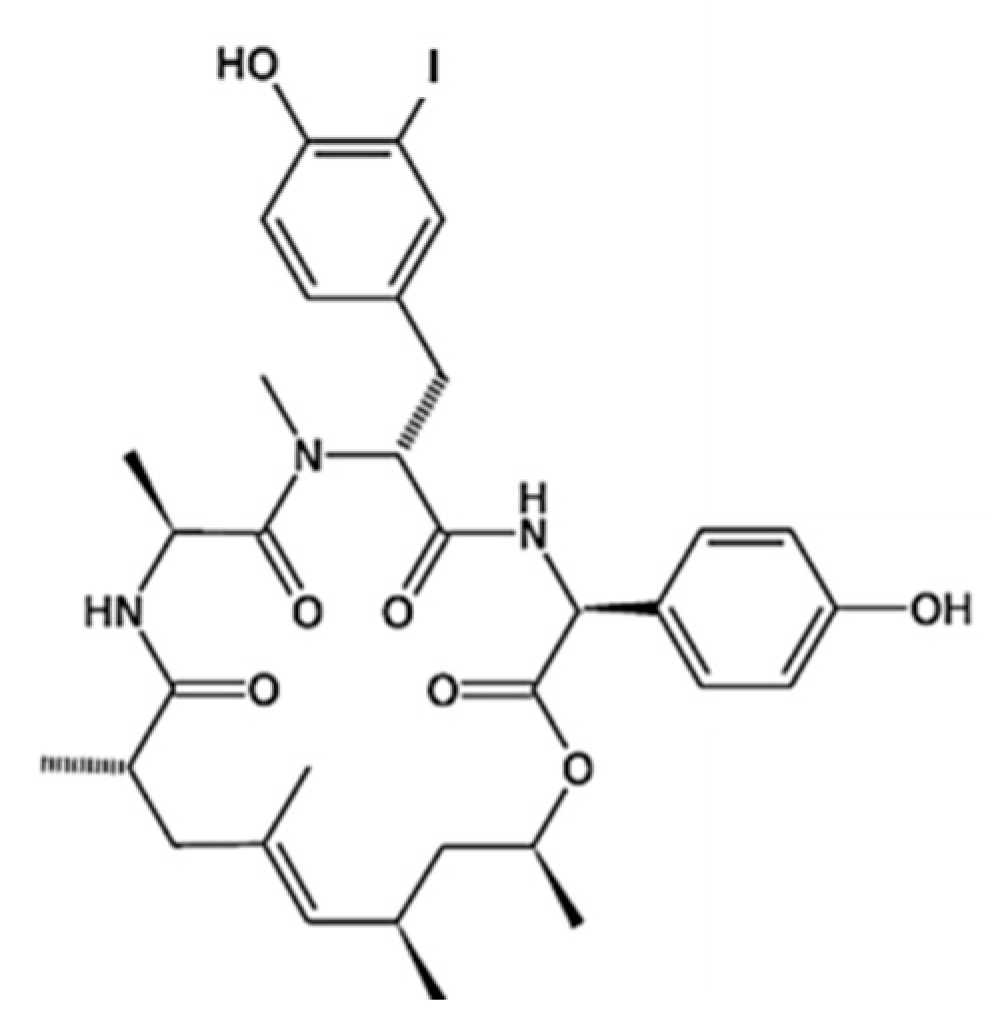
- Molina-Guijarro, J.M.; García, C.; Macías, Á.; García-Fernández, L.F.; Moreno, C.; Reyes, F.; Martínez-Leal, J.F.; Fernández, R.; Martínez, V.; Valenzuela, C.; et al. Elisidepsin interacts directly with glycosylceramides in the plasma membrane of tumor cells to induce necrotic cell death. PLoS ONE 2015, 10, e0140782. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Ocio, E.M.; Pandiella, A.; Maiso, P.; Gajate, C.; Garayoa, M.; Vilanova, D.; Montero, J.; Mitsiades, N.; McMullan, C.J.; et al. Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res. 2008, 68, 5216–5225. [Google Scholar] [CrossRef] [PubMed]
- Malaker, A.; Shah, A.I.A. Therapeutic potency of anticancer peptides derived from marine organisms. Int. J. Eng. Appl. Sci. 2013, 2, 53–65. [Google Scholar]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides-challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Z.; Wang, C.; Lang, L.; Zhou, Y.; Wang, H.; Shang, D.J. Design of potent, non-toxic anticancer peptides based on the structure of the antimicrobial peptide, temporin-1CEa. Arch. Pharm. Res. 2013, 36, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H. Marine peptides: Bioactivities and applications. Mar. Drugs 2015, 13, 4006–4043. [Google Scholar] [CrossRef] [PubMed]
- Thajuddin, N.; Subramanian, G. Cyanobacterial biodiversity and potential applications in biotechnology. Curr. Sci. 2005, 89, 47–57. [Google Scholar]
- Jha, R.K.; Zi-Rong, X. Biomedical compounds from marine organisms. Mar. Drugs 2004, 2, 123–146. [Google Scholar] [CrossRef]
- Sainis, I.; Fokas, D.; Vareli, K.; Tzakos, A.G.; Kounnis, V.; Briasoulis, E. Cyanobacterial cyclopeptides as lead compounds to novel targeted Cancer drugs. Mar. Drugs 2010, 8, 629–657. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Marine fungi: A source of potential anticancer compounds. Front. Microbiol. 2018, 8, 2536. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, I.; Kim, S.K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef] [PubMed]
- Evidente, A.; Kornienko, A.; Cimmino, A.; Andolfi, A.; Lefranc, F.; Mathieu, V.; Kiss, R. Fungal metabolites with anticancer activity. Nat. Prod. Rev. 2014, 31, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Laport, M.S.; Santos, D.C.; Muricy, G. Marine sponges: Potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 2009, 10, 86–105. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Seo, C.H.; Park, Y. Marine peptides and their anti-infective activities. Mar. Drugs 2015, 16, 618–654. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Fareed, S.; Ansari, S.; Khan, M.S. Marine natural products: A lead for anti-cancer. Indian J. Geo-Mar. Sci. 2012, 41, 27–39. [Google Scholar]
- Chakraborty, S.; Ghosh, U. Oceans: A store of house of drugs-A review. J. Pharm. Res. 2010, 3, 1293–1296. [Google Scholar]
- Venkatesan, J.; Anil, S.; Kim, S.K.; Shim, M.S. Marine fish proteins and peptides for cosmeceuticals: A review. Mar. Drugs 2017, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- Khora, S.S. Marine fish-derived bioactive peptides and proteins for human therapeutics. Int. J. Pharm. Pharm. Sci. 2013, 5, 31–37. [Google Scholar]
- Tan, L.T. Filamentous tropical marine cyanobacteria: A rich source of natural products for anticancer drug discovery. J. Appl. Phycol. 2010, 22, 659–676. [Google Scholar] [CrossRef]
- Ma, D.; Zou, B.; Cai, G.; Hu, X.; Liu, J.O. Total synthesis of the cyclodepsipeptide apratoxin a and its analogues and assessment of their biological activities. Chemistry 2006, 12, 7615–7626. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, M.; Maruthanayagam, V.; Sundararaman, M. A review of pharmacological and toxicological potentials of marine cyanobacterial metabolites. J. Appl. Toxicol. 2012, 32, 153–185. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Gutierrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin D, a potent cytotoxic cyclodepsipeptide from papua new guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rein, K.S. New peptides isolated from Lyngbya species: A review. Mar. Drugs 2010, 8, 1817–1837. [Google Scholar] [CrossRef] [PubMed]
- Leusch, H.; Chanda, S.K.; Raya, R.M.; deJesus, P.D.; Orth, A.P.; Walker, J.R.; Belmont, J.C.I.; Schultz, P.G. A functional genomics approach to the mode of action of Apratoxin A. Nat. Chem. Biol. 2006, 2, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, K.; Mutou, T.; Shibata, T.; Itoh, T.; Fujita, T.; Takada, N.; Hayamizu, K.; Takagi, M.; Irifune, T.; Kigoshi, H.; et al. Aurilide, a cytotoxic depsipeptide from the sea hare Dolabella auricularia: Isolation, structure determination, synthesis, and biological activity. Tetrahedron 2004, 60, 8509–8527. [Google Scholar] [CrossRef]
- Han, B.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Teruya, T.; Sasaki, H.; Fukazawa, H.; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2009, 11, 5062–5065. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barria, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.; Hirsch, C.; Sesin, D.; Flor, J.; Chartrain, M.; Fromtling, R.; Harris, G.; Salvatore, M.; Liesch, J.; Yudin, K. Pharmaceuticals from cultured algae. J. Ind. Microbiol. 1990, 5, 113–123. [Google Scholar] [CrossRef]
- Smith, C.D.; Zhang, X.; Mooberry, S.L.; Patterson, G.M.; Moore, R.E. Cryptophycin: A new antimicrotubule agent active against drug-resistant cells. Cancer Res. 1994, 54, 3779–3784. [Google Scholar] [PubMed]
- Lu, K.; Dempsey, J.; Schultz, R.M.; Shih, C.; Teicher, B.A. Cryptophycin-induced hyperphosphorylation of Bcl-2, cell cycle arrest and growth inhibition in human H460 NSCLC cells. Cancer Chemother. Pharmacol. 2001, 47, 170–178. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; del Campo, J.; Mellado, B.; Izquierdo, M.A.; Minarik, T.; Cirri, L.; Marini, L.; Perez-Gracia, J.L.; Scambia, G. A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum-resistant ovarian cancer. Int. J. Gynecol. Cancer 2006, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Edelman, M.J.; Gandara, D.R.; Hausner, P.; Israel, V.; Thornton, D.; DeSanto, J.; Doyle, L.A. Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer 2003, 39, 197–199. [Google Scholar] [CrossRef]
- Simmons, T.L.; Nogle, L.M.; Media, J.; Valeriote, F.A.; Mooberry, S.L.; Gerwick, W.H. Desmethoxymajusculamide C, a cyanobacterial depsipeptide with potent cytotoxicity in both cyclic and ring-opened forms. J. Nat. Prod. 2009, 72, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Ratnayake, R.; Abboud, K.A.; Paul, V.J.; Luesch, H. Grassypeptolides A-C, cytotoxic bis-thiazoline containing marine cyclodepsipeptides. J. Org. Chem. 2010, 75, 8012–8023. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Thimmaiah, M.; Shaala, L.A.; Hau, A.M.; Malmo, J.M.; Ishmael, J.E.; Youssef, D.T.; McPhail, K.L. Cyclic depsipeptides, grassypeptolides D and E and Ibu-epidemethoxylyngbyastatin 3, from a Red Sea Leptolyngbya cyanobacterium. J. Nat. Prod. 2011, 74, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Lee, P.P.F.; Tan, L.T. Hantupeptin A, a cytotoxic cyclic depsipeptide from a Singapore collection of Lyngbya majuscula. J. Nat. Prod. 2009, 72, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Tan, L.T.; Sitachitta, N. Nitrogen-containing metabolites from marine cyanobacteria. In The Alkaloids: Chemistry and Biology, 1st ed.; Cordell, G.A., Ed.; Academic: San Diego, CA, USA, 2001; Volume 57, pp. 75–184. [Google Scholar]
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar] [CrossRef] [PubMed]
- Marquez, B.L.; Watts, K.S.; Yokochi, A.; Roberts, M.A.; Verdier-Pinard, P.; Jimenez, J.I.; Hamel, E.; Scheuer, P.J.; Gerwick, W.H. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J. Nat. Prod. 2002, 65, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Mrozek, C.; Moghaddam, M.F.; Agarwal, S.K. Novel cytotoxic peptides from the tropical marine cyanobacterium Hormothamnion enteromorphoides. 1. Discovery, isolation and initial chemical and biological characterization of the hormothamnins from wild and cultured material. Experientia 1989, 45, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.I.; Vansach, T.; Yoshida, W.Y.; Sakamoto, B.; Porzgen, P.; Horgen, F.D. Halogenated fatty acid amides and cyclic depsipeptides from an eastern Caribbean collection of the cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2009, 72, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.; Tan, L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Park, H.; Al-Awadhi, F.H.; Liu, Y.; Kim, B.; Zeller, S.L.; Chen, Q.Y.; Hong, J.; Luesch, H. Modulation of Activity Profiles for Largazole-Based HDAC Inhibitors through Alteration of Prodrug Properties. ACS Med. Chem. Lett. 2014, 5, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yin, B.; Hu, Z.; Liao, C.; Liu, J.; Li, S.; Li, Z.; Nicklaus, M.C.; Zhou, G.; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Frankmolle, W.P.; Knubel, G.; Moore, R.E.; Patterson, G.M.L. Antifungal cyclic peptides from the terrestrial blue-green alga Anabeana laxa. 2. Structures of laxaphycins A, B, C, D, and E. J. Antibiot. 1992, 5, 1458–1466. [Google Scholar] [CrossRef]
- Bonnard, I.; Rolland, M.; Francisco, C.; Banaigs, B. Total structure and biological properties of laxaphycins A and B, cyclic lipopeptides from the marine cyanobacterium Lyngbya majuscula. Lett. Pept. Sci. 1997, 4, 289–292. [Google Scholar] [CrossRef]
- Bonnard, I.; Rolland, M.; Salmon, J.M.; Debiton, E.; Barthomeuf, C.; Banaigs, B. Total structure and inhibition of tumor cell proliferation of laxaphycins. J. Med. Chem. 2007, 50, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; McPhail, K.L.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Isolation and structure of five lyngbyabellin derivatives from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. Tetrahedron 2005, 61, 11723–11729. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Matthew, S.; Ross, C.; Rocca, J.R.; Paul, V.J.; Luesch, H. Lyngbyastatin 4, a dolastatin 13 analogue with elastase and chymotrypsin inhibitory activity from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2007, 70, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Tasiamide, a cytotoxic peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2002, 65, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. The isolation and structure elucidation of tasiamide B, a 4-amino-3-hydroxy-5-phenylpentanoic acid containing peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2003, 66, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, T.; Ma, Z.; Li, Y. Design, synthesis and biological evaluation of tasiamide analogues as tumor inhibitors. Mar. Drugs 2014, 12, 2308–2325. [Google Scholar] [CrossRef] [PubMed]
- Mevers, E.; Liu, W.T.; Engene, N.; Mohimani, H.; Byrum, T.; Pevzner, P.A.; Dorrestein, P.C.; Spadafora, C.; Gerwick, W.H. Cytotoxic veraguamides, alkynyl bromide-containing cyclic depsipeptides from the marine cyanobacterium cf. Oscillatoria margaritifera. J. Nat. Prod. 2011, 74, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Biggs, J.S.; Paul, V.J.; Luesch, H. Veraguamides A-G, cyclic hexadepsipeptides from a dolastatin 16-producing cyanobacterium Symploca cf. hydnoides from Guam. J. Nat. Prod. 2011, 74, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.X.; Crews, M.S.; Draskovic, M.; Sohn, J.; Johnson, T.A.; Tenney, K.; Valeriote, F.A.; Yao, X.J.; Bjeldanes, L.F.; Crews, P. Azonazine, a novel dipeptide from a Hawaiian marine sediment-derived fungus, Aspergillus insulicola. Org. Lett. 2010, 12, 4458–4461. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.S.; Vasko, R.C.; Lapera, S.A.; Johnson, V.A.; Sellers, R.P.; Lin, C.C.; Pan, C.M.; Davis, M.R.; Ardi, V.C.; McAlpine, S.R. A comprehensive study of Sansalvamide A derivatives: The structure-activity relationships of 78 derivatives in two pancreatic cancer cell lines. Bioorg. Med. Chem. 2009, 17, 5806–5825. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R.C.; Rodriguez, R.A.; Cunningham, C.N.; Ardi, V.C.; Agard, D.A.; McAlpine, S.R. Mechanistic Studies of Sansalvamide A-Amide: An Allosteric Modulator of Hsp90. ACS Med. Chem. Lett. 2010, 1, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lang, G.; Kajahn, I.; Schmaljohann, R.; Imhoff, J.F. Scopularides A and B, cyclodepsipeptides from a marine sponge-derived fungus, Scopulariopsis brevicaulis. J. Nat. Prod. 2008, 71, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, Z.; Wang, H. Cytotoxic natural products from marine sponge-derived microorganisms. Mar. Drugs 2017, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Aoki, S.; Ohyabu, N.; Kurosu, M.; Wang, W.; Kitagawa, I. Arenastatin A, a potent cytotoxic depsipeptide from the Okinawan marine sponge Dysidea arenaria. Tetrahedron Lett. 1994, 35, 7969–7972. [Google Scholar] [CrossRef]
- Koiso, Y.; Morita, K.; Kobayashi, M.; Wang, W.; Ohyabu, N.; Iwasaki, S. Effects of arenastatin A and its synthetic analogs on microtubule assembly. Chem. Biol. Interact. 1996, 102, 183–191. [Google Scholar] [CrossRef]
- Morita, K.; Koiso, Y.; Hashimoto, Y.; Kobayashi, M.; Wang, W.; Ohyabu, N.; Iwasaki, S. Interaction of arenastatin A with porcine brain tubulin. Biol. Pharm. Bull. 1997, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Tamura, S.; Koyama, K.; Sugimoto, M.; Maekawa, R.; Kobayashi, M. New analogue of arenastatin A, a potent cytotoxic spongean depsipeptide, with anti-tumor activity. Bioorg. Med. Chem. Lett. 2004, 14, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Kotoku, N.; Kato, T.; Narumi, F.; Ohtani, E.; Kamada, S.; Aoki, S.; Okada, N.; Nakagawa, S.; Kobayashi, M. Synthesis of 15,20-triamide analogue with polar substituent on the phenyl ring of arenastatin A, an extremely potent cytotoxic spongean depsipeptide. Bioorg. Med. Chem. 2006, 14, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- Aneiros, A.; Garateix, A. Bioactive peptides from marine sources: Pharmacological properties and isolation procedures. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 803, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, R.N.; Farias, J.J.; Tenney, K.; Mooberry, S.L.; Lobkovsky, E.; Clardy, J.; Crews, P. A further study of the cytotoxic constituents of a milnamide-producing sponge. Org. Lett. 2004, 6, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.E.; van Soest, R.; Andersen, R.J. New geodiamolides from the sponge Cymbastela sp. collected in Papua New Guinea. J. Nat. Prod. 1999, 62, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Freitas, V.; Rangel, M.; Bisson, L.; Jaeger, R.; Machado-Santelli, G. The geodiamolide H, derived from Brazilian sponge Geodia corticostylifera, regulates actin cytoskeleton, migration and invasion of breast cancer cells cultured in three-dimensional environment. J. Cell. Physiol. 2008, 216, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Gamble, W.R.; Durso, N.A.; Fuller, R.W.; Westergaard, C.K.; Johnson, T.R.; Sackett, D.L.; Hamel, E.; Cardellina, J.H., II; Boyd, M.R. Cytotoxic and tubulin-interactive hemiasterlins from Auletta sp. and Siphonochalina spp. sponges. Bioorg. Med. Chem. 1999, 7, 1611–1615. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug. Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Talpir, R.; Benayahu, Y.; Kashman, Y.; Pannell, L.; Schleyer, M. Hemiasterlin and geodiamolide TA; two new cytotoxic peptides from the marine sponge Hemiasterella minor (Kirkpatrick). Tetrahedron Lett. 1994, 35, 4453–4456. [Google Scholar] [CrossRef]
- Anderson, H.J.; Coleman, J.E.; Andersen, R.J.; Roberge, M. Cytotoxic peptides hemiasterlin, hemiasterlin A and hemiasterlin B induce mitotic arrest and abnormal spindle formation. Cancer Chemother. Pharmacol. 1997, 39, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Lima, C.M.; Bayraktar, S.; Macintyre, J.; Raez, L.; Flores, A.M.; Ferrell, A.; Rubin, E.H.; Poplin, E.A.; Tan, A.R.; Lucarelli, A.; et al. A phase 1 trial of E7974 administered on day 1 of a 21-day cycle in patients with advanced solid tumors. Cancer 2012, 118, 4262–4270. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Discafani, C.M.; Annable, T.; Beyer, C.; Musto, S.; Hari, M.; Tan, X.; Hardy, C.; Hernandez, R.; Baxter, M.; et al. HTI-286, a synthetic analogue of the tripeptide hemiasterlin, is a potent antimicrotubule agent that circumvents p-glycoprotein-mediated resistance in vitro and in vivo. Cancer Res. 2003, 63, 1838–1845. [Google Scholar] [PubMed]
- Hadaschik, B.A.; Ettinger, S.; Sowery, R.D.; Zoubeidi, A.; Andersen, R.J.; Roberge, M.; Gleave, M.E. Targeting prostate cancer with HTI-286, a synthetic analog of the marine sponge product hemiasterlin. Int. J. Cancer 2008, 122, 2368–2376. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Hadaschik, B.A.; Fazli, L.; Andersen, R.J.; Gleave, M.E.; So, A.I. Intravesical combination treatment with antisense oligonucleotides targeting heat shock protein-27 and HTI-286 as a novel strategy for high-grade bladder cancer. Mol. Cancer Ther. 2009, 8, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Zampella, A.; Sepe, V.; Luciano, P.; Bellotta, F.; Monti, M.; D’Auria, M.; Jepsen, T.; Petek, S.; Adeline, M.; Laprevote, O.; et al. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J. Org. Chem. 2008, 73, 5319–5327. [Google Scholar] [CrossRef] [PubMed]
- Braekman, J.C.; Daloze, D.; Moussiaux, B.; Riccio, R. Jaspamide from the marine sponge Jaspis johnstoni. J. Nat. Prod. 1987, 50, 994–995. [Google Scholar] [CrossRef]
- Odaka, C.; Sanders, M.L.; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immunol. 2000, 7, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Cioca, D.P.; Kitano, K. Induction of apoptosis and CD10/neutral endopeptidase expression by jaspamide in HL-60 line cells. Cell. Mol. Life Sci. 2002, 59, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Matsunaga, S.; Nakao, Y.; Furihata, K.; West, L.; Faulkner, D.J.; Fusetani, N. Koshikamide B, a cytotoxic peptide lactone from a marine sponge Theonella sp. J. Org. Chem. 2008, 73, 7889–7894. [Google Scholar] [CrossRef] [PubMed]
- Plaza, A.; Bifulco, G.; Masullo, M.; Lloyd, J.R.; Keffer, J.L.; Colin, P.L.; Hooper, J.N.A.; Bell, L.J.; Bewley, C.A. Mutremdamide A and koshikamides C–H, peptide inhibitors of HIV-1 entry from different Theonella species. J. Org. Chem. 2010, 75, 4344–4355. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Mangalindan, G.C.; Bojo, Z.P.; Antemano, R.R.; Rodriguez, N.O.; Concepcion, G.P.; Samson, S.C.; de Guzman, D.; Cruz, L.J.; Tasdemir, D.; et al. Microcionamides A and B, Bioactive Peptides from the Philippine Sponge Clathria (Thalysias) abietina. J. Org. Chem. 2004, 69, 4170–4176. [Google Scholar] [CrossRef] [PubMed]
- Fusetani, N.; Sugawara, T.; Matsunaga, S.; Hirota, H. Orbiculamide A: A novel cytotoxic cyclic peptide from a marine sponge Theonella sp. J. Am. Chem. Soc. 1991, 113, 7811–7812. [Google Scholar] [CrossRef]
- Ford, P.W.; Gustafson, K.R.; McKee, T.C.; Shigematsu, N.; Maurizi, L.K.; Pannell, L.K.; Williams, D.E.; de Silva, E.P.; Lassota, P.; Allen, T.M.; et al. Papuamides A-D, HIV-inhibitory and cytotoxic depsipeptides from the sponges Theonella mirabilis and Theonella swinhoei collected in Papua New Guinea. J. Am. Chem. Soc. 1999, 121, 5899–5909. [Google Scholar] [CrossRef]
- Prasad, P.; Aalbersberg, W.; Feussner, K.D.; Van Wagoner, R.M. Papuamides E and F, Cytotoxic Depsipeptides from the Marine Sponge Melophlus sp. Tetrahedron 2011, 67, 8529–8531. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Yi, Y.H.; Wu, H.M.; Xu, Q.Z.; Tang, H.F.; Zhou, D.Z.; Lin, H.W.; Wang, Z.H. Isolation and structure of the cytotoxic cycloheptapeptide Phakellistatin 13. J. Nat. Prod. 2002, 66, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Cichacz, Z.; Barkoczy, J.; Dorsaz, A.C.; Herald, D.L.; Williams, M.D.; Doubek, D.L.; Schmidt, J.M.; Tackett, L.P.; Brune, D.C.; et al. Isolation and structure of the marine sponge cell growth inhibitory cyclic peptide phakellistatin 1. J. Nat. Prod. 1993, 56, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Rodriquez, M.; Bruno, I.; Marzocco, S.; Autore, G.; Riccio, R.; Gomez-Paloma, L. Synthesis, structural aspects and cytotoxicity of the natural cyclopeptides yunnanins A, C and phakellistatins 1, 10. Tetrahedron 2003, 59, 10203–10211. [Google Scholar] [CrossRef]
- Williams, D.E.; Yu, K.; Behrisch, H.W.; Van Soest, R.; Andersen, R.J. Rolloamides A and B, cytotoxic cyclic heptapeptides isolated from the Caribbean marine sponge Eurypon laughlini. J. Nat. Prod. 2009, 72, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Raventos-Suarez, C.; Bifano, M.; Menendez, A.T.; Fairchild, C.R.; Faulkner, D.J. Scleritodermin A, a cytotoxic cyclic peptide from the lithistid sponge Scleritoderma nodosum. J. Nat. Prod. 2004, 67, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cui, Y.M.; Nan, F.J. Total synthesis of the originally proposed and revised structures of scleritodermin A. Org. Lett. 2008, 10, 3765–3768. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Chieze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef] [PubMed]
- Depenbrock, H.; Peter, R.; Faircloth, G.T.; Manzanares, I.; Jimeno, J.; Hanauske, A.R. In vitro activity of aplidine, a new marine-derived anti-cancer compound, on freshly explanted clonogenic human tumour cells and haematopoietic precursor cells. Br. J. Cancer 1998, 78, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Urdiales, J.L.; Morata, P.; de Castro, I.N.; Sánchez-Jiménez, F. Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 1996, 102, 31–37. [Google Scholar] [CrossRef]
- Garcia-Fernandez, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernandez-Sousa, J.M.; Munoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Alonso, M.J.; Gonzalez-Santiago, L.; Martinez, T.; Losada, A.; Galmarini, C.M.; Munoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542. [Google Scholar] [PubMed]
- Baudin, E.; Droz, J.P.; Paz-Ares, L.; van Oosterom, A.T.; Cullell-Young, M.; Schlumberger, M. Phase II study of plitidepsin 3-hour infusion every 2 weeks in patients with unresectable advanced medullary thyroid carcinoma. Am. J. Clin. Oncol. 2010, 33, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Cibeira, M.T.; Richardson, P.G.; Prosper, F.; Oriol, A.; de la Rubia, J.; Lahuerta, J.J.; Garcia-Sanz, R.; Extremera, S.; Szyldergemajn, S.; et al. Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. 2010, 16, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Ribrag, V.; Caballero, D.; Fermé, C.; Zucca, E.; Arranz, R.; Briones, J.; Gisselbrecht, C.; Salles, G.; Gianni, A.M.; Gomez, H.; et al. Multicenter phase II study of plitidepsin in patients with relapsed/refractory non-Hodgkin’s lymphoma. Haematologica 2013, 98, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Dumez, H.; Gallardo, E.; Culine, S.; Galceran, J.C.; Schoffski, P.; Droz, J.P.; Extremera, S.; Szyldergemajn, S.; Fléchon, A. Phase II study of biweekly plitidepsin as second-line therapy for advanced or metastatic transitional cell carcinoma of the urothelium. Mar. Drugs 2009, 7, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Broggini, M.; Marchini, S.V.; Galliera, E.; Borsotti, P.; Taraboletti, G.; Erba, E.; Sironi, M.; Jimeno, J.; Faircloth, G.T.; Giavazzi, R.; et al. Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (VEGF) secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human leukemia cells MOLT-4. Leukemia 2003, 17, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Advanced preclinical and clinical trials of natural products and related compounds from marine sources. Curr. Med. Chem. 2004, 11, 1693–1713. [Google Scholar] [CrossRef] [PubMed]
- Vera, M.D.; Joullie, M.M. Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 2002, 22, 102–145. [Google Scholar] [CrossRef] [PubMed]
- Hambley, T.W.; Hawkins, C.J.; Lavin, M.F.; van den Brenk, A.; Watters, D.J. Cycloxazoline: A cytotoxic cyclic hexapeptide from the ascidian Lissoclinum bistratum. Tetrahedron 1992, 48, 341–348. [Google Scholar] [CrossRef]
- Watters, D.J.; Beamish, H.J.; Marshall, K.A.; Gardiner, R.A.; Seymour, G.J.; Lavin, M.F. Accumulation of HL-60 leukemia cells in G2/M and inhibition of cytokinesis caused by two marine compounds, bistratene A and cycloxazoline. Cancer Chemother. Pharmacol. 1994, 33, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monserrate, Z.; Vervoort, H.C.; Bai, R.; Newman, D.J.; Howell, S.B.; Los, G.; Mullaney, J.T.; Williams, M.D.; Pettit, G.R.; Fenical, W.; et al. Diazonamide A and a synthetic structural analog: Disruptive effects on mitosis and cellular microtubules and analysis of their interactions with tubulin. Mol. Pharmacol. 2003, 63, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Lachia, M.; Moody, C.J. The synthetic challenge of diazonamide A, a macrocyclic indole bis-oxazole marine natural product. Nat. Prod. Rep. 2008, 25, 227–253. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Shioiri, T. Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem. Rev. 2005, 105, 4441–4482. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.; Bowden, B.; Coll, J.; Hockless, D.; Skelton, B.; White, A. Studies of Australian ascidians. Mollamide, a cytotoxic cyclic heptapeptide from the compound ascidian Didemnum molle. Aust. J. Chem. 1994, 47, 61–69. [Google Scholar] [CrossRef]
- Donia, M.S.; Wang, B.; Dunbar, D.C.; Desai, P.V.; Patny, A.; Avery, M.; Hamann, M.T. Mollamides B and C, Cyclic hexapeptides from the indonesian tunicate Didemnum molle. J. Nat. Prod. 2008, 71, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, H.; Fenical, W.; Epifanio, R.A. Tamandarins A and B: New cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000, 65, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Miller, C.; Venkatraman, S.; Fritch, P. Thiolysis of oxazolines-A new, selective method for the direct conversion of peptide oxazolines into thiazolines. Tetrahedron Lett. 1995, 36, 6395–6398. [Google Scholar] [CrossRef]
- Carroll, A.R.; Feng, Y.; Bowden, B.F.; Coll, J.C. Studies of Australian Ascidians. 5. Virenamides A–C, New Cytotoxic Linear Peptides from the Colonial Didemnid Ascidian Diplosoma virens. J. Org. Chem. 1996, 61, 4059–4061. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.C.; Fernandez, A.M.; Lassota, P.; Ireland, C.M.; Barrows, L.R. Inhibition of tubulin polymerization by vitilevuamide, a bicyclic marine peptide, at a site distinct from colchicine, the Vinca alkaloids, and dolastatin 10. Biochem. Pharmacol. 2002, 63, 707–715. [Google Scholar] [CrossRef]
- Von Angerer, E. Tubulin as a target for anticancer drugs. Curr. Opin. Drug Discov. Devel. 2000, 3, 575–584. [Google Scholar] [PubMed]
- Poncet, J. The dolastatins, a family of promising antineoplastic agents. Curr. Pharm. Des. 1999, 5, 139–162. [Google Scholar] [PubMed]
- Bai, R.; Friedman, S.J.; Pettit, G.R.; Hamel, E. Dolastatin-15, a potent antimitotic depsipeptide derived from Dolabella auricularia: Interaction with tubulin and effects on cellular microtubules. Biochem. Pharmacol. 1992, 43, 2637–2645. [Google Scholar] [CrossRef]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the Vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Pathak, S.; Multani, A.S.; Ozen, M.; Richardson, M.A.; Newman, R.A. Dolastatin-10 induces polyploidy, telomeric associations and apoptosis in a murine melanoma cell line. Oncol. Rep. 1998, 5, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Endo, Y.; Shimada, N.; Natsume, T.; Sasaki, T.; Kobayashi, M. Antiangiogenic activity of TZT-1027 (soblidotin) on chick chorioallantoic membrane and human umbilical vein endothelial cells. In Vivo 2007, 21, 297–304. [Google Scholar] [PubMed]
- Riely, G.J.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K.; Kris, M.G.; Rizvi, N.A. A phase 2 study of TZT1027, administered weekly to patients with advanced non-small cell lung cancer following treatment with platinum-based chemotherapy. Lung Cancer 2007, 55, 181–185. [Google Scholar] [CrossRef] [PubMed]
- García-Rocha, M.; Bonay, P.; Avila, J. The antitumoral compound Kahalalide F acts on cell lysosomes. Cancer Lett. 1996, 99, 43–50. [Google Scholar] [CrossRef]
- Faircloth, G.T.; Smith, B.; Grant, W. Selective antitumor activity of Kahalalide F, a marine derived cyclic depsipeptide. Proc. Am. Assoc. Cancer Res. 2001, 42, 1140. [Google Scholar]
- Rademaker-Lakhai, J.M.; Horenblas, S.; Meinhardt, W.; Stokvis, E.; de Reijke, T.M.; Jimeno, J.M.; Lopez-Lazaro, L.; Lopez Martin, J.A.; Beijnen, J.H.; Schellens, J.H. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin. Cancer Res. 2005, 11, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Wosikowski, K.; Schuurhuis, D.; Johnson, K.; Paull, K.D.; Myers, T.G.; Weinstein, J.N.; Bates, S.E. Identification of epidermal growth factor receptor and c-erbB2 pathway inhibitors by correlation with gene expression patterns. J. Natl. Cancer Inst. 1997, 89, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Gonzalez, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Munoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003, 2, 863–872. [Google Scholar] [PubMed]
- Sewell, J.M.; Mayer, I.; Langdon, S.P.; Smyth, J.F.; Jodrell, D.I.; Guichard, S.M. The mechanism of action of kahalalide F: Variable cell permeability in human hepatoma cell lines. Eur. J. Cancer 2005, 41, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, M.L.; Rodriguez, J.A.; Jimeno, J.; Kruyt, F.A.; Giaccone, G. Kahalalide F induces necrosis-like cell death that involves depletion of erbB3 and inhibition of akt signaling. Mol. Pharmacol. 2005, 68, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Martín-Algarra, S.; Espinosa, E.; Rubió, J.; López, J.J.L.; Manzano, J.L.; Carrión, L.A.; Plazaola, A.; Tanovic, A.; Paz-Ares, L. Phase II study of weekly Kahalalide F in patients with advanced malignant melanoma. Eur. J. Cancer 2009, 45, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Corte´s-Funes, H.; Casado, E.; Pardo, B.; Lo´pez-Martı´n, A.; Cuadra, C.; Tabernero, J.; Coronado, C.; García, M.; Soto Matos-Pita, A.; et al. Phase I study of weekly kahalalide F as prolonged infusionin patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 72, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Serova, M.; de Gramont, A.; Bieche, I.; Riveiro, M.E.; Galmarini, C.M.; Aracil, M.; Jimeno, J.; Faivre, S.; Raymond, E. Predictive factors of sensitivity to elisidepsin, a novel kahalalide F-derived marine compound. Mar. Drugs 2013, 11, 944–959. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Arguelaguet, E.; Pons, B.; Aracil, M.; Jimeno, J.; Somoza, R.; Mares, R.; Ramon, Y.C.S.; Hernandez-Losa, J. ErbB3 expression predicts sensitivity to elisidepsin treatment: In vitro synergism with cisplatin, paclitaxel and gemcitabine in lung, breast and colon cancer cell lines. Int. J. Oncol. 2012, 41, 317–324. [Google Scholar] [PubMed]
- Teixido, C.; Mares, R.; Aracil, M.; Ramon y Cajal, S.; Hernandez-Losa, J. Epithelial-mesenchymal transition markers and her3 expression are predictors of elisidepsin treatment response in breast and pancreatic cancer cell lines. PLoS ONE 2013, 8, e53645. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.; Anthoney, A.; Metges, J.P.; Alsin, M.; Gonçalves, A.; Brown, J.; Montagut, C.; Gunzer, K.; Laus, G.; Iglesias-Dios, J.L.; et al. Phase Ib/II study of elisidepsin in metastatic or advanced gastroesophageal cancer (IMAGE trial). Cancer Chemother. Pharmacol. 2016, 77, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Aracil, M.; Jimeno, J.; Perez-Soler, R.; Zou, Y. Molecular pharmacodynamics of PM02734 (elisidepsin) as single agent and in combination with erlotinib; synergistic activity in human non-small cell lung cancer cell lines and xenograft models. Eur. J. Cancer 2009, 45, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Wesson, K.J.; Hamann, M.T. Keenamide A, a bioactive cyclic peptide from the marine mollusk Pleurobranchus forskalii. J. Nat. Prod. 1996, 59, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Yoshida, W.Y.; Takada, Y.; Kimura, J.; Yang, L.; Mooberry, S.L.; Scheuer, P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004, 67, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. ω-Conotoxin MVIIA: From Marine Snail Venom to Analgesic Drug. In Drugs from the Sea; Fusetani, N., Ed.; Karger: Basel, Switzerland, 2000; pp. 74–85. [Google Scholar]
- Olivera, B.M. Conus peptides: Biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef] [PubMed]
- Minich, S.S. Brentuximab vedotin: A new age in the treatment of Hodgkin lymphoma and anaplastic large cell lymphoma. Ann. Pharmacother. 2012, 46, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.; Sapra, P.; Hurvitz, S.A.; Senter, P.; Wahl, A.; Schutten, M.; Shah, D.K.; Haddish-Berhane, N.; Kabbarah, O. Antibody-drug conjugates: An emerging modality for the treatment of cancer. Ann. N. Y. Acad. Sci. 2014, 1321, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Lin, L.C.; Tzen, J.T.; Chen, J.Y. Pardaxin-induced apoptosis enhances antitumor activity in HeLa cells. Peptides 2011, 32, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Y.; Lin, C.N.; Chiou, M.T.; Yu, C.Y.; Chen, J.Y.; Chien, C.H. The antimicrobial peptide pardaxin exerts potent anti-tumor activity against canine perianal gland adenoma. Oncotarget 2015, 10, 2290–2301. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Wei, R.-B.; Luo, H.-Y.; Yang, Z.-S. Isolation and identification of an antiproliferative peptide derived from heated products of peptic hydrolysates of half-fin anchovy (Setipinna taty). J. Funct. Foods 2014, 10, 104–111. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Dennison, S.R.; Whittaker, M.; Harris, F.; Phoenix, D.A. Anticancer α-helical peptides and structure/function relationships underpinning their interactions with tumour cell membranes. Curr. Protein Pept. Sci. 2006, 7, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.B.; Wang, X.F.; Wang, H.Y.; Liu, Y.; Chen, Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Cancer Ther. 2011, 10, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Míguez, A.; Gutiérrez-Jácome, A.; Pérez-Pérez, M.; Pérez-Rodríguez, G.; Catalán-García, S.; Fdez-Riverola, F.; Lourenço, A.; Sánchez, B. From amino acid sequence to bioactivity: The biomedical potential of antitumor peptides. Protein Sci. 2016, 25, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Peptide | Natural Sources | Class/Types | Mode of Action/Investigative Status | Growth Inhibition Concentration (Cell Line) | Ref. |
|---|---|---|---|---|---|
| Apratoxin A–D (1–4) | Cyanobacteria: Lyngbya majuscula, L. sordida | Cyclic depsipeptide | Induction of G1 phase cell cycle arrest and apoptosis/in vitro only | IC50: 0.36 nM (LoVo), 0.52 nM (KB), 2.6 nM (H-460) | [29,30,31,32,33,34,35] |
| Aurilide (5), B (6), C (7) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide | Cytotoxicity /in vitro only | LC50: 40–130 nM (NCI-H460), 10–50 nM (neuro-2a), GI50 < 10 nM (NCI 60 cell line panel) | [36,37] |
| Bisebromoamide (8) | Cyanobacteria: Lyngbya majuscula | Linear peptide | Activation of ERK pathway/in vitro only | IC50: 0.04 μg/mL (HeLa S3), GI50 = 40 nM (JFCR39 cell line panel) | [38] |
| Coibamide A (9) | Cyanobacteria: Leptolyngbya sp. | Cyclic depsipeptide | Cancer cell proliferation inhibition/in vitro only | LC50 < 23 nM (NCI-H460, neuro-2a) GI50 < 7.6 nM (NCI 60 cell line panel) | [39] |
| Cryptophycin (10) | Cyanobacteria: Nostoc sp. | Depsipeptide | Apoptosis and microtubule inhibition /in vitro only (Cryptophycin-52: Phase II human clinical trial) | IC50 < 50 pM (MDR tumor cell lines), | [40,41,42,43,44] |
| Desmethoxymajusculamide C (11) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide | Tubulin polymerization Inhibition/in vitro only | IC50: 20 nM (HCT-116), | [45] |
| Grassypeptolide A–E (12–16) | Cyanobacteria: Lyngbya confervoides | Cyclic depsipeptide | Induction of G2/M phase cell cycle arrest /in vitro only | IC50: 192–335 nM (HeLa), 407–599 nM (neuro-2a) | [46,47] |
| Hantupeptin A (17) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide | Cytotoxicity /in vitro only | IC50: 32 nM (MOLT-4), 4 μM (MCF-7) | [48,49,50] |
| Hectochlorin (18) | Cyanobacteria: Lyngbya majuscula | Lipopeptide | Hyperpolymerization /in vitro only | IC50: 20 nM (CA46), 300 nM (PtK2) | [51] |
| Hormothamnin A (19) | Cyanobacteria: Hormothamnion enteromorphoides | Cyclic undecapeptide | Cytotoxicity /in vitro only | IC50: 0.13–0.72 μg/mL (SW1271, A529, B16-F10, HCT-116)) | [52] |
| Itralamide A (20), B (21) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide | Antiproliferative activity/in vitro only | IC50: 6 μM (HEK293) | [53] |
| Lagunamide A–C (22–24) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide | Antiproliferative activities and apoptosis/in vitro only | IC50: 6.4–24.4 nM (P388) | [54,55] |
| Largazole (25) | Cyanobacteria: Symploca sp. | Cyclic depsipeptide | Stimulation of histone hyperacetylation in the tumor/in vitro only | GI50: 7.7 nM (MDA-MB-231), 122 nM (NMuMG), 55 nM (U2OS), 480 nM (NIH3T3) | [56,57,58,59,60] |
| Laxaphycin A (26), B (27) | Cyanobacteria: Lyngbya majuscula | Cyclic peptide | Antiproliferative activities/in vitro only | IC50 < 2 μM (CEM-WT) | [61,62,63] |
| Lyngbyabellin A (28), E (29), B (30) | Cyanobacteria: Lyngbya majuscula | Lipopeptides | Cytoskeletal actin disruption/in vitro only | IC50: 0.03–0.1 μg/mL (KB), IC50: 0.5–0.83 μg/mL (LoVo), LC50: 0.4–1.2 μg/mL (NCI-H460, neuro-2a) | [64,65] |
| Lyngbyastatin 4–7 (31–34) | Cyanobacteria: Lyngbya sp. | Depsipeptide | Porcine pancreatic elastase inhibition /in vitro only | IC50: 120–210 μM (elastase inhibition) | [66,67] |
| Symplocamide A (35) | Cyanobacteria: Symploca sp. | Cyclic depsipeptide | Proteasome inhibition/in vitro only | IC50: 40 nM (NCI-H460), 29 nM (neuro-2a) | [68] |
| Tasiamide (36), B (37) | Cyanobacteria: Symploca sp. | Linear peptide | Cytotoxicity /in vitro only | IC50: 0.48 μg/mL (KB), 3.47 μg/mL (LoVo) | [69,70,71] |
| Veraguamide A (38), D (39), E (40) | Cyanobacteria: Oscillatoria margaritifera, Symploca cf. Hydnoides sp. | Cyclic depsipeptide | Cytotoxicity /in vitro only | LC50: 141 nM (H-460), IC50: 0.5–1.5 μM (HT29, HeLa) | [72,73] |
| Azonazine (41) | Fungus: Aspergillus insulicola | Hexacyclic dipeptide | Cytotoxicity /in vitro only | IC50 < 15 ng/mL (HCT-116) | [74] |
| Sansalvamide A (42) | Fungus: the genus Fusarium | Cyclic depsipeptide | Apoptosis and inhibition of topoisomerase I /in vitro only | IC50: 4.5 μg/mL (HT29) | [75,76] |
| Scopularides A (43), B (44) | Fungus: Scopulariopsis brevicaulis | Cyclic depsipeptide | Cytotoxicity /in vitro only | IC50: 10 μg/mL (Colo357, Panc89, HT29) | [77] |
| Arenastatin A (45) | Sponge: Dysidea arenaria | Cyclic depsipeptide | Inhibition of microtubule assembly /in vitro only | IC50: 5 pg/mL (KB) | [78,79,80,81,82,83] |
| Discodermin A–H (46–53) | Sponge: Discodermia kiiensis | Tetra- decapeptide | Membrane permeabilization /in vitro only | IC50: 0.02–20 μg/mL (P388, A549) | [84] |
| Geodiamolide H (54) | Sponge: Geodia corticostylifera | Cyclic depsipeptide | Antiproliferative activity /in vitro only | G100: 18.6 nM (OV Car-4), | [85,86,87] |
| Hemiasterlin (55), A (56), C (57) | Sponge: Auletta sp., Siphonochalina sp. | Linear tripeptide | Inhibition of tubulin polymerization /Phase I human clinical trial, (HT1286: Phase I) | IC50: 0.0484–0.269 nM (PC3), 0.404–10.3 nM (NFF), | [88,89,90,91,92,93,94,95] |
| Homophymine A–E (58–62), A1–E1 (63–67) | Sponge: Homophymia sp. | Cyclic depsipeptide | Activation of caspase-3 and 7/in vitro only | IC50: 2–100 nM (MCF7/MCF7R, HCT116/HCT15, HL60/HL60R) | [96] |
| Jaspamide (68) | Sponge: Jaspis johnstoni | Cyclic depsipeptide | Activation of caspase-3, depression of Bcl-2 protein expression /in vitro only | IC50 : 0.04 ng/mL (P388) | [97,98,99] |
| Koshikamide B (69), F–H (70–72) | Sponge: Theonella sp. | Peptide lactone | Cytotoxicity /in vitro only | IC50: 0.45–2.3 μM (P388), 5.5–10 μM (HCT-116) | [100,101] |
| Microcionamide A (73), B (74) | Sponge: Clathria (Thalysias) abietina | Cyclic heptapeptide | Cytotoxicity /in vitro only | IC50: 125–177 nM (MCF-7), 98–172 μM (SKBR-3) | [102] |
| Orbiculamide A (75) | Sponge: Theonella sp. | Cyclic peptide | Cytotoxicity /in vitro only | IC50: 4.7 μg/mL (P388) | [103] |
| Papuamide A–F (76–81) | Sponge: Theonella mirabilis, T. swinhoei | Cyclic depsipeptide | Cytotoxicity and inhibition of infection /in vitro only | IC50: 0.75 ng/mL (human cell line panel) | [83,104,105] |
| Phakellistatin 1 (82), 13 (83) | Sponge: Phakellia carteri | Cyclic heptapeptides | Antiproliferative activity/in vitro only | ED50: 7.5 ug/mL (P388), IC50: 0.75 ng/mL (BEL-7404) | [106,107,108] |
| Rolloamide A (84) | Sponge: Eurypon laughlini | Cyclic heptapeptides | Tubulin polymerization inhibition/in vitro only | IC50: 0.4–5.8 μM (SKBR3, A2780) | [109] |
| Scleritodermin A (85) | Sponge: Scleritoderm nodosum | Cyclic peptide | Microtubule assembly inhibition/in vitro only | IC50 < 2 μM (HCT116, SKBR3, A2780) | [110,111] |
| Aplidin (plitidepsin, 86) | Tunicate: Aplidium albicans | Cyclic depsipeptide | Activation of JNK and p38 MAPK/Phase III human clinical trial | IC50: 0.2–27 nM (CFU-GEMM, CFU-GM, BFU-E) | [112,113,114,115,116,117,118,119,120,121] |
| Didemnin B (87) | Tunicate: Trididemnum solidum | Cyclic depsipeptide | Apoptosis/Phase II human clinical trial | IC50: 2 ng/mL (L1210) | [122,123,124] |
| Cycloxazoline (88) | Ascidia: Lissoclinum bistratum | Cyclic hexapeptide | Apoptosis/in vitro only | IC50: 0.5 μg/mL (MRC5CV1, T24) | [125] |
| Diazonamide A (89) | Ascidia: Diazona angulata | Macrocyclic peptide | Tubulin polymerization inhibition/in vitro only | IC50: 2–5 nM (CA46, MCF7, PC3, A549) | [126,127] |
| Mollamide B (90), C (91) | Ascidia: Didemnum molle | Cyclic depsipeptide | Antiproliferative activity/in vitro only | IC50: 1 μg/mL (P388), 1 μg/mL (A549, HT29) | [128,129,130] |
| Tamandarin A (92), B (93) | Ascidia: Didemnum sp. | Cyclic depsipeptide | Cytotoxicity /in vitro only | IC50: 1.79 μg/mL (BX-PC3), 1.36 μg/mL (DU145), 0.99 μg/mL (UMSCC10b) | [131] |
| Trunkamide A (94) | Ascidia: Lissoclinum sp. | Cyclic peptide | Cytotoxicity /in vitro only | IC50: 0.5 μg/mL (P388, A549, HT29), 1.0 μg/mL (MEL-28) | [132] |
| Virenamides A–C (95–97) | Ascidia: Diplosoma virens | Linear tripeptides | Apoptosis /in vitro only | IC50: 5–10 μg/mL (P388, A549, HT29, CV1) | [133] |
| Vitilevuamide (98) | Ascidia: Didemnum cuculiferum, Polysyncranton lithostrotum | Bicyclic depsipeptide | Tubulin polymerization inhibition/in vitro only | IC50: 6–311 nM (P388, A549, HT29) | [122,134] |
| Dolastatin 10 (99), 15 (100) | Mollusk: Dolabella auricularia | Linear peptide | Microtubule assembly inhibition and Bcl-2 phosphorylation /Human clinical trial Dolastatin 10: phase II (TZT-1027: Phase III) Dolastatin 15: preclinical (ILX651: Phase II, LU-103793: Phase I) | IC50: 50–5000 pM (CA46), 0.5–3 nM (L1210) | [135,136,137,138,139,140,141] |
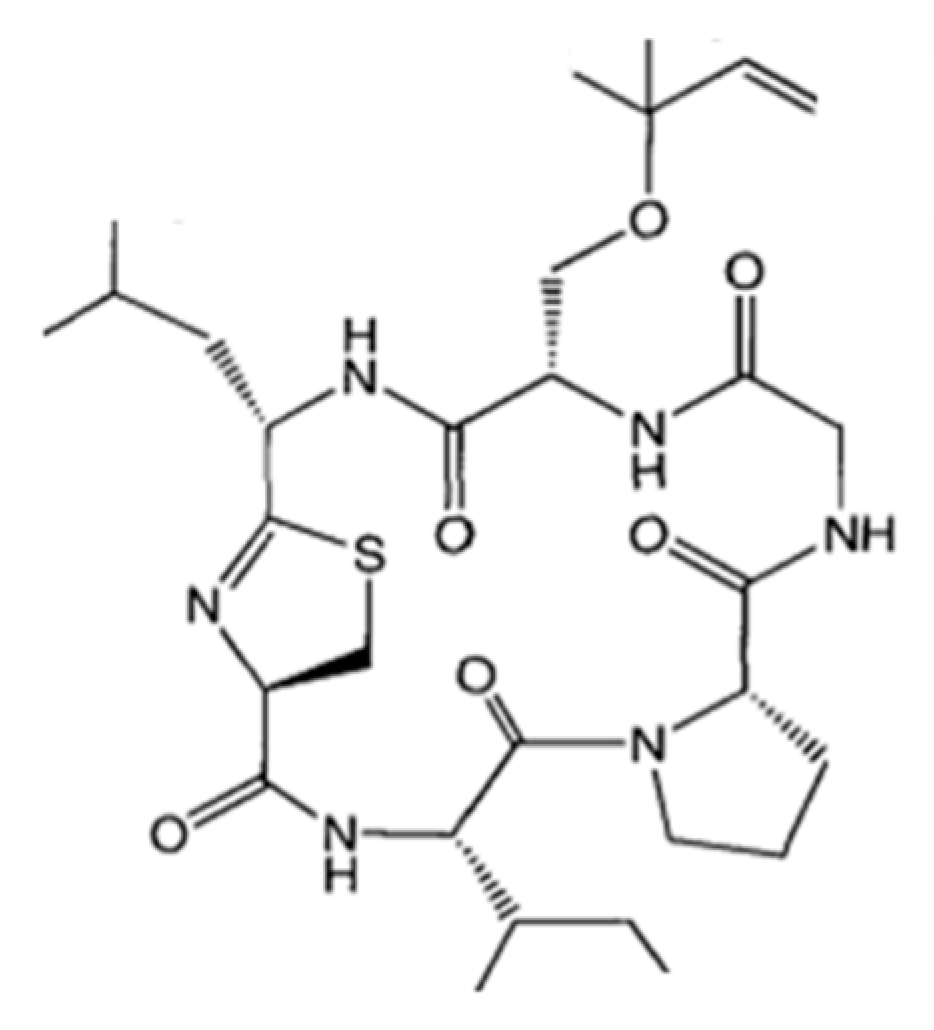
| Kahalalide F (101) | Mollusk: Elysia rufescens | Depsipeptide | ErbB3 protein and PI3K-Akt pathway involved in necrosis induction, apoptosis /Phase I human clinical trial (Elisidepsin: phase II) | IC50: 0.162–0.288 μM (colon), 0.135 μM (A549), 0.162 μM (H5578T), 0.479 μM (HS-578T) | [142,143,144,145,146,147,148,149,150,151,152,153,154,155] |
| Keenamide A (102) | Mollusk: Pleurobranchus forskalii | Cyclic hexapeptide | Cytotoxicity /in vitro only | IC50: 2.5 μg/mL (P388, A549, MEL-20), 5 μg/mL (HT29) | [156] |
| Kulokekahilide-2 (103) | Mollusk: Philinopsis speciosa | Cyclic depsipeptide | Cytotoxicity /in vitro only | IC50: 4.2–59.1 nM (P388, SKOV-3, MDA-MB-435, A-10) | [157] |
| Ziconotide (104) | Mollusk: Conus magus | Linear peptide | Selective N-type calcium channel blocker/FDA approved | IC50: 100 nM (HEK), 10 nM (IMR32) | [158,159,160,161] |
| Pardaxin (105) | Fish: Pardachirus marmoratus | Linear peptide | Caspase-dependent and ROS-mediated Apoptosis /active in animal | IC90: 13 μg/mL (NH-11) | [162,163] |
| YALRAH (106) | Fish: Setipinna taty | Linear peptide | Antiproliferative activity/in vitro only | IC50: 11.1 μM (PC3) | [164] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.K.; Choi, M.-C.; Seo, C.H.; Park, Y. Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides. Int. J. Mol. Sci. 2018, 19, 919. https://doi.org/10.3390/ijms19030919
Kang HK, Choi M-C, Seo CH, Park Y. Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides. International Journal of Molecular Sciences. 2018; 19(3):919. https://doi.org/10.3390/ijms19030919
Chicago/Turabian StyleKang, Hee Kyoung, Moon-Chang Choi, Chang Ho Seo, and Yoonkyung Park. 2018. "Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides" International Journal of Molecular Sciences 19, no. 3: 919. https://doi.org/10.3390/ijms19030919
APA StyleKang, H. K., Choi, M.-C., Seo, C. H., & Park, Y. (2018). Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides. International Journal of Molecular Sciences, 19(3), 919. https://doi.org/10.3390/ijms19030919