Health Risks of Hypovitaminosis D: A Review of New Molecular Insights


Abstract
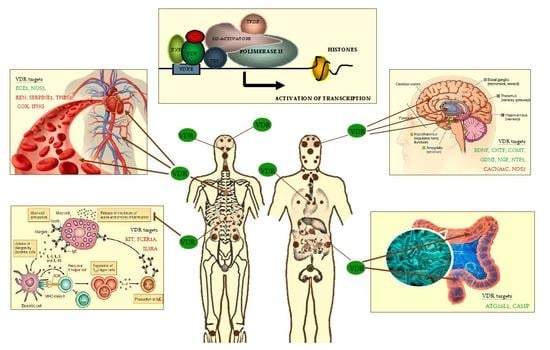
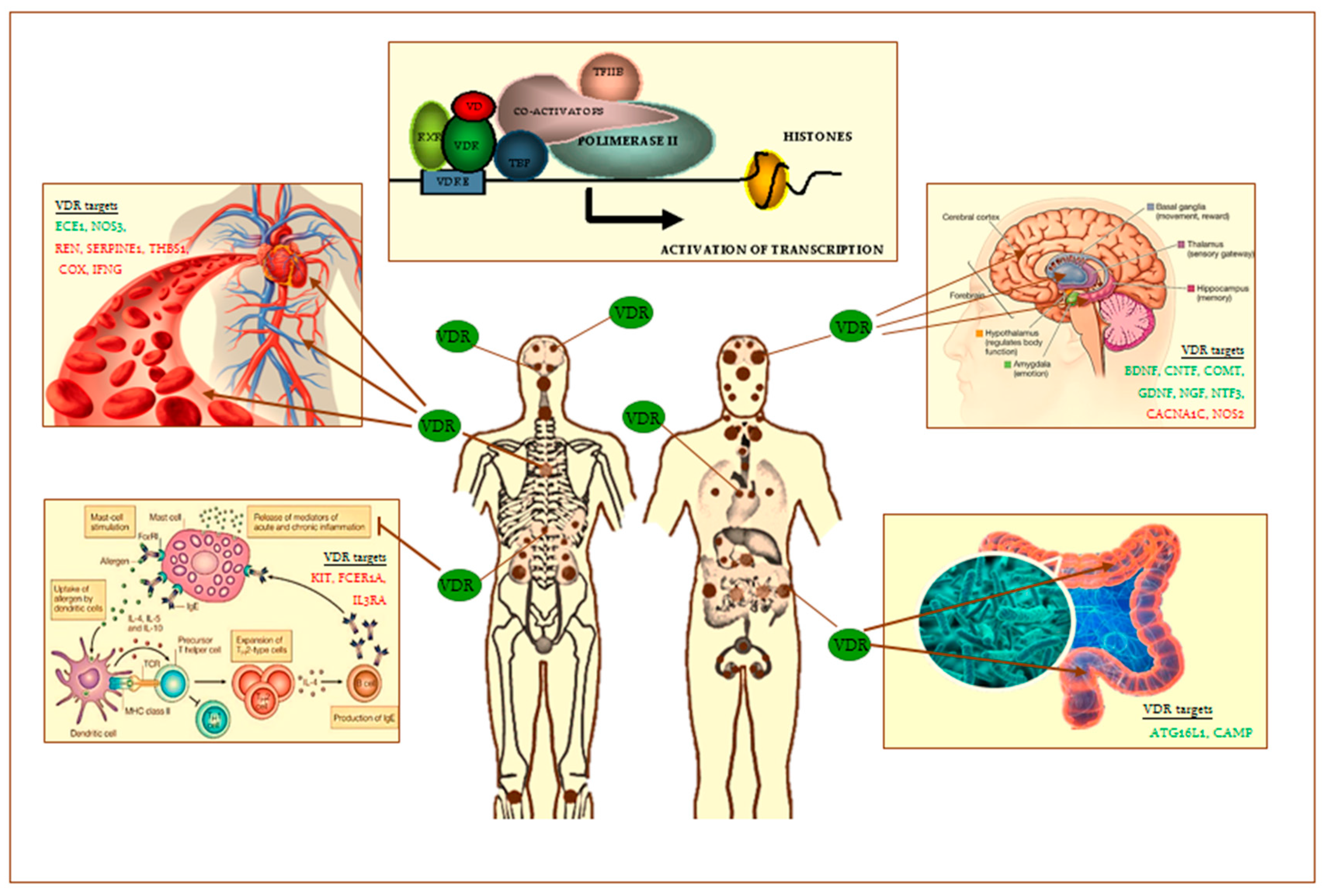
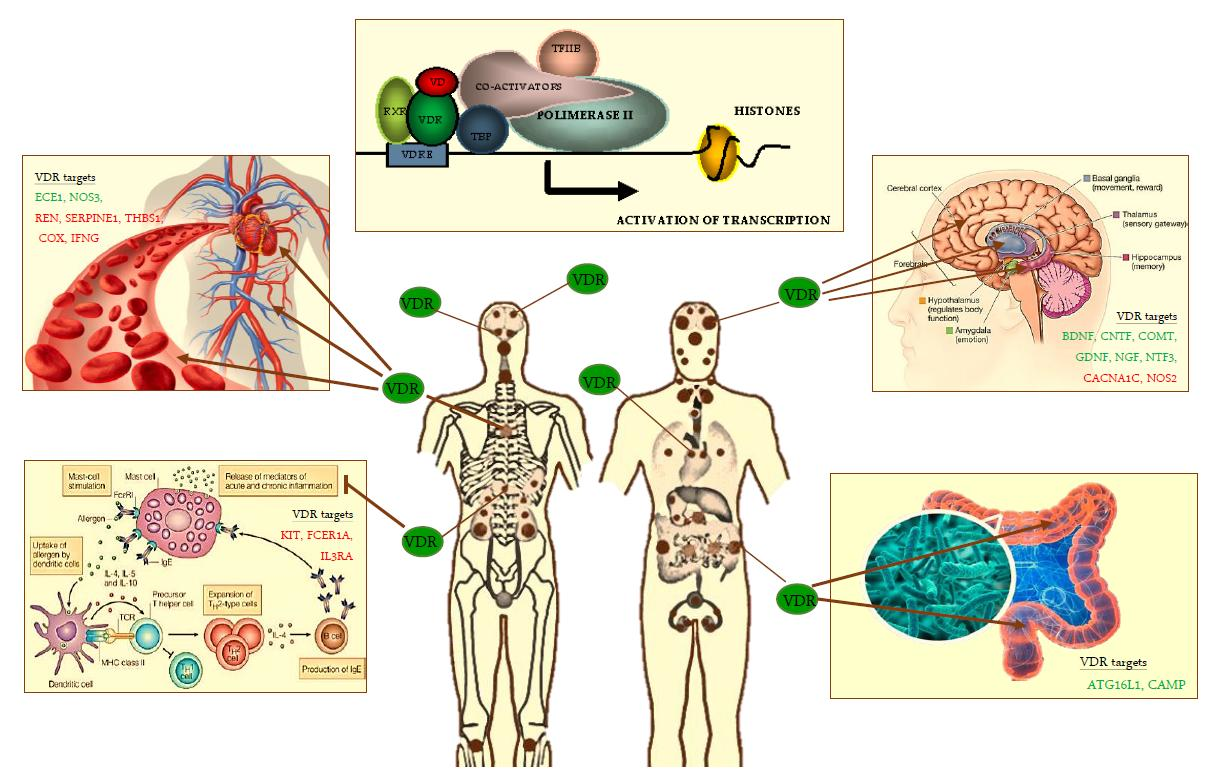{kind=link}
{kind=link}
1. Introduction
2. Vitamin D Analogs
3. Vitamin D Genomic, Epigenomic and Non-Genomic Actions
4. Regulation of Redox/Detoxification Metabolism by Vitamin D
5. Vitamin D, its Analogs and Cardiovascular Disorders
6. Vitamin D and Neurological Disorders
6.1. Vitamin D, Cognitive Impairment, Vascular Dementia and AD
6.2. Vitamin D, Dopaminergic Signaling and PD
6.3. Vitamin D and Neuroinflammation
6.4. Vitamin D and Stroke
7. Vitamin D and Immune System
7.1. Hypovitaminos D and Mast Cell Activation
7.2. Hypovitaminosis D and Hematological Malignancies
8. Vitamin D and the Gastrointestinal Microbioma
9. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoseinzadeh, E.; Taha, P.; Wei, C.; Godini, H.; Ashraf, G.M.; Taghavi, M.; Miri, M. The impact of air pollutants, UV exposure and geographic location on vitamin D deficiency. Food Chem. Toxicol. 2018, 113, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Gorman, S.; Black, L.; Neale, R.E. Clinical, Research, and Public Health Implications of Poor Measurement of Vitamin D Status. J. AOAC Int. 2017, 100, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Schramm, S.; Lahner, H.; Jöckel, K.H.; Erbel, R.; Führer, D.; Moebus, S.; Heinz Nixdorf Recall Study Group. Impact of season and different vitamin D thresholds on prevalence of vitamin D deficiency in epidemiological cohorts—A note of caution. Endocrine 2017, 56, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Gaksch, M.; Jorde, R.; Grimnes, G.; Joakimsen, R.; Schirmer, H.; Wilsgaard, T.; Mathiesen, E.B.; Njølstad, I.; Løchen, M.L.; März, W.; et al. Vitamin D and mortality: Individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLoS ONE 2017, 12, e0170791. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Grübler, M.; Gaksch, M.; Schwetz, V.; Trummer, C.; Hartaigh, B.Ó.; Verheyen, N.; Tomaschitz, A.; März, W. Vitamin D and Mortality. Anticancer Res. 2016, 36, 1379–1387. [Google Scholar] [PubMed]
- Herrmann, M.; Farrell, C.L.; Pusceddu, I.; Fabregat-Cabello, N.; Cavalier, E. Assessment of vitamin D status—A changing landscape. Clin. Chem. Lab. Med. 2017, 55, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Makishima, M. Therapeutic application of vitamin D receptor ligands: An updated patent review. Expert Opin. Ther. Pat. 2015, 25, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Stokes, C.S.; Lammert, F. Vitamin D supplementation: Less controversy, more guidance needed. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.W.; Meyer, M.B.; Lee, S.-M.; Onal, M.; Benkusky, N.A. The vitamin D receptor: Contemporary genomic approaches reveal new basic and translational insights. J. Clin. Investig. 2017, 127, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.W.; Benkusky, N.A.; Meyer, M.B.; Lee, S.M.; St John, H.; Carlson, A.; Onal, M.; Shamsuzzaman, S. Genomic determinants of vitamin D-regulated gene expression. Vitam. Horm. 2016, 100, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Goeman, F.; De Nicola, F.; D’Onorio, F.; De Meo, P.; Pallocca, M.; Elmi, B.; Castrignanò, T.; Pesole, G.; Strano, S.; Blandino, G.; Fanciulli, M.; et al. VDR primary targets by genome-wide transcriptional profiling. J. Steroid Biochem. Mol. Biol. 2014, 143, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Molecular endocrinology of vitamin D on the epigenome level. Mol. Cell. Endocrinol. 2017, 453, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Hii, C.S.; Ferrante, A. The non-genomic actions of vitamin, D. Nutrients 2016, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signalling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Virgili, N.; Mancera, P.; Wappenhans, B.; Sorrosal, G.; Biber, K.; Pugliese, M.; Espinosa-Parrilla, J.F. KATP channel opener diazoxide prevents neurodegeneration: A new mechanism of action via antioxidative pathway activation. PLoS ONE 2013, 8, e75189. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Daffu, G.; del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int. J. Mol. Sci. 2013, 14, 19891–19910. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. The redox proteome. J. Biol. Chem. 2013, 288, 26512–26520. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone Klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Zeldich, E.; Chen, C.-D.; Colvin, T.A.; Bove-Fenderson, E.A.; Liang, J.; Tucker Zhou, T.B.; Harris, D.A.; Abraham, C.R. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J. Biol. Chem. 2014, 289, 24700–24715. [Google Scholar] [CrossRef] [PubMed]
- Trehan, N.; Afonso, L.; Levine, D.L.; Levy, P.D. Vitamin D Deficiency, Supplementation, and Cardiovascular Health. Crit. Pathw. Cardiol. 2017, 16, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Shapses, S.A.; Manson, J.E. Vitamin D and prevention of cardiovascular disease and diabetes: Why the evidence falls short. JAMA 2011, 305, 2565–2566. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Manson, J.E.; Song, Y.; Sesso, H.D. Systematic review: Vitamin D and calcium supplementation in prevention of cardiovascular events. Ann. Intern. Med. 2010, 152, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Avenell, A.; MacLennan, G.S.; Jenkinson, D.J.; McPherson, G.C.; McDonald, A.M.; Pant, P.R.; Grant, A.M.; Campbell, M.K.; Anderson, F.H.; Cooper, C.; et al. Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D3 and/or calcium (RECORD Trial). J. Clin. Endocrinol. Metab. 2012, 97, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Cremer, A.; Tambosco, C.; Corcuff, J.B.; Boulestreau, R.; Gaillard, P.; Lainé, M.; Papaioannou, G.; Gosse, P. Investigating the association of vitamin D with blood pressure and the renin-angiotensin-aldosterone system in hypertensive subjects: A cross-sectional prospective study. J. Hum. Hypertens. 2018, 32, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tomaschitz, A.; Pilz, S.; Ritz, E.; Grammer, T.; Drechsler, C.; Boehm, B.O.; März, W. Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin. Chim. Acta 2010, 411, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Al Mheid, I.; Quyyumi, A.A. Vitamin D and Cardiovascular Disease: Controversy Unresolved. J. Am. Coll. Cardiol. 2017, 70, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Chen, X.; Cheng, L.; Rao, M.; Chen, K.; Zhang, N.; Meng, J.; Li, M.; Liu, Z.Q.; Yang, P.C. Vitamin D receptor restricts Th2-biased inflammation in the heart. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef]
- Salum, E.; Kals, J.; Kampus, P.; Salum, T.; Zilmer, K.; Aunapuu, M.; Arend, A.; Eha, J.; Zilmer, M. Vitamin D reduces deposition of advanced glycation end-products in the aortic wall and systemic oxidative stress in diabetic rats. Diabetes. Res. Clin. Pract. 2013, 100, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I.; Marginean, O. Links between Vitamin D Deficiency and Cardiovascular Diseases. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Santoro, D.; Caccamo, D.; Lucisano, S.; Buemi, M.; Sebekova, K.; Teta, D.; De Nicola, L. Interplay of vitamin D, erythropoiesis, and the renin-angiotensin system. Biomed. Res. Int. 2015, 2015, 145828. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xu, C. Physiology and Pathophysiology of the Intrarenal Renin-Angiotensin System: An Update. J. Am. Soc. Nephrol. 2017, 28, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Legarth, C.; Grimm, D.; Wehland, M.; Bauer, J.; Krüger, M. The Impact of Vitamin D in the Treatment of Essential Hypertension. Int. J. Mol. Sci. 2018, 19, 455. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.M.; Norris, K.C.; Artaza, J.N. Vitamin D and Cardiac Differentiation. Vitam. Horm. 2016, 100, 299–320. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, M.; Manzanares, E.; Bas, M.; Martín-Nunes, L.; Val-Blasco, A.; Jesús Larriba, M.; Fernández-Velasco, M.; Delgado, C. Calcitriol (1,25-dihydroxyvitamin D(3)) increases L-type calcium current via protein kinase A signaling and modulates calcium cycling and contractility in isolated mouse ventricular myocytes. Heart Rhythm. 2017, 14, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Delansorne, R.; Man, R.Y.; Svenningsen, P.; Vanhoutte, P.M. Chronic treatment with vitamin D lowers arterial blood pressure and reduces endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1226–H1234. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Ireland, S.; Graeme Houston, J.; Gandy, S.J.; Waugh, S.; Macdonald, T.M.; Mackenzie, I.S.; Struthers, A.D. Vitamin D therapy to reduce blood pressure and left ventricular hypertrophy in resistant hypertension: Randomized, controlled trial. Hypertension 2014, 63, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.J.; Mousa, A.; Ebeling, P.R.; Scott, D.; de Courten, B. Effects of vitamin D supplementation on inflammatory markers in heart failure: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 1169. [Google Scholar] [CrossRef] [PubMed]
- Franczyk, A.; Stolarz-Skrzypek, K.; Wesołowska, A.; Czarnecka, A. Vitamin D and Vitamin D Receptor Activators in Treatment of Hypertension and Cardiovascular Disease. Cardiovasc. Haematol. Dis. Drug Targets 2014, 14, 34–44. [Google Scholar] [CrossRef]
- Sezer, S.; Tutal, E.; Bal, Z.; Uyar, M.E.; Bal, U.; Cakir, U.; Acar, N.O.; Haberal, M. Differential influence of vitamin D analogs on left ventricular mass index in maintenance hemodialysis patients. Int. J. Artif. Organs. 2014, 37, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Barrio-Vázquez, S.; Naves-Díaz, M.; Carrillo-López, N.; Rodríguez, I.; Fernández-Vázquez, A.; Valdivielso, J.M.; Thadhani, R.; Cannata-Andía, J.B. Vitamin D receptor activation, left ventricular hypertrophy and myocardial fibrosis. Nephrol. Dial. Transplant. 2013, 28, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Carrillo-López, N.; Naves-Díaz, M.; Solache-Berrocal, G.; Martínez-Arias, L.; Rodrigues-Díez, R.R.; Fernández-Vázquez, A.; Martínez-Salgado, C.; Ruiz-Ortega, M.; Dusso, A.; et al. Regulation of miR-29b and miR-30c by vitamin D receptor activators contributes to attenuate uraemia-induced cardiac fibrosis. Nephrol. Dial. Transplant. 2017, 32, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Domínguez-Pimentel, V.; Muros-de-Fuentes, M.; Mora-Fernández, C.; Martín-Núñez, E.; Cazaña-Pérez, V.; Navarro-González, J.F. Beneficial effects of selective vitamin D receptor activation by paricalcitol in chronic kidney disease. Curr. Drug Targets 2014, 15, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Rasmussen, K.; Rasmussen, L.M.; Bruunsgaard, H.; Brandi, L. The influence of vitamin D analogs on calcification modulators, N-terminal pro-B-type natriuretic peptide and inflammatory markers in hemodialysis patients: A randomized crossover study. BMC Nephrol. 2014, 15, 130. [Google Scholar] [CrossRef] [PubMed]
- Giakoumis, M.; Tsioufis, C.; Dimitriadis, K.; Sonikian, M.; Kasiakogias, A.; Andrikou, E.; Kalos, T.; Konstantinidis, D.; Filis, K.; Petras, D.; et al. Effects of oral paricalcitol therapy on arterial stiffness and osteopontin in hypertensive patients with chronic kidney disease and secondary hyperparathyroidism. Hell. J. Cardiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mpandzou, G.; Aït Ben Haddou, E.; Regragui, W.; Benomar, A.; Yahyaoui, M. Vitamin D deficiency and its role in neurological conditions: A review. Rev. Neurol. (Paris) 2016, 172, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Burne, T.H.J.; McGrath, J.J. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front. Neuroendocrinol. 2013, 34, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the brain: Genomic and non-genomic actions. Mol. Cell. Endocrinol. 2017, 453, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Gooch, H.; Groves, N.J.; Sah, P.; Burne, T.H.; Eyles, D.W.; McGrath, J.J. Vitamin D and the brain: Key questions for future research. J. Steroid Biochem. Mol. Biol. 2015, 148, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Tong, L.; Peng, J.B.; Jin, X.H.; Wei, D.; Xu, B.K.; Wang, Z.Y. Changes in the expression of the vitamin D receptor and LVSCC-A1C in the rat hippocampus submitted to single prolonged stress. Mol. Med. Rep. 2014, 9, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Cui, J.; Jin, F.; Cui, Y.; Li, R.; Jiang, X.; Tian, Y.; Wang, K.; Jiang, P.; Gao, J. Induction of the Vitamin D Receptor Attenuates Autophagy Dysfunction-Mediated Cell Death Following Traumatic Brain Injury. Cell. Physiol. Biochem. 2017, 42, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Song, S.; Cui, J.; Feng, Y.; Gao, J.; Jiang, P. Vitamin D Receptor Activation Influences NADPH Oxidase (NOX(2)) Activity and Protects against Neurological Deficits and Apoptosis in a Rat Model of Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Dursun, E.; Yilmazer, S. The effects of vitamin D receptor silencing on the expression of LVSCC-A1C and LVSCC-A1D and the release of NGF in cortical neurons. PLoS ONE 2011, 6, e17553. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of Estrogen and Other Sex Hormones in Brain Aging. Neuroprotection and DNA Repair. Front. Aging Neurosci. 2017, 9, 430–451. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, P.; Bhattarai, J.P.; Kim, M.S.; Han, S.K. Non-genomic action of vitamin D3 on N-methyl-d-aspartate and kainate receptor-mediated actions in juvenile gonadotrophin releasing hormone neurons. Reprod. Fertil. Dev. 2017, 29, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, M.; Talaei, S.A.; Djazayeri, A.; Salami, M. Vitamin D supplementation restores suppressed synaptic plasticity in Alzheimer’s disease. Nutr. Neurosci. 2014, 17, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Buitrago, C.; Boland, R. Caveolae and caveolin-1 are implicated 1 in 1alpha,25(OH)2-vitamin D3-dependent modulation of Src, MAPK cascades and VDR localization in skeletal muscle cells. J. Steroid Biochem. Mol. Biol. 2010, 121, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Boyan, B.D.; Sylvia, V.L.; Dean, D.D.; Del Toro, F.; Schwartz, Z. Differential regulation of growth plate chondrocytes by 1alpha,25-(OH)2D3 and 24R,25-(OH)2D3 involves cell-maturation-specific membrane-receptor-activated phospholipid metabolism. Crit. Rev. Oral Biol. Med. 2002, 13, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Doroudi, M.; Cheung, J.; Grozier, A.L.; Schwartz, Z.; Boyan, B.D. Plasma membrane PDIA3 and VDR interact to elicit rapid responses to 1alpha,25(OH)2D3. Cell. Signal. 2013, 25, 2362–2373. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, L.; Goulart, P.B.; Goncalves, R.; Pierozan, P.; Winkelmann-Duarte, E.C.; Woehl, V.M.; Pessoa-Pureur, R.; Silva, F.R.; Zamoner, A. 1alpha,25-dihydroxyvitamin D3 mechanism of action: Modulation of L-type calcium channels leading to calcium uptake and intermediate filament phosphorylation in cerebral cortex of young rats. Biochim. Biophys. Acta 2012, 1823, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. The molecular biology and pathophysiology of vascular calcification. Postgrad. Med. 2014, 126, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Xie, S.X.; Kling, M.A.; Toledo, J.B.; Wolk, D.A.; Lee, E.B.; Van Deerlin, V.; Lee, V.M.; Trojanowski, J.Q.; Arnold, S.E. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 2012, 135 Pt 12, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Buell, J.S.; Dawson-Hughes, B.; Scott, T.M.; Weiner, D.E.; Dallal, G.E.; Qui, W.Q.; Bergethon, P.; Rosenberg, I.H.; Folstein, M.F.; Patz, S.; et al. 25-Hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology 2010, 74, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Fantino, B.; Le Gall, D.; Schott, A.M.; Berrut, G.; Beauchet, O. Severe vitamin D deficiency is associated with advanced-stage dementia in geriatric inpatients. J. Am. Geriatr. Soc. 2011, 59, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaft, J.; Koek, H.L.; Dijkstra, E.; Verhaar, H.J.; van der Schouw, Y.T.; Emmelot-Vonk, M.H. The association between vitamin D and cognition: A systematic review. Ageing Res. Rev. 2013, 12, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Vitamin D deficiency is associated with increased risk of Alzheimer’s disease and dementia: Evidence from meta-analysis. Nutr. J. 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Lökk, J.; Solomon, A.; Mangialasche, F.; Miralbell, J.; Spulber, G.; Annerbo, S.; Andreasen, N.; Winblad, B.; Cedazo-Minguez, A.; et al. Vitamin D in relation to cognitive impairment, cerebrospinal fluid biomarkers, and brain volumes. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Montero-Odasso, M.; Llewellyn, D.J.; Richard-Devantoy, S.; Duque, G.; Beauchet, O. Meta-analysis of memory and executive dysfunctions in relation to vitamin D. J. Alzheimers. Dis. 2013, 37, 147–171. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Rolland, Y.; Schott, A.M.; Blain, H.; Vellas, B.; Herrmann, F.R.; Beauchet, O. Higher vitamin D dietary intake is associated with lower risk of Alzheimer’s disease: A 7-year follow-up. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hara, K.; Van Baaren, J.M.; Price, J.C.; Beecham, G.W.; Gallins, P.J.; Whitehead, P.L.; Wang, G.; Lu, C.; Slifer, M.A.; et al. Vitamin D receptor and Alzheimer’s disease: A genetic andfunctional study. Neurobiol. Aging 2012, 33, 1844.e1–1844.e9. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Song, G.G. Vitamin D receptor polymorphisms and susceptibility to Parkinson’s disease and Alzheimer’s disease: A meta-analysis. Neurol. Sci. 2014, 35, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Shah, J. Role of Vitamin D in Amyloid clearance via LRP-1 upregulation in Alzheimer’s disease: A potential therapeutic target? J. Chem. Neuroanat. 2017, 85, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Dursun, E.; Yilmazer, S. Vitamin D inquiry in hippocampal neurons: Consequences of vitamin D-VDR pathway disruption on calcium channel and the vitamin D requirement. Neurol. Sci. 2013, 34, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.X.; He, L.Y.; Zhang, M.; Wang, F.; Liu, F.; Peng, W.X. 1,25-Dihydroxyvitamin D3 regulates expression of LRP1 and RAGE in vitro and in vivo, enhancing Abeta1-40 brain-to-blood efflux and peripheral uptake transport. Neuroscience 2016, 322, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Landel, V.; Millet, P.; Baranger, K.; Loriod, B.; Féron, F. Vitamin D interacts withEsr1 and Igf1 to regulate molecular pathways relevant to Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Pelekanos, M.; Liu, P.Y.; Burne, T.H.; McGrath, J.J.; Eyles, D.W. The vitamin D receptor in dopamine neurons; its presence in human substantianigra and its ontogenesis in rat midbrain. Neuroscience 2013, 236, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Orme, R.P.; Middleditch, C.; Waite, L.; Fricker, R.A. The Role of Vitamin D3 in the Development and Neuroprotection of Midbrain Dopamine Neurons. Vitam. Horm. 2016, 100, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Rimmelzwaan, L.M.; van Schoor, N.M.; Lips, P.; Berendse, H.W.; Eekhoff, E.M. Systematic Review of the Relationship between Vitamin D and Parkinson’s Disease. J. Parkinsons Dis. 2016, 6, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Luan, W.; Hammond, L.A.; Cotter, E.; Osborne, G.W.; Alexander, S.A.; Nink, V.; Cui, X.; Eyles, D.W. Developmental Vitamin D (DVD) Deficiency Reduces Nurr1 and TH Expression in Post-mitotic Dopamine Neurons in Rat Mesencephalon. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pertile, R.A.; Cui, X.; Eyles, D.W. Vitamin D signaling and the differentiation of developing dopamine systems. Neuroscience 2016, 333, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Orme, R.P.; Bhangal, M.S.; Fricker, R.A. Calcitriol imparts neuroprotection in vitro to midbrain dopaminergic neurons by upregulating GDNF expression. PLoS ONE 2013, 8, e62040. [Google Scholar] [CrossRef] [PubMed]
- Gatto, N.M.; Paul, K.C.; Sinsheimer, J.S.; Bronstein, J.M.; Bordelon, Y.; Rausch, R.; Ritz, B. Vitamin D receptor gene polymorphisms and cognitive decline in Parkinson’s disease. J. Neurol. Sci. 2016, 370, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Rcom-H’cheo-Gauthier, A.N.; Meedeniya, A.C.; Pountney, D.L. Calcipotriol inhibitsα-synuclein aggregation in SH-SY5Y neuroblastoma cells by aCalbindin-D28k-dependent mechanism. J. Neurochem. 2017, 141, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Briones, T.L.; Darwish, H. Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J. Neuroinflamm. 2012, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Boontanrart, M.; Hall, S.D.; Spanier, J.A.; Hayes, C.E.; Olson, J.K. Vitamin D3 alters microglia immune activation by an IL-10 dependent SOCS3 mechanism. J. Neuroimmunol. 2016, 292, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Djukic, M.; Onken, M.L.; Schutze, S.; Redlich, S.; Gotz, A.; Hanisch, U.K.; Bertsch, T.; Ribes, S.; Hanenberg, A.; Schneider, S.; et al. Vitamin D deficiency reduces the immune response, phagocytosis rate, and intracellular killing rate of microglial cells. Infect. Immun. 2014, 82, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Guéry, J.C. Estrogens and inflammatory autoimmune diseases. Jt. Bone Spine 2012, 79, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chen, J.; Zheng, C.; Wu, J.; Cheng, Y.; Zhu, S.; Lin, C.; Cao, Q.; Zhu, J.; Jin, T. 1,25-dihydroxyvitamin D3 -induced dendritic cells suppress experimental autoimmune encephalomyelitis by increasing proportions of the regulatory lymphocytes and reducing T helper type 1 and type 17 cells. Immunology 2017, 152, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, H.A.; Rasouli, J.; Ciric, B.; Wei, D.; Rostami, A.; Zhang, G.X. 1,25-Dihydroxyvitamin D3 suppressed experimental autoimmune encephalomyelitis through both immunomodulation and oligodendrocyte maturation. Exp. Mol. Pathol. 2017, 102, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Azizi, S.V.; Arabi, Z.; Ahangar-Parvin, R.; Mohammadi-Kordkhayli, M.; Larussa, T.; Khatami, F.; Nemati, M. Vitamin D down-regulates the expression of some Th17 cell-related cytokines, key inflammatory chemokines, and chemokine receptorsin experimental autoimmune encephalomyelitis. Nutr. Neurosci. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nystad, A.E.; Wergeland, S.; Aksnes, L.; Myhr, K.M.; Bø, L.; Torkildsen, O. Effect ofhigh-dose 1.25 dihydroxyvitamin D3 on remyelination in the cuprizone model. APMIS 2014, 122, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, D.F.; Lehmann, M.F.; Oliveira, S.R.; Flauzino, T.; Delongui, F.; de Araújo, M.C.; Dichi, I.; Delfino, V.D.; Mezzaroba, L.; Simão, A.N.; et al. Vitamin D deficiency is associated with acute ischemic stroke, C-reactive protein, and short-term outcome. Metab. Brain Dis. 2017, 32, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D prevents hypoxia/reoxygenation-induced blood-brain barrier disruption via vitamin D receptor-mediated NF-kB signaling pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [PubMed]
- Enkhjargal, B.; McBride, D.W.; Manaenko, A.; Reis, C.; Sakai, Y.; Tang, J.; Zhang, J.H. Intranasal administration of vitamin D attenuates blood-brain barrier disruption through endogenous up-regulation of osteopontin and activation of CD44/P-gp glycosylation signaling after subarachnoid hemorrhage in rats. J. Cereb. Blood Flow Metab. 2017, 37, 2555–2566. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, I.; Pawliczak, R. The active metabolite of vitamin D3 as a potent immunomodulator. Scand. J. Immunol. 2016, 83, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Wöbke, T.K.; Sorg, B.L.; Steinhilber, D. Vitamin D in inflammatory diseases. Front. Physiol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Li, X.X.; Qiu, S.Q.; Yu, Y.; Li, M.G.; Yang, L.T.; Li, L.J.; Wang, S.; Zheng, P.Y.; Liu, Z.G.; et al. Vitamin D contributes to mast cell stabilization. Allergy 2017, 72, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Finn, D.F.; Walsh, J.J. Twenty-first century mast cell stabilizers. Br. J. Pharmacol. 2013, 170, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, M.; Destefanis, M.; Morena, D.; Foglizzo, V.; Forneris, M.; Pescarmona, G.; Silvagno, F. The vitamin D receptor inhibits the respiratory chain, contributing to the metabolic switch that is essential for cancer cell proliferation. PLoS ONE 2014, 9, e115816. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Jansson, B.; Bonelli, F. Modulation of inflammatory and immune responses by vitamin D. J. Autoimmun. 2017, 85, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Vanherwegen, A.S.; Gysemans, C.; Mathieu, C. Regulation of Immune Function by Vitamin D and Its Use in Diseases of Immunity. Endocrinol. Metab. Clin. N. Am. 2017, 46, 1061–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, G.; He, X.; Gao, J.; Wang, R.; Wang, Y.; Zhao, W. Serum 25-hydroxyvitamin D levels and prognosis in hematological malignancies: A systematic review and meta-analysis. Cell. Physiol. Biochem. 2015, 35, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Bruns, H.; Büttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.H.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra47. [Google Scholar] [CrossRef] [PubMed]
- Kozielewicz, P.; Grafton, G.; Kutner, A.; Curnow, S.J.; Gordon, J.; Barnes, N.M. Novel vitamin D analogues; cytotoxic and anti-proliferative activity against a diffuse large B-cell lymphoma cell line and B-cells from healthy donors. J. Steroid Biochem. Mol. Biol. 2016, 164, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kulling, P.M.; Olson, K.C.; Olson, T.L.; Feith, D.J.; Loughran, T.P., Jr. Vitamin D in hematological disorders and malignancies. Eur. J. Haematol. 2017, 98, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Barbalho, S.M.; Goulart, R.A.; Gasparini, R.G. Associations between inflammatory bowel diseases and vitamin D. Crit. Rev. Food Sci. Nutr. 2017, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.E.; Williams, E.A.; Corfe, B.M. Vitamin D status in irritable bowel syndrome and the impact of supplementation on symptoms: What do we know and what do we need to know? Eur. J. Clin. Nutr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Ma, Y.Y.; Zhu, J.; Zuo, S.; Zhang, J.L.; Chen, Z.Y.; Chen, G.W.; Wang, X.; Pan, Y.S.; Liu, Y.C.; et al. Protective effect of 1,25-dihydroxyvitamin D3 on ethanol-induced intestinal barrier injury both in vitro and in vivo. Toxicol. Lett. 2015, 237, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Assa, A.; Vong, L.; Pinnell, L.J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D deficiency promotes epithelial barrier dysfunction and intestinal inflammation. J. Infect. Dis. 2014, 210, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Stio, M.; Retico, L.; Annese, V.; Bonanomi, A.G. Vitamin D regulates the tight-junction protein expression in active ulcerative colitis. Scand. J. Gastroenterol. 2016, 51, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; McDaniel, K.; Bora, S.; Chen, J.; James, J. Vitamin D, immune regulation, the microbiota, and inflammatory bowel disease. Exp. Biol. Med. (Maywood) 2014, 239, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Bashir, M.; Prietl, B.; Tauschmann, M.; Mautner, S.I.; Kump, P.K.; Treiber, G.; Wurm, P.; Gorkiewicz, G.; Högenauer, C.; Pieber, T.R. Effects of high doses of vitamin D3 on mucosa-associated gut microbiome vary between regions of the human gastrointestinal tract. Eur. J. Nutr. 2016, 55, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Sun, J. Vitamin D/VDR, Probiotics, and Gastrointestinal Diseases. Curr. Med. Chem. 2017, 24, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thingholm, L.B.; Skiecevičienė, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide host-microbiota association analysis of 1812 individuals identifies vitamin D receptor genetic variation and other host factors shaping the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Floch, M.H. The Role of Prebiotics and Probiotics in Gastrointestinal Disease. Gastroenterol. Clin. N. Am. 2018, 47, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.C.; Chang, C.H.; Su, C.H.; Yu, B.; Hsu, Y.M. Pteris. multifida, Cortex phellodendri, and probiotics attenuated inflammatory status and immunity in mice with a Salmonella enterica serovar Typhimurium infection. Biosci. Biotechnol. Biochem. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Oral supplementation with probiotic, L. reuteri NCIMB 30242 increases mean circulating 25-hydroxyvitamin D: A post hoc analysis of a randomized controlled trial. J. Clin. Endocrinol. Metab. 2013, 98, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yoon, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Wan, J.; Petrof, E.O.; Claud, E.C.; Chen, D.; Sun, J. Vitamin D receptor pathway is required for probiotic protection in colitis. Am. J. Physiol. Gastroint. Liver Physiol. 2015, 309, G341–G349. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caccamo, D.; Ricca, S.; Currò, M.; Ientile, R. Health Risks of Hypovitaminosis D: A Review of New Molecular Insights. Int. J. Mol. Sci. 2018, 19, 892. https://doi.org/10.3390/ijms19030892
Caccamo D, Ricca S, Currò M, Ientile R. Health Risks of Hypovitaminosis D: A Review of New Molecular Insights. International Journal of Molecular Sciences. 2018; 19(3):892. https://doi.org/10.3390/ijms19030892
Chicago/Turabian StyleCaccamo, Daniela, Sergio Ricca, Monica Currò, and Riccardo Ientile. 2018. "Health Risks of Hypovitaminosis D: A Review of New Molecular Insights" International Journal of Molecular Sciences 19, no. 3: 892. https://doi.org/10.3390/ijms19030892
APA StyleCaccamo, D., Ricca, S., Currò, M., & Ientile, R. (2018). Health Risks of Hypovitaminosis D: A Review of New Molecular Insights. International Journal of Molecular Sciences, 19(3), 892. https://doi.org/10.3390/ijms19030892