Refining the Results of a Classical SELEX Experiment by Expanding the Sequence Data Set of an Aptamer Pool Selected for Protein A



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
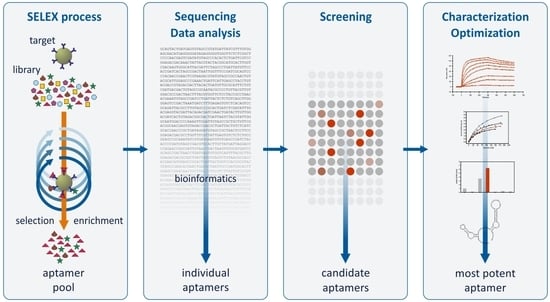
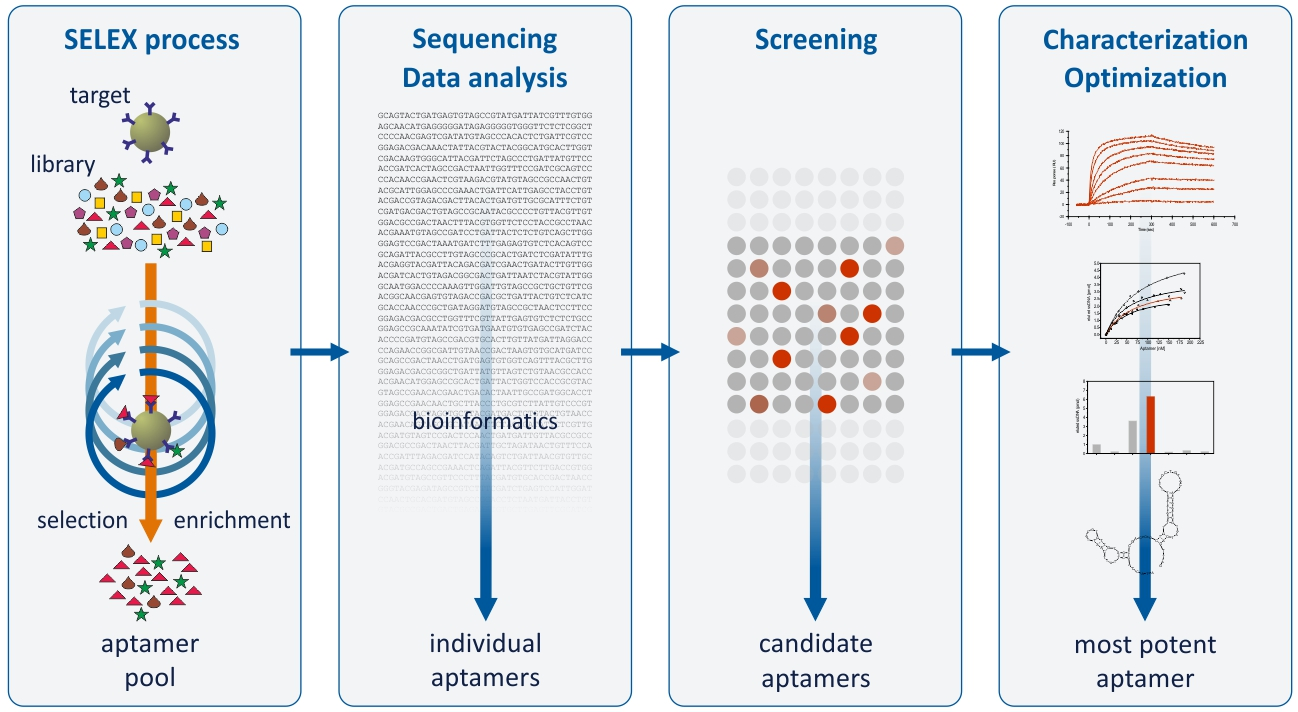
1. Introduction
2. Results and Discussion
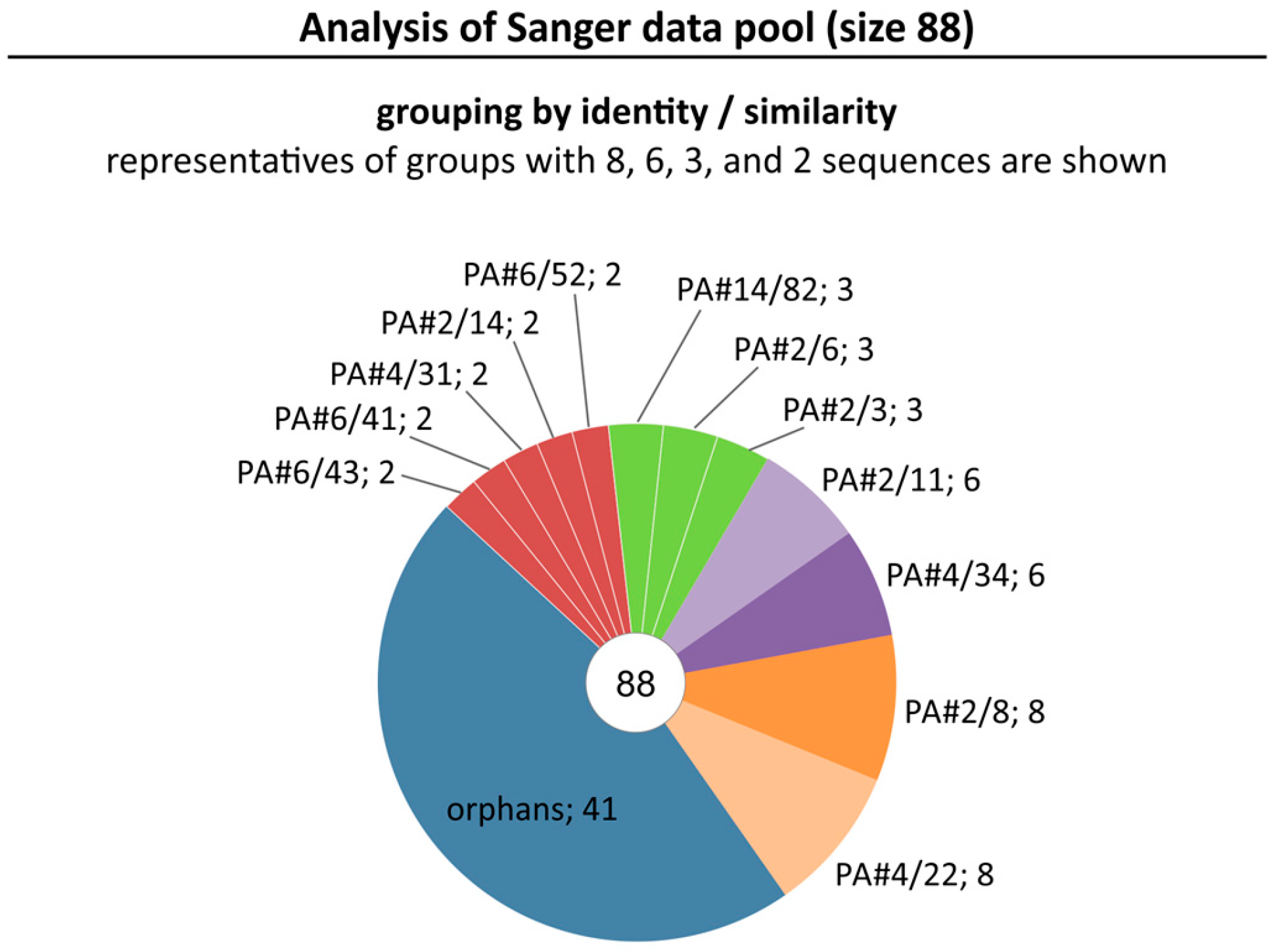
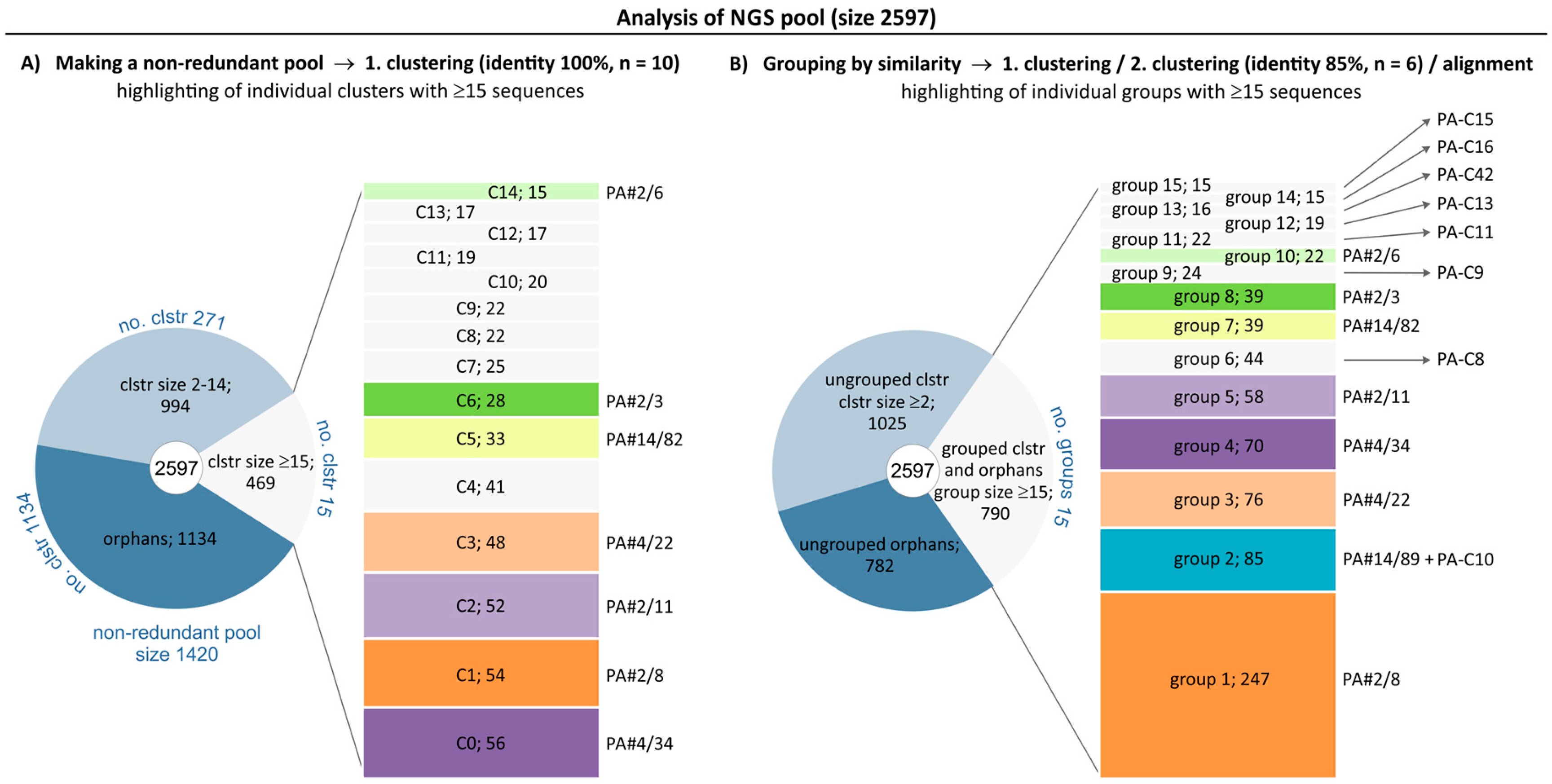
2.1. Broadening the Sequence Data Set of the Aptamer Pool Selected for Protein A
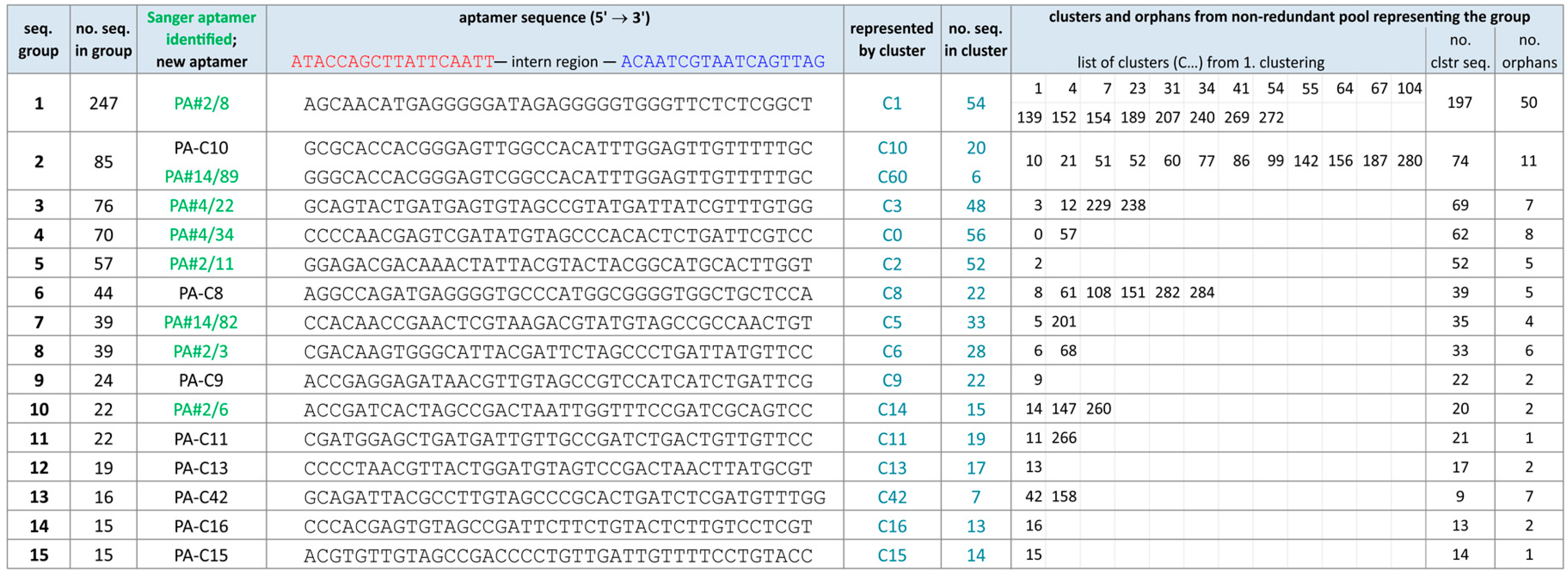
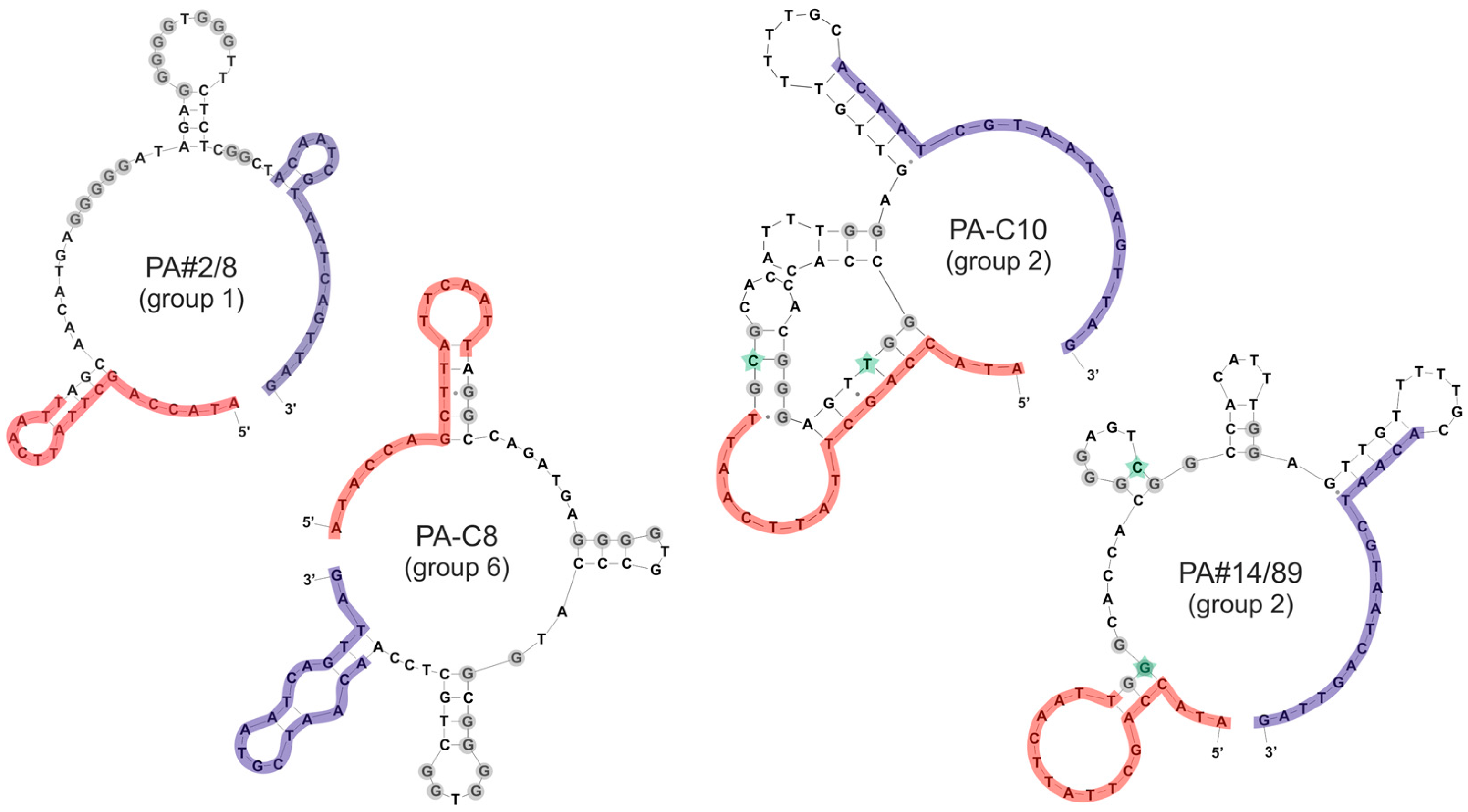
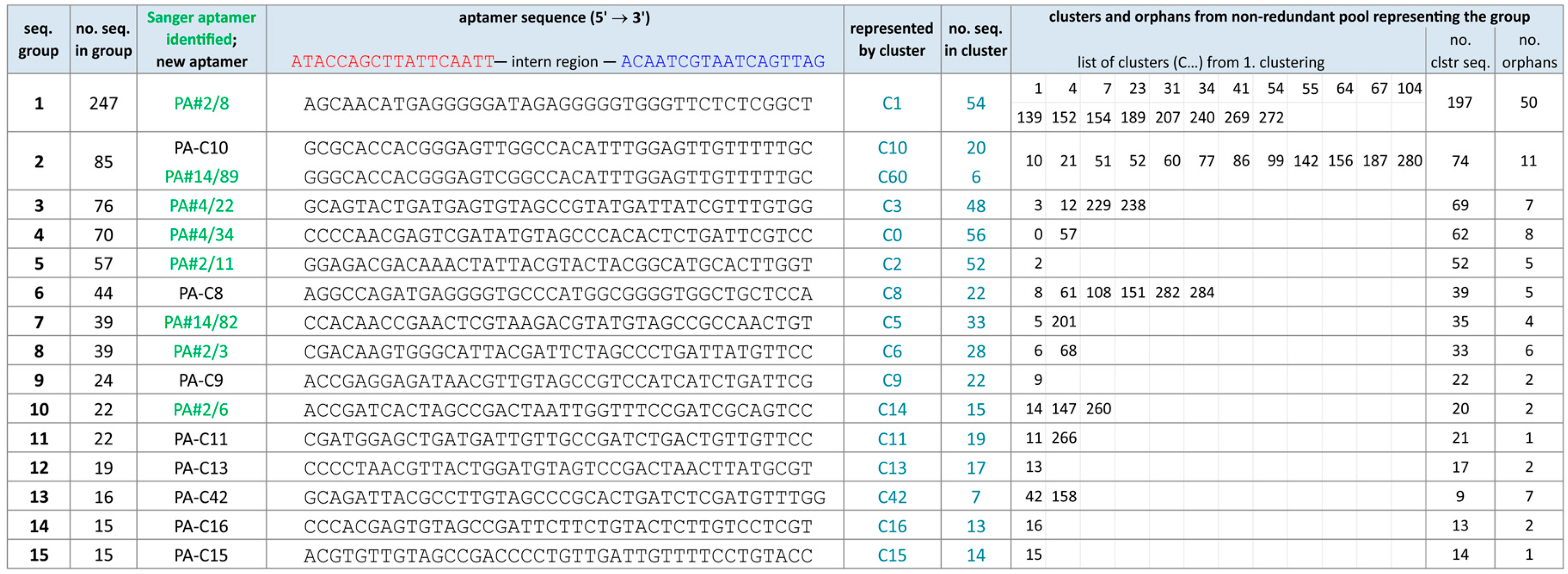
2.2. Group Complexities and Consensus Sequences
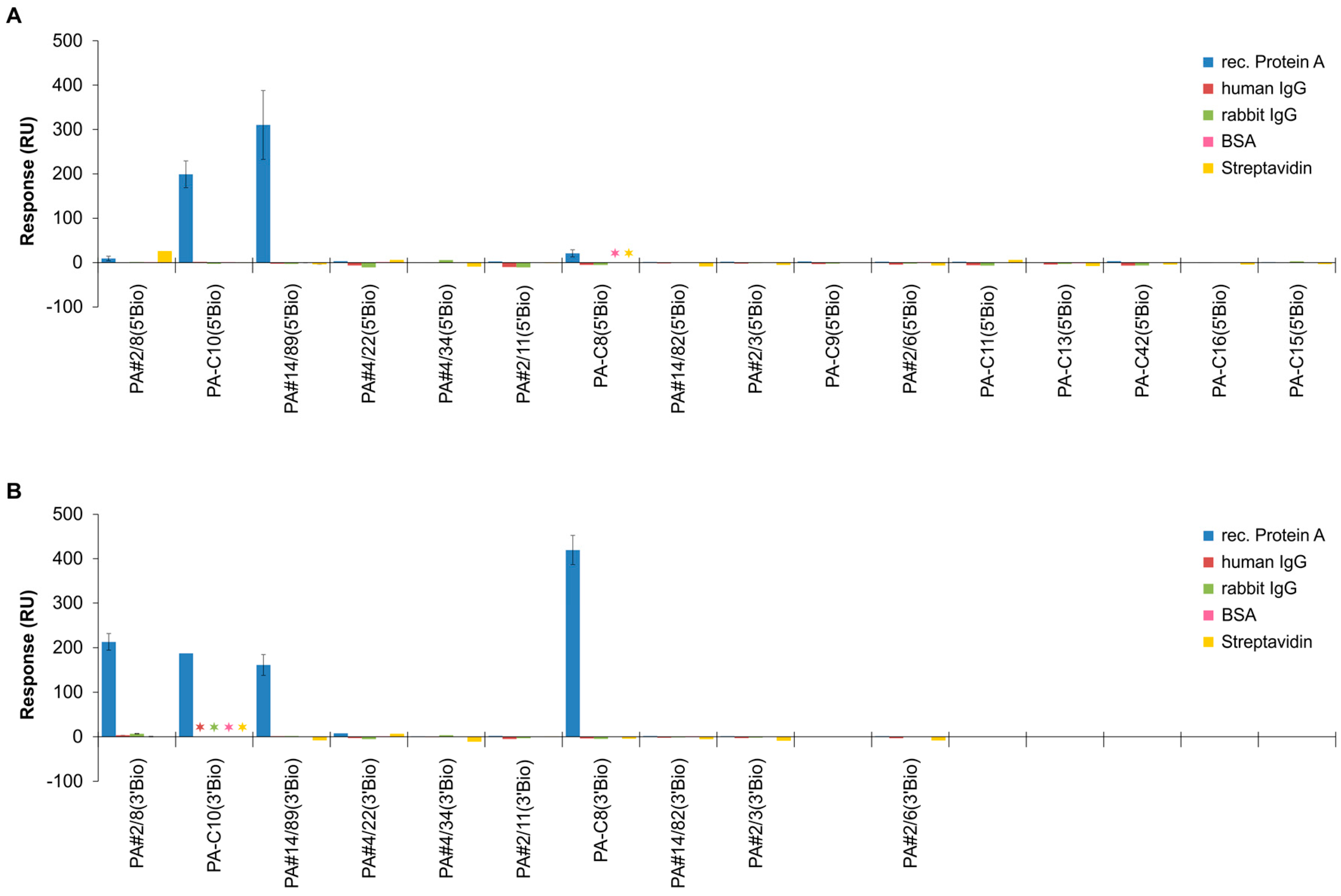
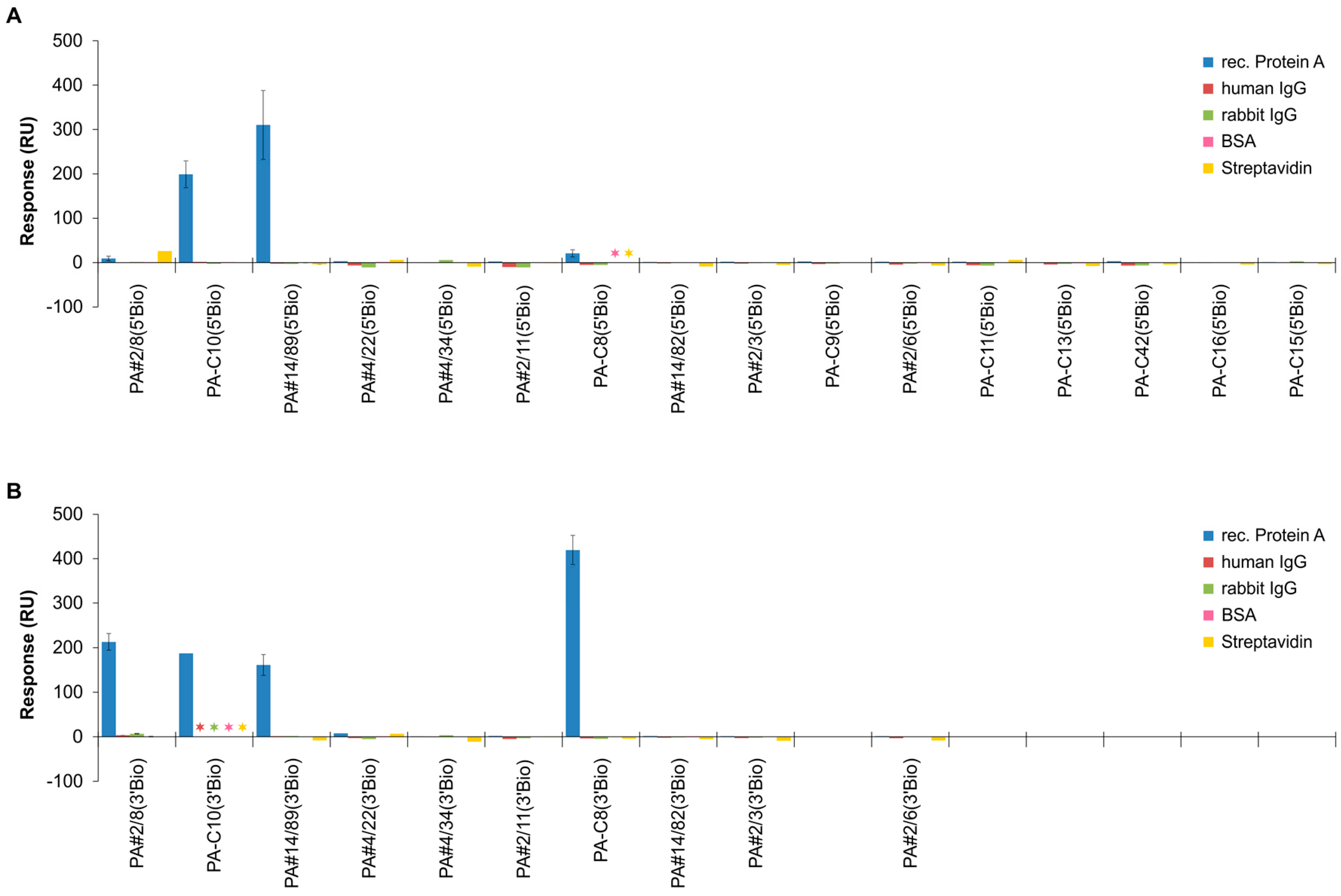
2.3. Functional Screening of Identified Aptamer Groups
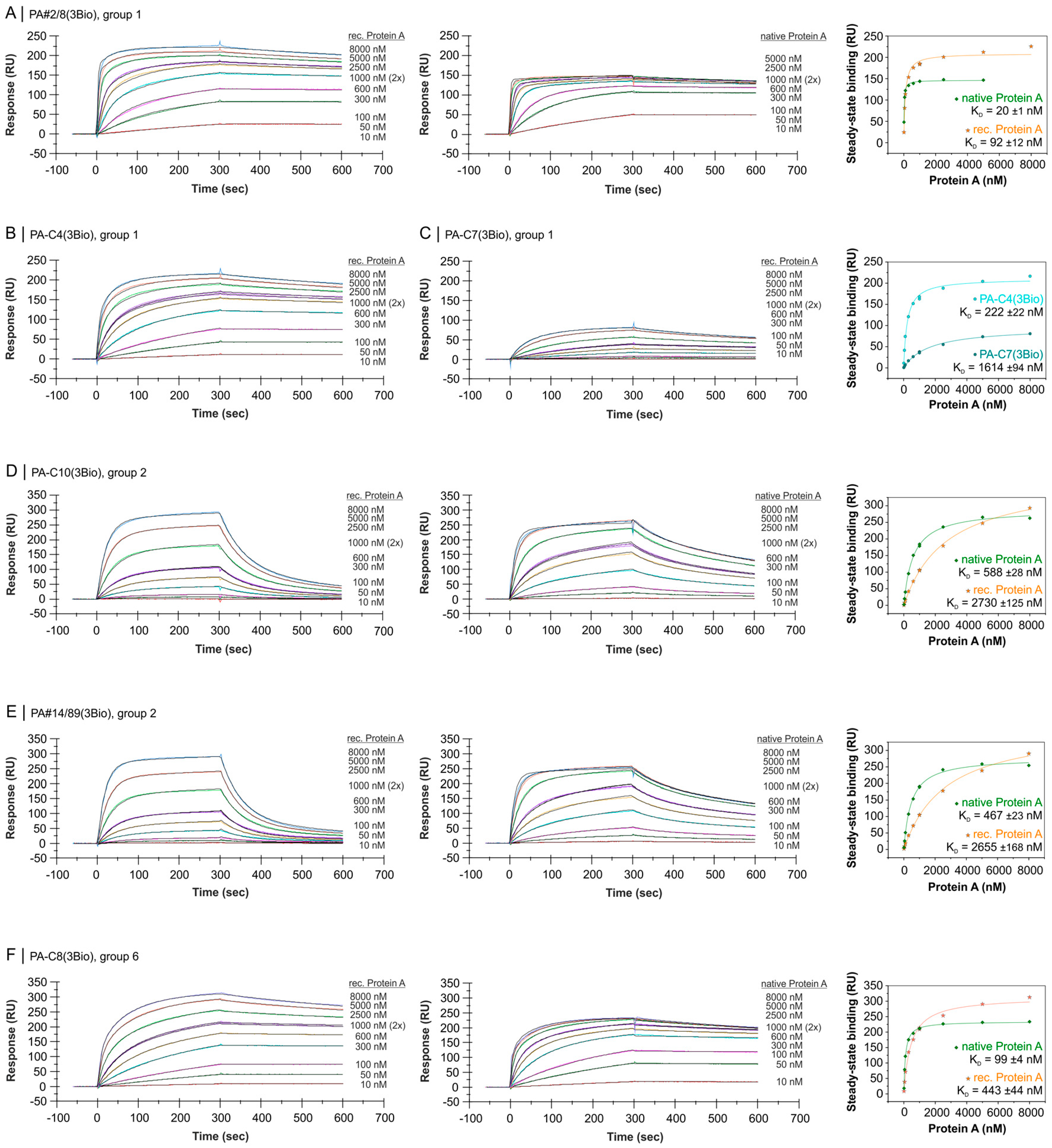
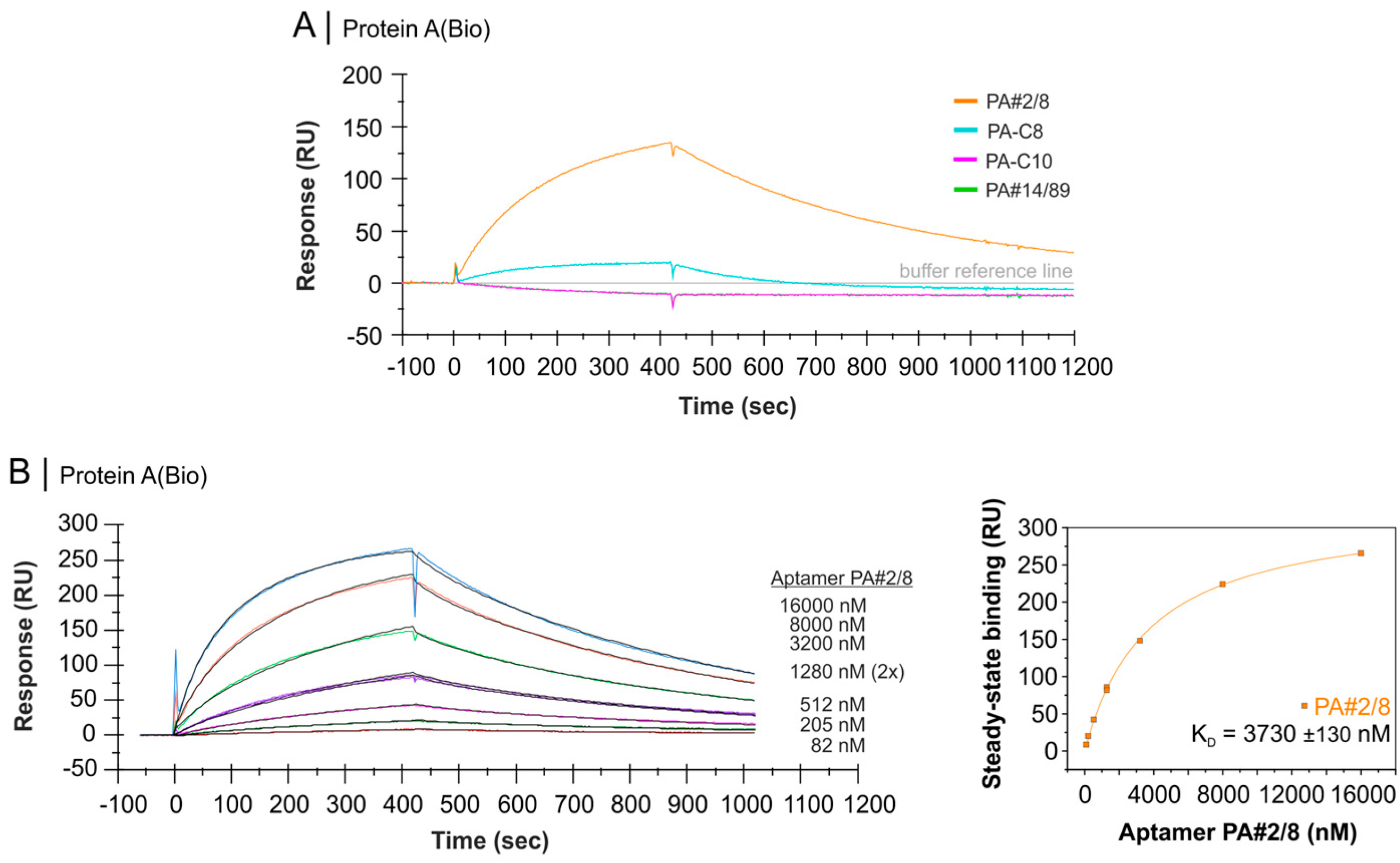
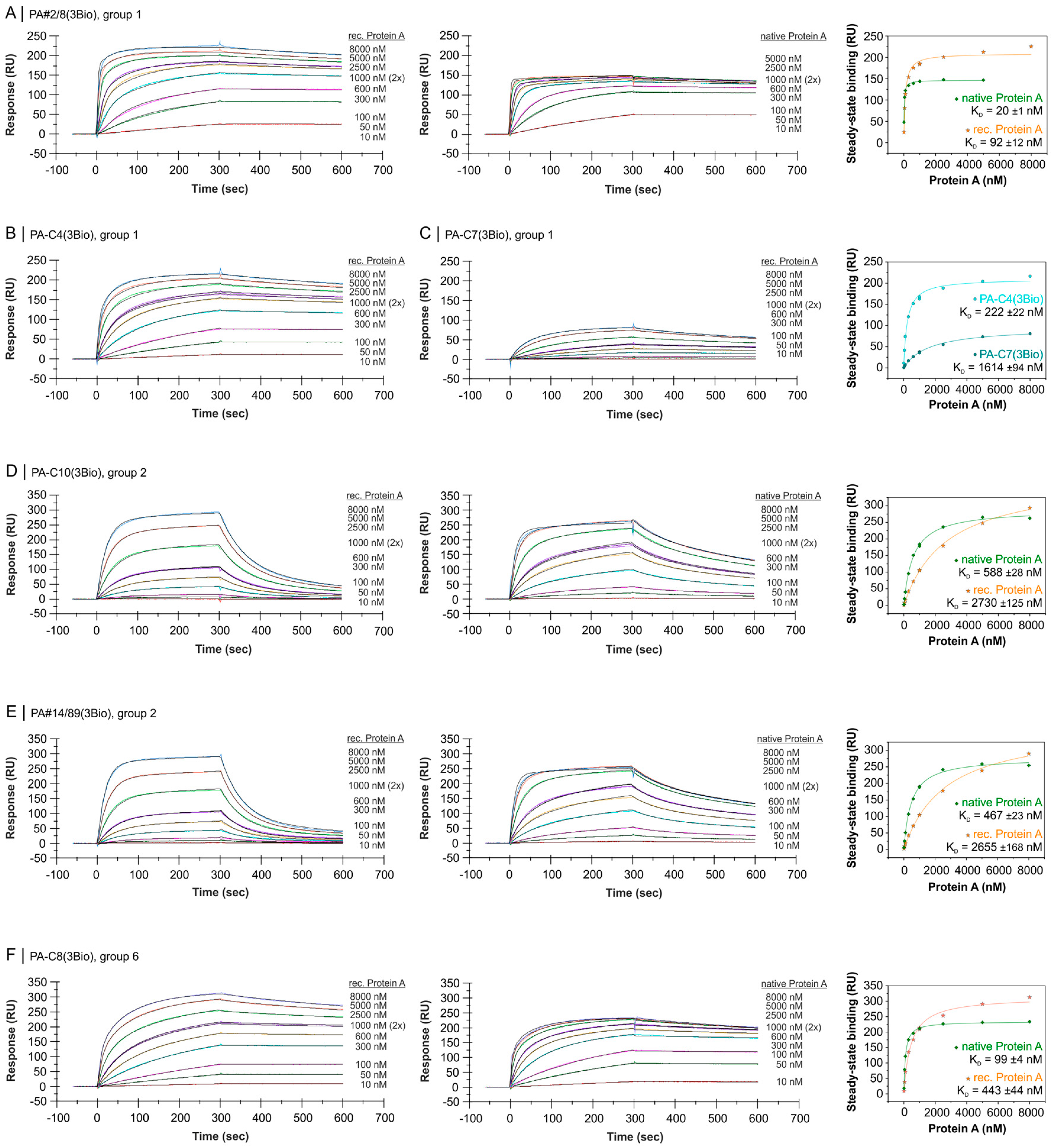
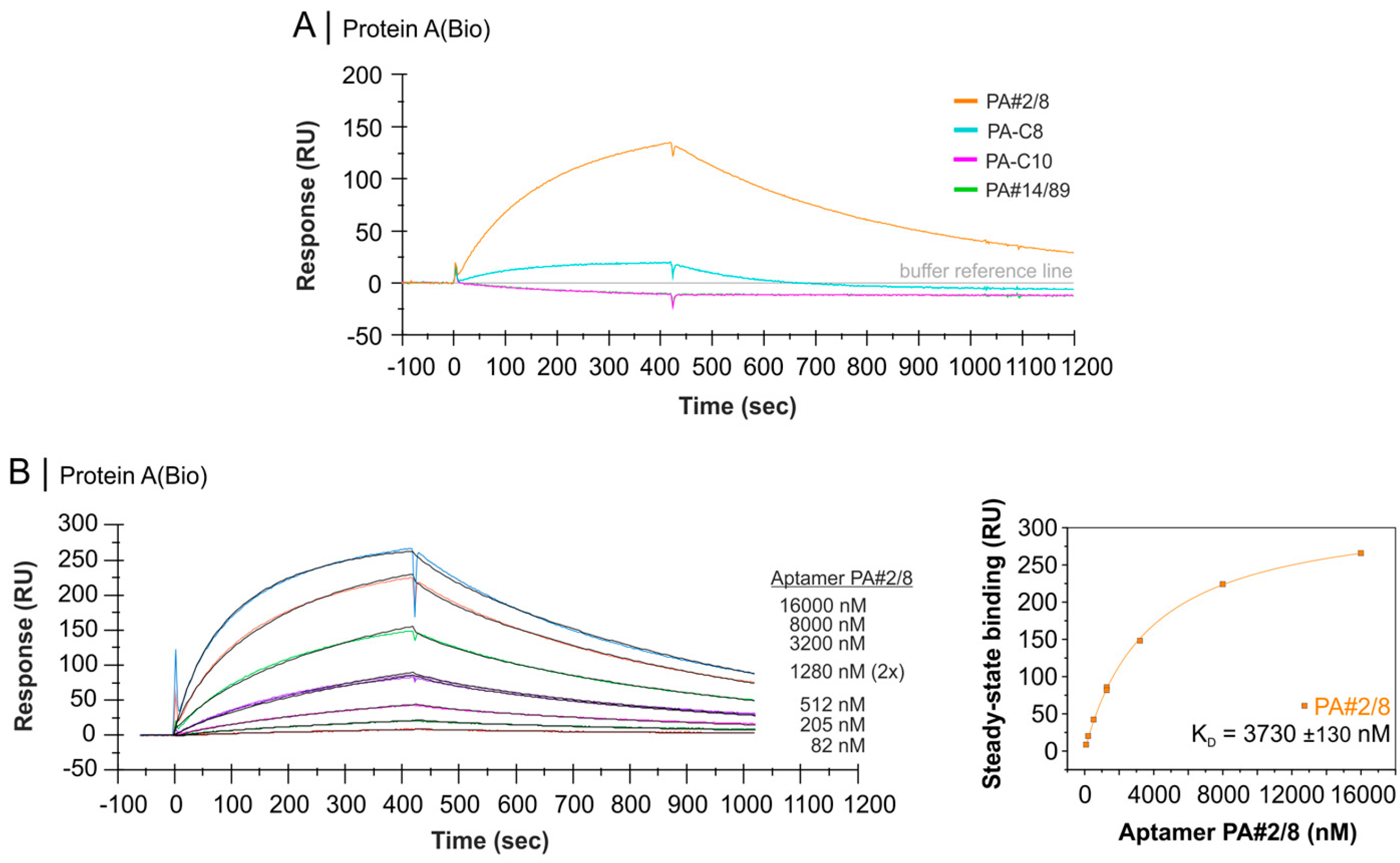
2.4. Comparative Affinity Studies of Protein A-Targeting Aptamers
3. Materials and Methods
3.1. Materials
3.2. Next Generation Sequencing (Roche 454 GS FLX System)
3.3. Bioinformatics Analysis
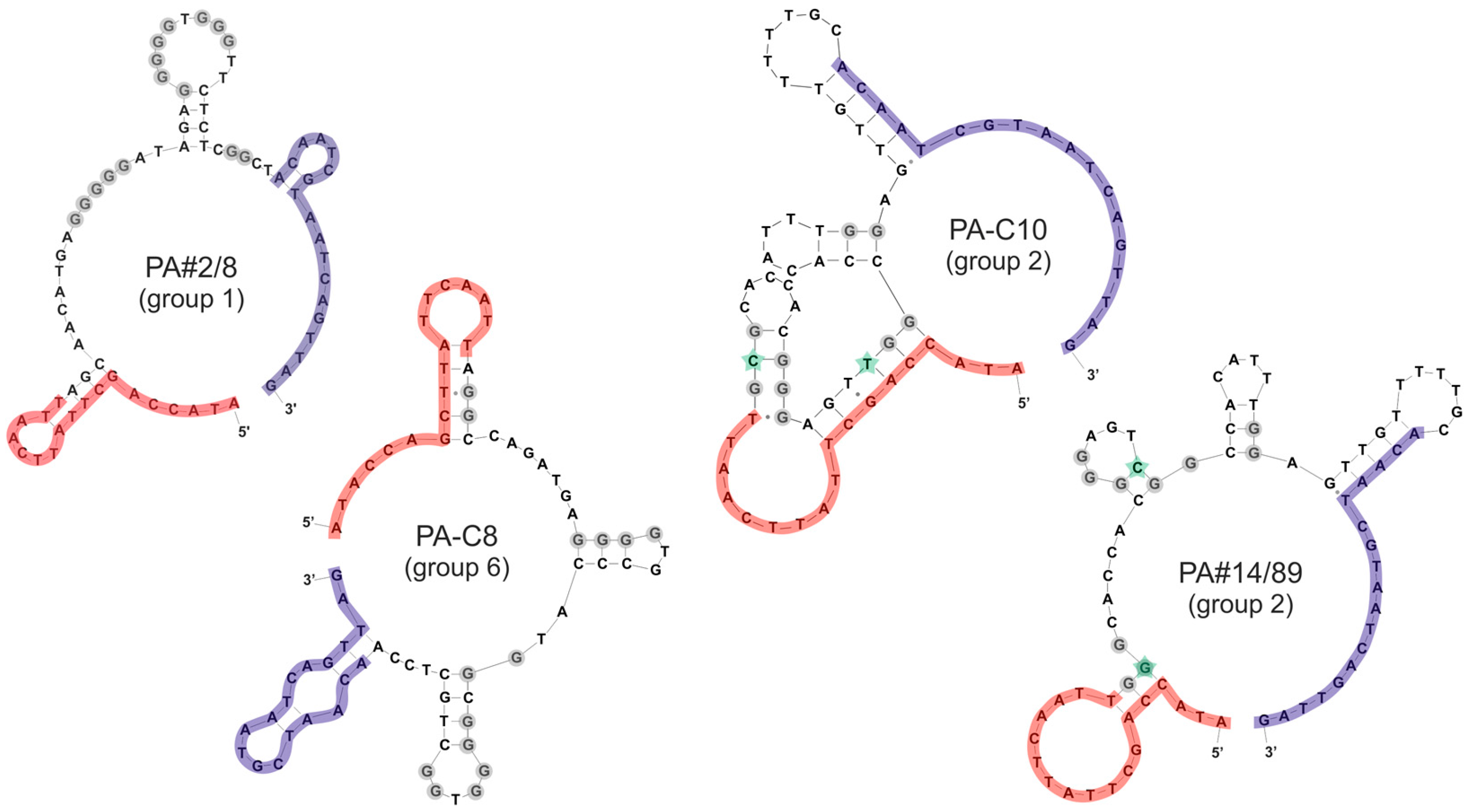
3.4. Secondary Structure Prediction
3.5. SPR-Based Analyses (Biacore X100)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BSA | Bovine Serum Albumin |
| CMA | Conserved Motif Accumulation |
| DNA | Deoxyribonucleic Acid |
| ELONA | Enzyme-linked Oligonucleotide Assay |
| HSA | Human Serum Albumin |
| IgG | Immunoglobulin |
| KD | Equilibrium Dissociation Constant |
| LFA | Lateral Flow Assay |
| NGS | Next Generation Sequencing |
| nt | Nucleotide |
| PCR | Polymerase Chain Reaction |
| RNA | Ribonucleic Acid |
| RT | Room Temperature |
| RU | Resonance Units |
| SELEX | Systematic Evolution of Ligands by Exponential Enrichment |
| S. aureus | Staphylococcus aureus |
| SPR | Surface Plasmon Resonance Spectroscopy |
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, A.; Kalra, P.; Bansal, V.; Bruno, J.G.; Sharma, T.K. Aptamer-based point-of-care diagnostic platforms. Sens. Actuators B Chem. 2017, 246, 535–553. [Google Scholar] [CrossRef]
- Zhang, H.M.; Zhou, L.J.; Zhu, Z.; Yang, C.Y. Recent Progress in Aptamer-Based Functional Probes for Bioanalysis and Biomedicine. Chem. Eur. J. 2016, 22, 9886–9900. [Google Scholar] [CrossRef] [PubMed]
- Ilgu, M.; Nilsen-Hamilton, M. Aptamers in analytics. Analyst 2016, 141, 1551–1568. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.B.; Gu, M.B. Aptamer-based sandwich-type biosensors. J. Biol. Eng. 2017, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Radom, F.; Jurek, P.M.; Mazurek, M.P.; Otlewski, J.; Jeleń, F. Aptamers: Molecules of great potential. Biotechnol. Adv. 2013, 31, 1260–1274. [Google Scholar] [CrossRef] [PubMed]
- Darmostuk, M.; Rimpelova, S.; Gbelcova, H.; Ruml, T. Current approaches in SELEX: An update to aptamer selection technology. Biotechnol. Adv. 2015, 33, 1141–1161. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX-A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Szeitner, Z.; András, J.; Gyurcsányi, R.E.; Mészáros, T. Is less more? Lessons from aptamer selection strategies. J. Pharm. Biomed. Anal. 2014, 101, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozer, A.; Pagano, J.M.; Lis, J.T. New Technologies Provide Quantum Changes in the Scale, Speed, and Success of SELEX Methods and Aptamer Characterization. Mol. Ther. Nucleic Acids 2014, 3, e183. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Z.; Yu, Y.; Wang, M.; Li, J.; Zhang, Z.; Liu, J.; Wu, X.; Lu, A.; Zhang, G.; Zhang, B. Recent Advances in SELEX Technology and Aptamer Applications in Biomedicine. Int. J. Mol. Sci. 2017, 18, 2142. [Google Scholar] [CrossRef] [PubMed]
- Blind, M.; Blank, M. Aptamer Selection Technology and Recent Advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, S.K.; Bowser, M.T. Microfluidic methods for aptamer selection and characterization. Analyst 2018, 143, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zu, Y. A Highlight of Recent Advances in Aptamer Technology and Its Application. Molecules 2015, 20, 11959–11980. [Google Scholar] [CrossRef] [PubMed]
- Jijakli, K.; Khraiwesh, B.; Fu, W.Q.; Luo, L.M.; Alzahmi, A.; Koussa, J.; Chaiboonchoe, A.; Kirmizialtin, S.; Yen, L.S.; Salehi-Ashtiani, K. The in vitro selection world. Methods 2016, 106, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Quang, N.N.; Perret, G.; Ducongé, F. Applications of High-Throughput Sequencing for In Vitro Selection and Characterization of Aptamers. Pharmaceuticals 2016, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX Process with Next-Generation Sequencing. PLoS ONE 2011, 6, e29604. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, A.; Fraser, L.; Liang, S.; Shiu, S.; Tanner, J. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2516. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.X.; Zheng, X.; Jiao, B.H.; Wang, L.H. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016, 408, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Stobberingh, E.E. The evolution of Staphylococcus aureus. Infect. Genet. Evolut. 2008, 8, 747–763. [Google Scholar] [CrossRef] [PubMed]
- Stryjewski, M.E.; Corey, G.R. Methicillin-Resistant Staphylococcus aureus: An Evolving Pathogen. Clin. Infect. Dis. 2014, 58, S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Community-associated MRSA: What makes them special? Int. J. Med. Microbiol. 2013, 303, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Kadariya, J.; Smith, T.C.; Thapaliya, D. Staphylococcus aureus and Staphylococcal Food-Borne Disease: An Ongoing Challenge in Public Health. BioMed Res. Int. 2014, 2014, 827965. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wei, C.; Zhong, J.; Jin, S. Research advance in rapid detection of foodborne Staphylococcus aureus. Biotechnol. Biotechnol. Equip. 2016, 30, 827–833. [Google Scholar] [CrossRef]
- Law, J.W.; Ab Mutalib, N.S.; Chan, K.G.; Lee, L.H. Rapid methods for the detection of foodborne bacterial pathogens: Principles, applications, advantages and limitations. Front. Microbiol. 2014, 5, 770. [Google Scholar] [CrossRef] [PubMed]
- Shahdordizadeh, M.; Taghdisi, S.M.; Ansari, N.; Alebooye Langroodi, F.; Abnous, K.; Ramezani, M. Aptamer based biosensors for detection of Staphylococcus aureus. Sens. Actuators B Chem. 2017, 241, 619–635. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Schubert, T.; Strehlitz, B. In Vitro Selection and Interaction Studies of a DNA Aptamer Targeting Protein A. PLoS ONE 2015, 10, e0134403. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Krafčiková, P.; Víglaský, V.; Strehlitz, B. G-quadruplex aptamer targeting Protein A and its capability to detect Staphylococcus aureus demonstrated by ELONA. Sci. Rep. 2016, 6, 33812. [Google Scholar] [CrossRef] [PubMed]
- Reich, P.; Stoltenburg, R.; Strehlitz, B.; Frense, D.; Beckmann, D. Development of An Impedimetric Aptasensor for the Detection of Staphylococcus aureus. Int. J. Mol. Sci. 2017, 18, 2484. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Huber, J.A.; Morrison, H.G.; Sogin, M.L.; Welch, D.M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007, 8, R143. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.; Baillie, G.; Watson, S.J.; Kellam, P.; Rambaut, A.; Robertson, D.L. Analysis of high-depth sequence data for studying viral diversity: A comparison of next generation sequencing platforms using Segminator II. BMC Bioinform. 2012, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, S.; Obubuafo, A.; McCarley, R.L.; Soper, S.A.; Spivak, D.A. Effect of Linker Structure on Surface Density of Aptamer Monolayers and Their Corresponding Protein Binding Efficiency. Anal. Chem. 2008, 80, 9630–9634. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.R.; Liu, S.; Yu, G.; Libby, K.; Cubicciotti, R.; Colyer, C.L. Using capillary electrophoresis to characterize polymeric particles. J. Chromatogr. 2016, 1463, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.-G.; Kökpinar, Ö.; Friehs, K.; Stahl, F.; Scheper, T. Systematic Investigation of Optimal Aptamer Immobilization for Protein−Microarray Applications. Anal. Chem. 2008, 80, 7372–7378. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Stewart, C.A.; McNamara, J.O., II; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378. [Google Scholar] [CrossRef] [PubMed]
- Hoon, S.; Zhou, B.; Janda, K.D.; Brenner, S.; Scolnick, J. Aptamer selection by high-throughput sequencing and informatic analysis. BioTechniques 2011, 51, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Durney, B.C.; Crihfield, C.L.; Holland, L.A. Capillary electrophoresis applied to DNA: Determining and harnessing sequence and structure to advance bioanalyses (2009–2014). Anal. Bioanal. Chem. 2015, 407, 6923–6938. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.R.; Gagliano, J.; Xiao, J.; Libby, K.; Saito, S.; Yu, G.; Cubicciotti, R.; Macosko, J.; Colyer, C.L.; Guthold, M.; et al. Combining capillary electrophoresis and next-generation sequencing for aptamer selection. Anal. Bioanal. Chem. 2015, 407, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Stuart, C.H.; Riley, K.R.; Boyacioglu, O.; Herpai, D.M.; Debinski, W.; Qasem, S.; Marini, F.C.; Colyer, C.L.; Gmeiner, W.H. Selection of a Novel Aptamer Against Vitronectin Using Capillary Electrophoresis and Next Generation Sequencing. Mol. Ther. Nucleic Acids 2016, 5, e386. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, S.; de Girolamo, A.; DeRosa, M.C.; McKeague, M.; Schena, R.; Catucci, L.; Pascale, M. Screening and Identification of DNA Aptamers to Tyramine Using In Vitro Selection and High-Throughput Sequencing. ACS Comb. Sci. 2016, 18, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Hervas, S.; Villanueva, H.; Lozano, T.; Rabal, O.; Oyarzabal, J.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; López-Díaz de Cerio, A.; et al. Identification of LAG3 high affinity aptamers by HT-SELEX and Conserved Motif Accumulation (CMA). PLoS ONE 2017, 12, e0185169. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; de Girolamo, A.; Valenzano, S.; Pascale, M.; Ruscito, A.; Velu, R.; Frost, N.R.; Hill, K.; Smith, M.; McConnell, E.M.; et al. Comprehensive Analytical Comparison of Strategies Used for Small Molecule Aptamer Evaluation. Anal. Chem. 2015, 87, 8608–8612. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Niu, B.; Fu, L.; Sun, S.; Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinform. 2010, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Peyret, N. Prediction of Nucleic Acid Hybridization: Parameters and Algorithms. Ph.D. Thesis, Department of Chemistry, Wayne State University, Detroit, MI, USA, 2000. [Google Scholar]
- SantaLucia, J., Jr. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoltenburg, R.; Strehlitz, B. Refining the Results of a Classical SELEX Experiment by Expanding the Sequence Data Set of an Aptamer Pool Selected for Protein A. Int. J. Mol. Sci. 2018, 19, 642. https://doi.org/10.3390/ijms19020642
Stoltenburg R, Strehlitz B. Refining the Results of a Classical SELEX Experiment by Expanding the Sequence Data Set of an Aptamer Pool Selected for Protein A. International Journal of Molecular Sciences. 2018; 19(2):642. https://doi.org/10.3390/ijms19020642
Chicago/Turabian StyleStoltenburg, Regina, and Beate Strehlitz. 2018. "Refining the Results of a Classical SELEX Experiment by Expanding the Sequence Data Set of an Aptamer Pool Selected for Protein A" International Journal of Molecular Sciences 19, no. 2: 642. https://doi.org/10.3390/ijms19020642
APA StyleStoltenburg, R., & Strehlitz, B. (2018). Refining the Results of a Classical SELEX Experiment by Expanding the Sequence Data Set of an Aptamer Pool Selected for Protein A. International Journal of Molecular Sciences, 19(2), 642. https://doi.org/10.3390/ijms19020642