Impact of Arginine to Cysteine Mutations in Collagen II on Protein Secretion and Cell Survival

 , ,
, ,
Abstract
1. Introduction
2. Results
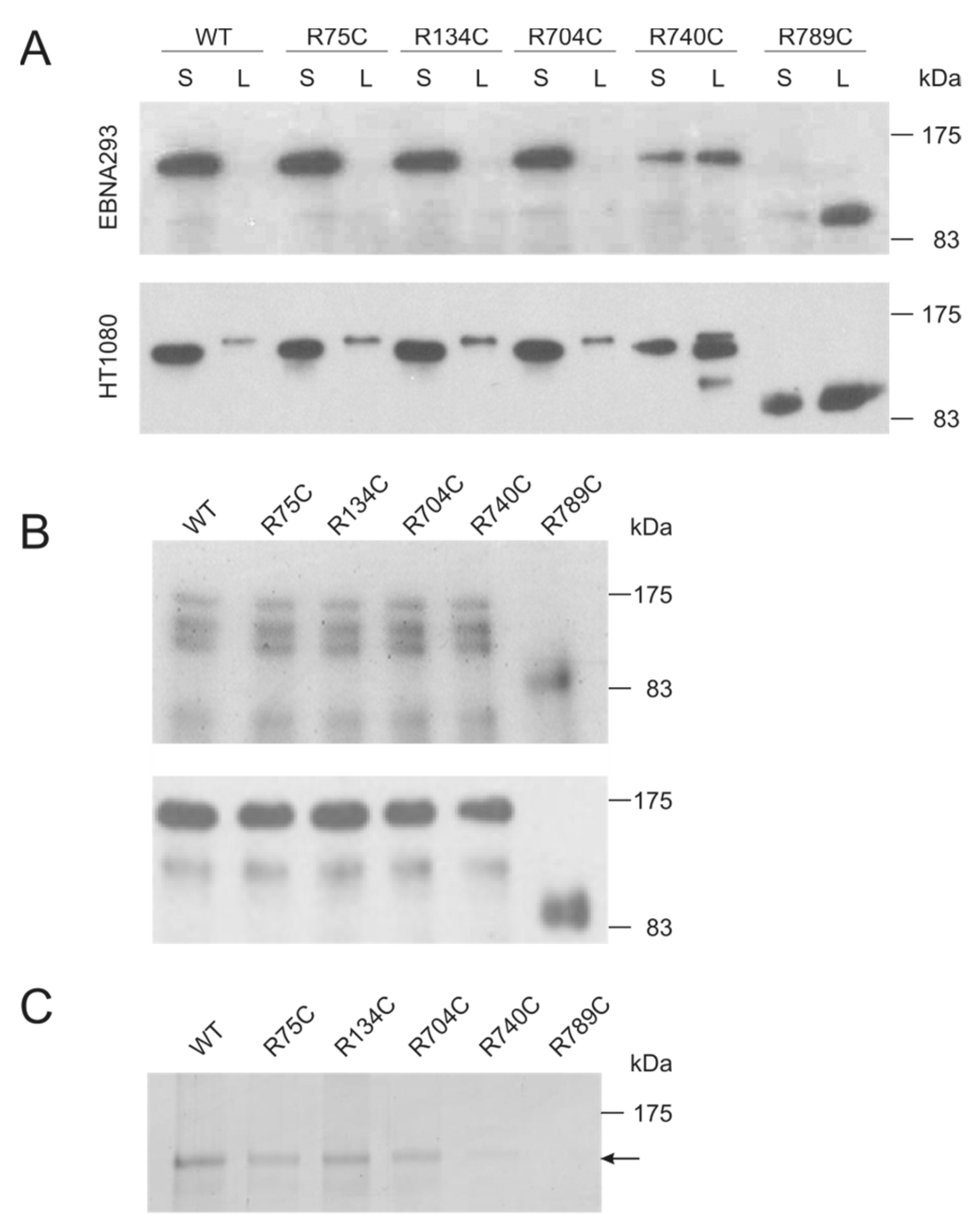
2.1. Collagen II Mutations towards the C Terminus Affect Secretion and Increase Susceptibility to Proteases
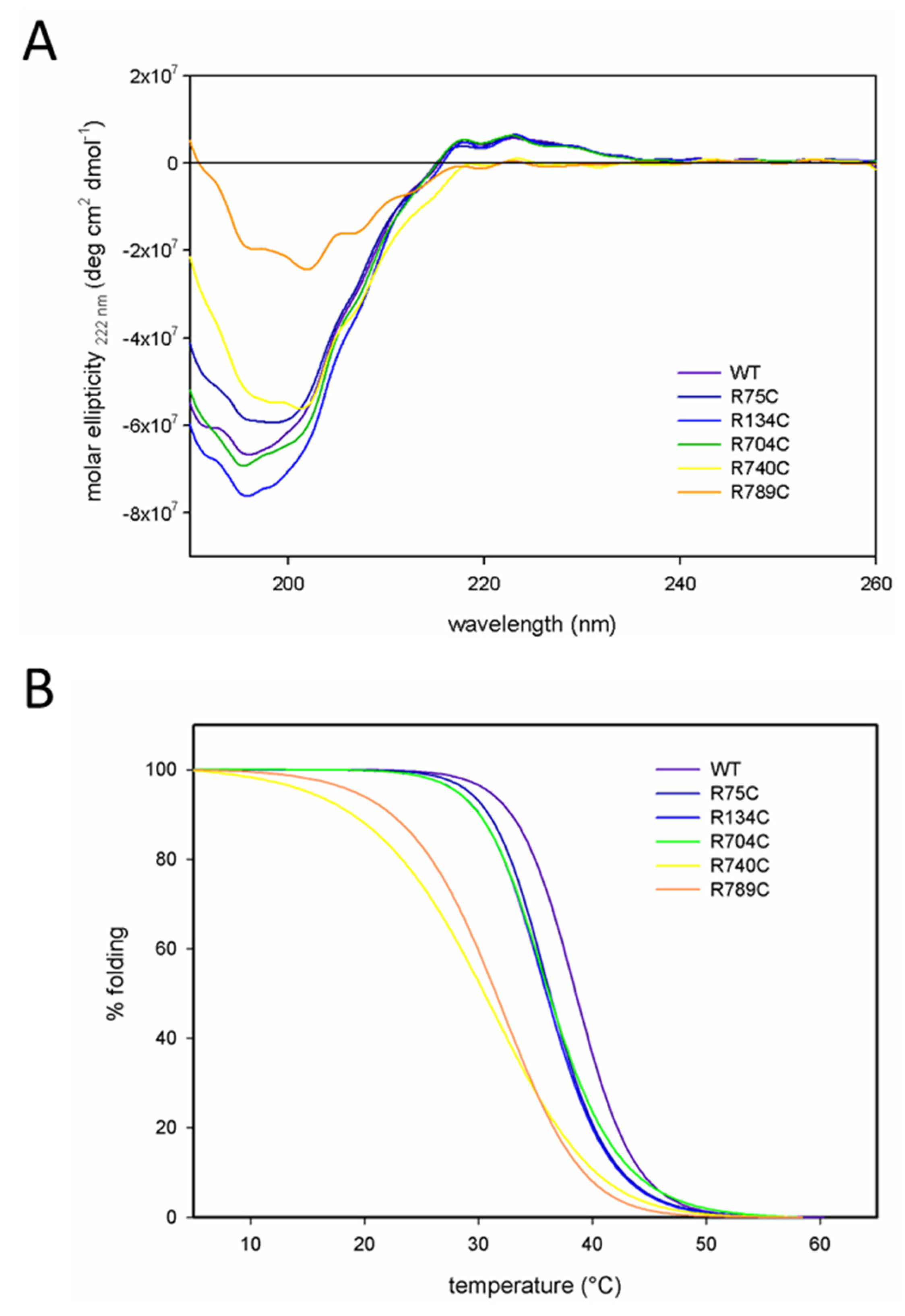
2.2. C-Terminal Mutations (R740C and R789C) Lead to Thermal Instability of the Triple Helix
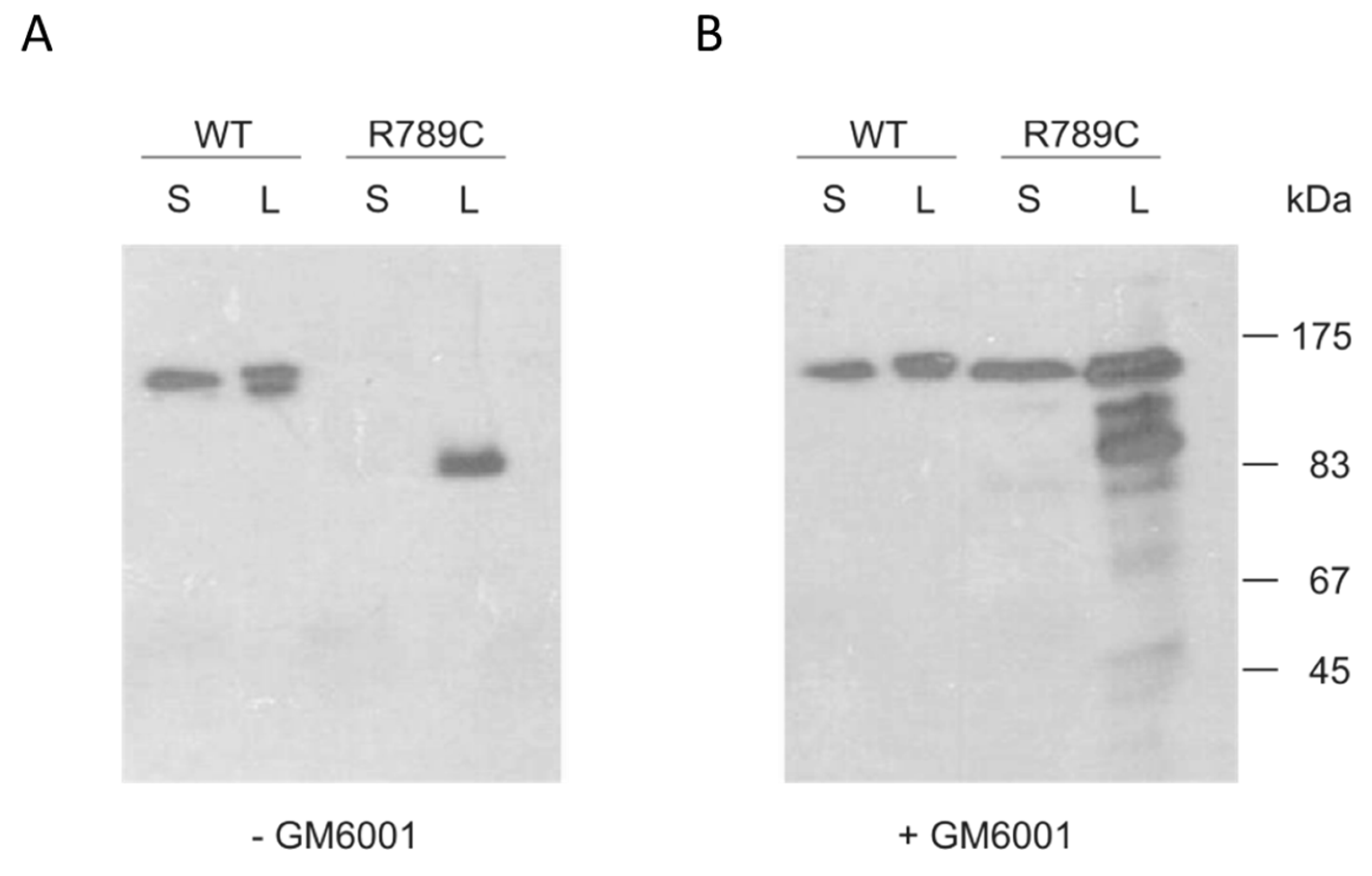
2.3. Inhibition of MMP Activity Prevents Cleavage of R789C
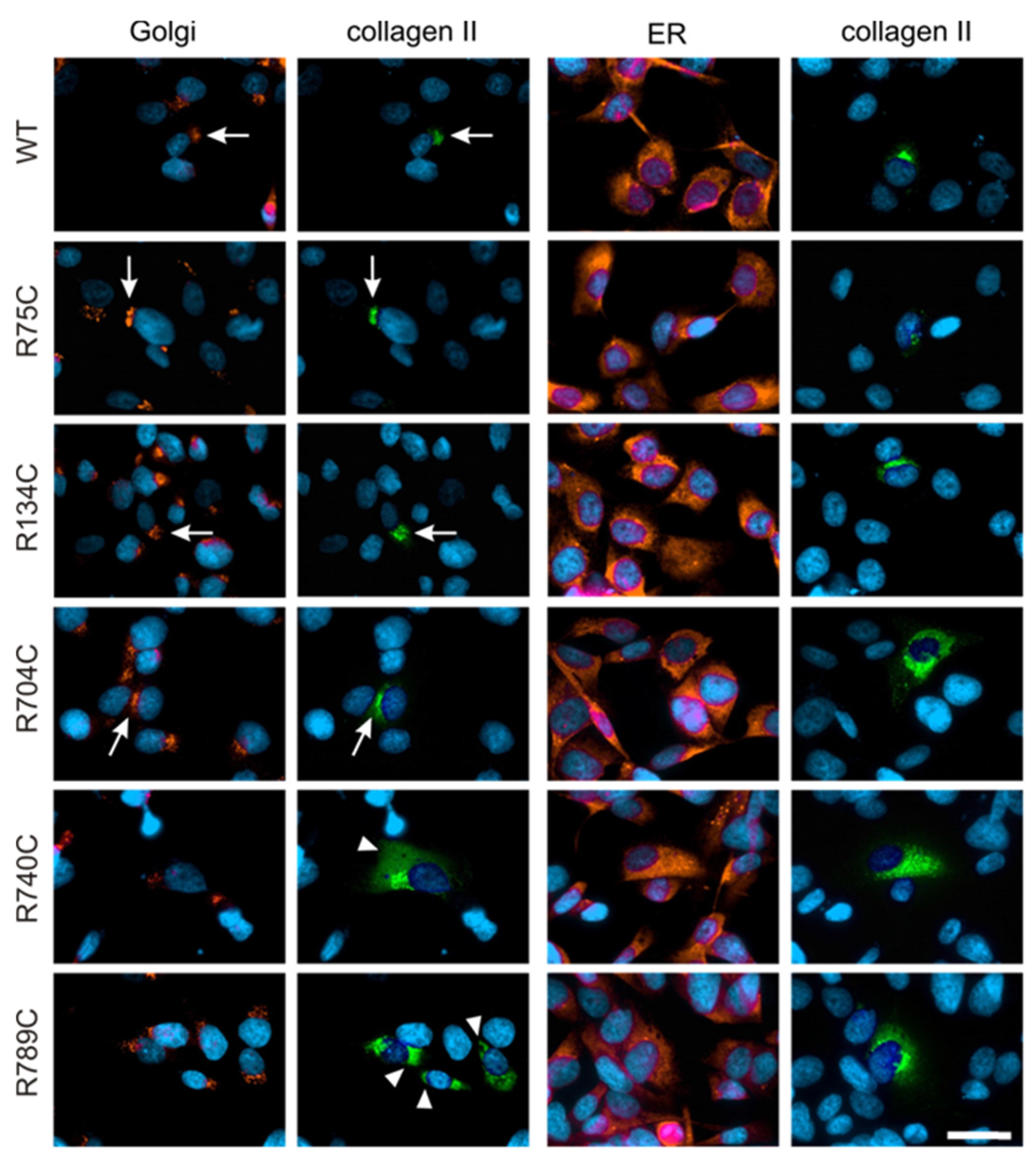
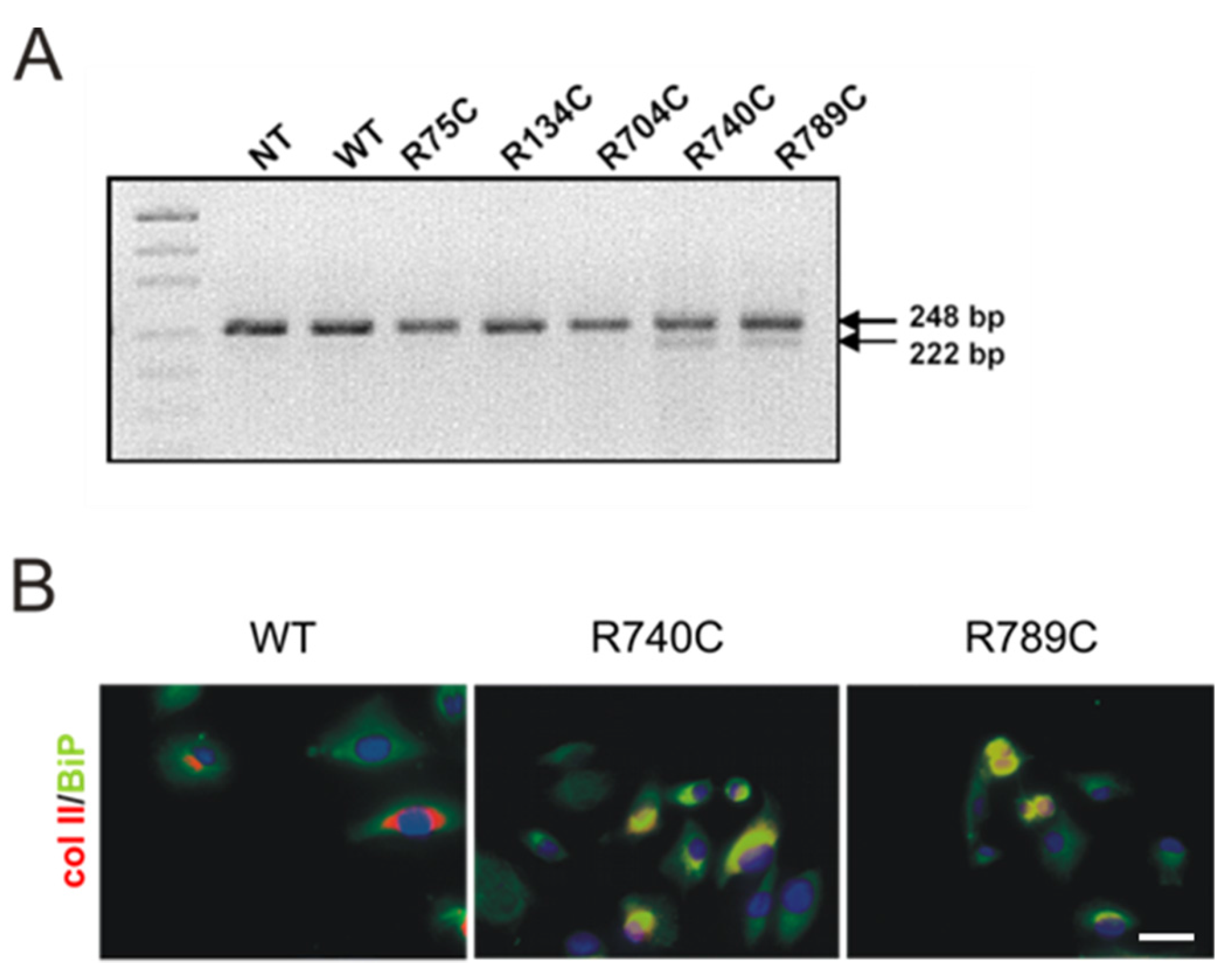
2.4. Effect of Mutations on Intracellular Collagen Trafficking in Transfected HT1080 Cells
2.5. C-Terminal Mutations Induce Activation of the Unfolded Protein Response Due to Accumulation of Misfolded Collagen II Proteins
Expression and Association of Chaperones with Mutant Collagen II Proteins
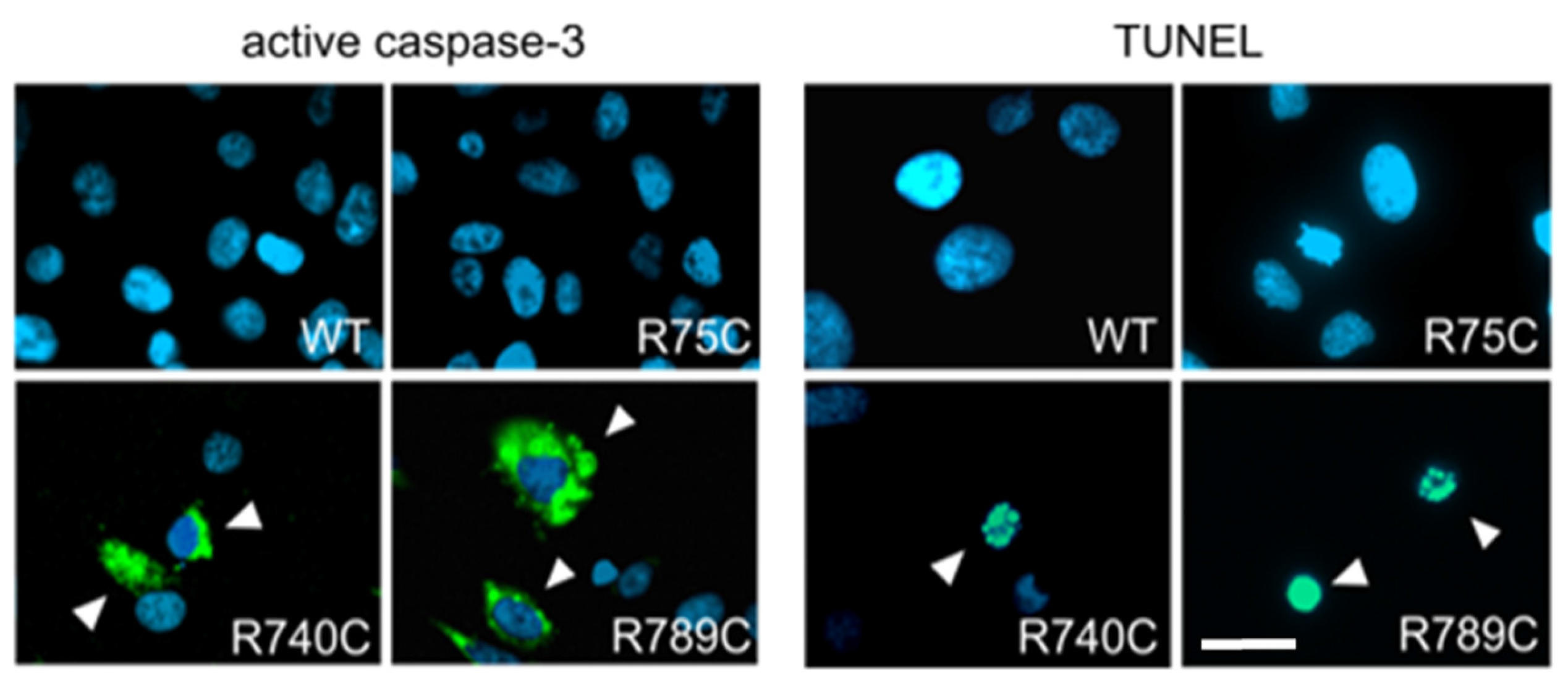
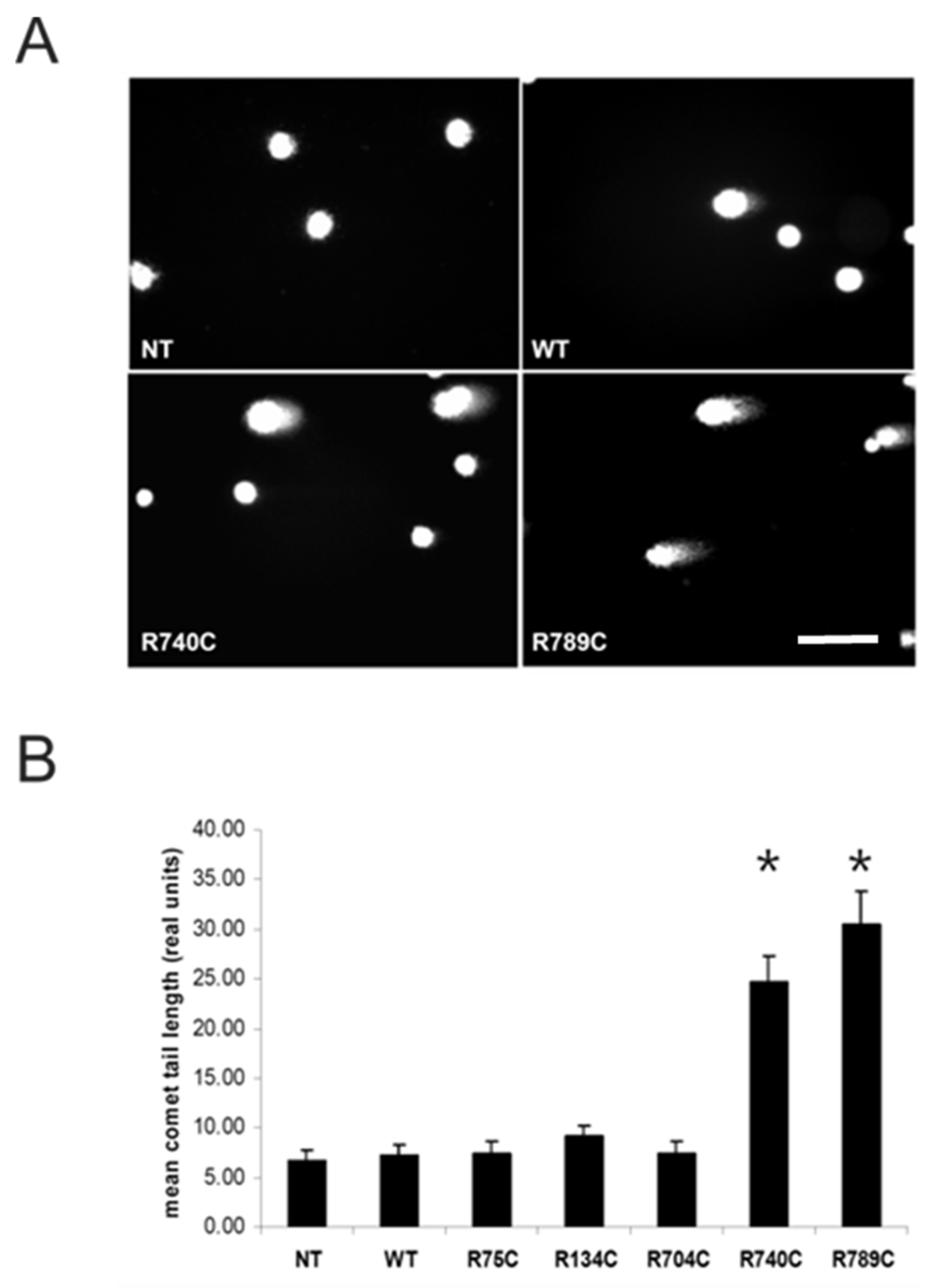
2.6. Intracellular Accumulation of Misfolded Proteins Induces Apoptosis in Cells Expressing Mutant Collagens
3. Discussion
4. Materials and Methods
4.1. Collagen II cDNA, Site-Directed Mutagenesis and Removal of Endogenous Signal Peptide
4.2. Cell Culture and Transfection
4.3. Purification of Collagen II Proteins
4.4. CD Spectroscopy and Melting Curves
4.5. SDS-PAGE and Immunoblotting
4.6. Trypsin Digestion
4.7. Inhibition of Matrix Metalloproteinase Cleavage
4.8. Reverse Transcription-PCR for Analysis of Spliced XBP1 Transcripts
4.9. Immunofluorescence Staining of HT1080 Cells
4.10. Nick Labelling
4.11. Comet Assay or Single Cell Gel Electrophoresis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Myllyharju, J.; Kivirikko, K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Brewton, R.G.; Mayne, R. Mammalian vitreous humor contains networks of hyaluronan molecules: Electron microscopic analysis using the hyaluronan-binding region (G1) of aggrecan and link protein. Exp. Cell Res. 1992, 198, 237–249. [Google Scholar] [CrossRef]
- Kivirikko, K.I. Collagens and their abnormalities in a wide spectrum of diseases. Ann. Med. 1993, 25, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, R. The human type I collagen mutation database. Nucleic Acids Res. 1997, 25, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, R. The human collagen mutation database 1998. Nucleic Acids Res. 1998, 26, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Bulleid, N.J.; John, D.C.; Kadler, K.E. Recombinant expression systems for the production of collagen. Biochem. Soc. Trans. 2000, 28, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Canty, E.G.; Kadler, K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005, 118, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, V.; Erlmann, P. The pathway of collagen secretion. Annu. Rev. Cell Dev. Biol. 2015, 31, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Bolund, L.; Bross, P. Protein misfolding, aggregation, and degradation in disease. Mol. Biotechnol. 2005, 31, 141–150. [Google Scholar] [CrossRef]
- Walmsley, A.R.; Batten, M.R.; Lad, U.; Bulleid, N.J. Intracellular retention of procollagen within the endoplasmic reticulum is mediated by prolyl 4-hydroxylase. J. Biol. Chem. 1999, 274, 14884–14892. [Google Scholar] [CrossRef] [PubMed]
- Mundlos, S.; Olsen, B.R. Heritable diseases of the skeleton. Part II: Molecular insights into skeletal development-matrix components and their homeostasis. FASEB J. 1997, 11, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J.; Kivirikko, K.I. Collagens and collagen-related diseases. Ann. Med. 2001, 33, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Vikkula, M.; Metsaranta, M.; Ala-Kokko, L. Type II collagen mutations in rare and common cartilage diseases. Ann. Med. 1994, 26, 107–114. [Google Scholar] [PubMed]
- Barat-Houari, M.; Sarrabay, G.; Gatinois, V.; Fabre, A.; Dumont, B.; Genevieve, D.; Touitou, I. Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies. Hum. Mutat. 2016, 37, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hoornaert, K.P.; Vereecke, I.; Dewinter, C.; Rosenberg, T.; Beemer, F.A.; Leroy, J.G.; Bendix, L.; Björck, E.; Bonduelle, M.; Boute, O.; et al. Stickler syndrome caused by COL2A1 mutations: Genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. 2010, 18, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Hoornaert, K.P.; Dewinter, C.; Vereecke, I.; Beemer, F.A.; Courtens, W.; Fryer, A.; Fryssira, H.; Lees, M.; Müllner-Eidenböck, A.; Rimoin, D.L.; et al. The phenotypic spectrum in patients with arginine to cysteine mutations in the COL2A1 gene. J. Med. Genet. 2006, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.J.; Baguley, D.M.; Yates, J.R.; Lane, C.; Nicol, M.; Harper, P.S.; Scott, J.D.; Snead, M.P. Variation in the vitreous phenotype of stickler syndrome can be caused by different amino acid substitutions in the x position of the type II collagen gly-x-y triple helix. Am. J. Hum. Genet. 2000, 67, 1083–1094. [Google Scholar] [PubMed]
- Williams, C.J.; Considine, E.L.; Knowlton, R.G.; Reginato, A.; Neumann, G.; Harrison, D.; Buxton, P.; Jimenez, S.; Prockop, D.J. Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg75→cys mutation in the procollagen type Ii gene (COL2A1). Hum. Genet. 1993, 92, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Ballo, R.; Beighton, P.H.; Ramesar, R.S. Stickler-like syndrome due to a dominant negative mutation in the COL2A1 gene. Am. J. Med. Genet. A 1998, 80, 6–11. [Google Scholar] [CrossRef]
- Richards, A.J.; McNinch, A.; Martin, H.; Oakhill, K.; Rai, H.; Waller, S.; Treacy, B.; Whittaker, J.; Meredith, S.; Poulson, A.; et al. Stickler syndrome and the vitreous phenotype: Mutations in COL2A1 and COL11A1. Hum. Mutat. 2010, 31, E1461–E1471. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Taylor, T.K.; Cole, W.G. Characterization of an arginine 789 to cysteine substitution in alpha 1 (ii) collagen chains of a patient with spondyloepiphyseal dysplasia. J. Biol. Chem. 1993, 268, 15238–15245. [Google Scholar] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [PubMed]
- Bruckner-Tuderman, L.; Bruckner, P. Genetic diseases of the extracellular matrix: More than just connective tissue disorders. J. Mol. Med. 1998, 76, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Bonafe, L.; Cormier-Daire, V.; Hall, C.; Lachman, R.; Mortier, G.; Mundlos, S.; Nishimura, G.; Sangiorgi, L.; Savarirayan, R.; Sillence, D.; et al. Nosology and classification of genetic skeletal disorders: 2015 Revision. Am. J. Med. Genet. A 2015, 167A, 2869–2892. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Rucker, E.; Steplewski, A.; McAdams, E.; Brittingham, R.J.; Alabyeva, T.; Fertala, A. Guilty by association: Some collagen II mutants alter the formation of ECM as a result of atypical interaction with fibronectin. J. Mol. Biol. 2005, 352, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Steplewski, A.; Ito, H.; Rucker, E.; Brittingham, R.J.; Alabyeva, T.; Gandhi, M.; Ko, F.K.; Birk, D.E.; Jimenez, S.A.; Fertala, A. Position of single amino acid substitutions in the collagen triple helix determines their effect on structure of collagen fibrils. J. Struct. Biol. 2004, 148, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Steplewski, A.; Majsterek, I.; McAdams, E.; Rucker, E.; Brittingham, R.J.; Ito, H.; Hirai, K.; Adachi, E.; Jimenez, S.A.; Fertala, A. Thermostability gradient in the collagen triple helix reveals its multi-domain structure. J. Mol. Biol. 2004, 338, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.V.; Fertala, A.; Sieron, A.L.; Hattori, H.; Mechling, D.; Bächinger, H.P.; Prockop, D.J. Recombinant procollagen II: Deletion of D period segments identifies sequences that are required for helix stabilization and generates a temperature-sensitive N-proteinase cleavage site. J. Biol. Chem. 1998, 273, 31822–31828. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Hayashi, T.; Bächinger, H.P. Hydroxylation-induced stabilization of the collagen triple helix. Further characterization of peptides with 4(R)-hydroxyproline in the XAA position. J. Biol. Chem. 2003, 278, 32373–32379. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Pöschl, E.; Turnay, J.; Baik, J.; Pihlajaniemi, T.; Frischholz, S.; von der Mark, K. Coexpression of alpha and beta subunits of prolyl 4-hydroxylase stabilizes the triple helix of recombinant human type X collagen. Biochem. J. 2000, 352, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.A.; Wilkin, D.J.; Kim, H.J.; Wilcox, W.R.; Lachman, R.S.; Rimoin, D.L.; Cohn, D.H.; Eyre, D.R. Structurally abnormal type II collagen in a severe form of Kniest dysplasia caused by an exon 24 skipping mutation. J. Biol. Chem. 1998, 273, 4761–4768. [Google Scholar] [CrossRef] [PubMed]
- Superti-Furga, A.; Steinmann, B.; Ramirez, F.; Byers, P.H. Molecular defects of type III procollagen in Ehlers-Danlos syndrome type IV. Hum. Genet. 1989, 82, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Väisänen, L.; Has, C.; Franzke, C.; Hurskainen, T.; Tuomi, M.L.; Bruckner-Tuderman, L.; Tasanen, K. Molecular mechanisms of junctional epidermolysis bullosa: Col 15 domain mutations decrease the thermal stability of collagen XVII. J. Investig. Dermatol. 2005, 125, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Freije, J.M.; Diez-Itza, I.; Balbin, M.; Sanchez, L.M.; Blasco, R.; Tolivia, J.; Lopez-Otin, C. Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J. Biol. Chem. 1994, 269, 16766–16773. [Google Scholar] [PubMed]
- Knauper, V.; Lopez-Otin, C.; Smith, B.; Knight, G.; Murphy, G. Biochemical characterization of human collagenase-3. J. Biol. Chem. 1996, 271, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Billinghurst, R.C.; Dahlberg, L.; Ionescu, M.; Reiner, A.; Bourne, R.; Rorabeck, C.; Mitchell, P.; Hambor, J.; Diekmann, O.; Tschesche, H.; et al. Enhanced cleavage of type ii collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Investig. 1997, 99, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, P.; Prockop, D.J. Proteolytic enzymes as probes for the triple-helical conformation of procollagen. Anal Biochem. 1981, 110, 360–368. [Google Scholar] [CrossRef]
- Cabral, W.A.; Chernoff, E.J.; Marini, J.C. G76e substitution in type I collagen is the first nonlethal glutamic acid substitution in the alpha1(i) chain and alters folding of the N-terminal end of the helix. Mol. Genet. Metab. 2001, 72, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Galicka, A.; Wolczynski, S.; Gindzienski, A. Studies on type I collagen in skin fibroblasts cultured from twins with lethal osteogenesis imperfecta. Acta Biochim. Pol. 2003, 50, 481–488. [Google Scholar] [PubMed]
- Majsterek, I.; McAdams, E.; Adachi, E.; Dhume, S.T.; Fertala, A. Prospects and limitations of the rational engineering of fibrillar collagens. Protein Sci. 2003, 12, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Salminen, H.J.; Saamanen, A.M.; Vankemmelbeke, M.N.; Auho, P.K.; Perala, M.P.; Vuorio, E.I. Differential expression patterns of matrix metalloproteinases and their inhibitors during development of osteoarthritis in a transgenic mouse model. Ann. Rheum. Dis. 2002, 61, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Tchetina, E.V.; Squires, G.; Poole, A.R. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol. 2005, 32, 876–886. [Google Scholar] [PubMed]
- Wu, W.; Billinghurst, R.C.; Pidoux, I.; Antoniou, J.; Zukor, D.; Tanzer, M.; Poole, A.R. Sites of collagenase cleavage and denaturation of type II collagen in aging and osteoarthritic articular cartilage and their relationship to the distribution of matrix metalloproteinase 1 and matrix metalloproteinase 13. Arthritis Rheum. 2002, 46, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Fertala, A.; Sieron, A.L.; Ganguly, A.; Li, S.W.; Ala-Kokko, L.; Anumula, K.R.; Prockop, D.J. Synthesis of recombinant human procollagen II in a stably transfected tumour cell line (ht1080). Biochem. J. 1994, 298 Pt 1, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Steplewski, A.; Brittingham, R.; Jimenez, S.A.; Fertala, A. Single amino acid substitutions in the c-terminus of collagen II alter its affinity for collagen ix. Biochem. Biophys. Res. Commun. 2005, 335, 749–755. [Google Scholar] [CrossRef] [PubMed]
- John, D.C.; Grant, M.E.; Bulleid, N.J. Cell-free synthesis and assembly of prolyl 4-hydroxylase: The role of the beta-subunit (PDI) in preventing misfolding and aggregation of the alpha-subunit. EMBO J. 1993, 12, 1587–1595. [Google Scholar] [PubMed]
- Kivirikko, K.I.; Myllyharju, J. Prolyl 4-hydroxylases and their protein disulfide isomerase subunit. Matrix Biol. 1998, 16, 357–368. [Google Scholar] [CrossRef]
- Hecht, J.T.; Makitie, O.; Hayes, E.; Haynes, R.; Susic, M.; Montufar-Solis, D.; Duke, P.J.; Cole, W.G. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J. Orthop. Res. 2004, 22, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Vranka, J.; Mokashi, A.; Keene, D.R.; Tufa, S.; Corson, G.; Sussman, M.; Horton, W.A.; Maddox, K.; Sakai, L.; Bachinger, H.P. Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biol. 2001, 20, 439–450. [Google Scholar] [CrossRef]
- Rajpar, M.H.; McDermott, B.; Kung, L.; Eardley, R.; Knowles, L.; Heeran, M.; Thornton, D.J.; Wilson, R.; Bateman, J.F.; Poulsom, R.; et al. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet. 2009, 5, e1000691. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Freddi, S.; Chan, D.; Cheah, K.S.; Bateman, J.F. Misfolding of collagen x chains harboring schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J. Biol. Chem. 2005, 280, 15544–15552. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of chop/gadd153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Chessler, S.D.; Byers, P.H. BiP binds type I procollagen pro alpha chains with mutations in the carboxyl-terminal propeptide synthesized by cells from patients with osteogenesis imperfecta. J. Biol. Chem. 1993, 268, 18226–18233. [Google Scholar] [PubMed]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Hintze, V.; Steplewski, A.; Ito, H.; Jensen, D.A.; Rodeck, U.; Fertala, A. Cells expressing partially unfolded R789C/p.R989C type II procollagen mutant associated with spondyloepiphyseal dysplasia undergo apoptosis. Hum. Mutat. 2008, 29, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, K.G.; Maddox, B.K.; Bann, J.G.; Boswell, B.A.; Keene, D.R.; Garofalo, S.; Horton, W.A. Y-position collagen II mutation disrupts cartilage formation and skeletal development in a transgenic mouse model of spondyloepiphyseal dysplasia. J. Bone Miner. Res. 2002, 17, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.T.; Hayes, E.; Haynes, R.; Cole, W.G. Comp mutations, chondrocyte function and cartilage matrix. Matrix Biol. 2005, 23, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Dinser, R.; Zaucke, F.; Kreppel, F.; Hultenby, K.; Kochanek, S.; Paulsson, M.; Maurer, P. Pseudoachondroplasia is caused through both intra- and extracellular pathogenic pathways. J. Clin. Investig. 2002, 110, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Becker, A.; Schmitz, A.; Weirich, C.; Paulsson, M.; Zaucke, F.; Dinser, R. Disruption of extracellular matrix structure may cause pseudoachondroplasia phenotypes in the absence of impaired cartilage oligomeric matrix protein secretion. J. Biol. Chem. 2006, 281, 32587–35295. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.F.; Moeller, I.; Hannagan, M.; Chan, D.; Cole, W.G. Characterization of three osteogenesis imperfecta collagen alpha 1(i) glycine to serine mutations demonstrating a position-dependent gradient of phenotypic severity. Biochem. J. 1992, 288, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Jakkula, E.; Mäkitie, O.; Czarny-Ratajczak, M.; Jackson, G.C.; Damignani, R.; Susic, M.; Briggs, M.D.; Cole, W.G.; Ala-Kokko, L. Mutations in the known genes are not the major cause of MED; distinctive phenotypic entities among patients with no identified mutations. Eur. J. Hum. Genet. 2005, 13, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Bonadio, J.; Byers, P.H. Subtle structural alterations in the chains of type I procollagen produce osteogenesis imperfecta type II. Nature 1985, 316, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H. Brittle bones-fragile molecules: Disorders of collagen gene structure and expression. Trends Genet. 1990, 6, 293–300. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G.; Prockop, D.J. Mutations in collagen genes: Causes of rare and some common diseases in humans. FASEB J. 1991, 5, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Kohfeldt, E.; Maurer, P.; Vannahme, C.; Timpl, R. Properties of the extracellular calcium binding module of the proteoglycan testican. FEBS Lett. 1997, 414, 557–561. [Google Scholar] [CrossRef]
- Wuttke, M.; Muller, S.; Nitsche, D.P.; Paulsson, M.; Hanisch, F.G.; Maurer, P. Structural characterization of human recombinant and bone-derived bone sialoprotein. Functional implications for cell attachment and hydroxyapatite binding. J. Biol. Chem. 2001, 276, 36839–36848. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Melting Temperature (Tm) |
|---|---|
| WT | 38.6 °C |
| R75C | 36.2 °C |
| R134C | 36.1 °C |
| R704C | 36.1 °C |
| R740C | 30.2 °C |
| R789C | 31.5 °C |
| Primer | Sequence |
|---|---|
| 75F | 5′-GGTCCTCAGGGTGCTTGTGGTTTCCCAGG-3′ |
| 75R | 5′-CCTGGGAAACCACAAGCACCCTGAGGACC-3′ |
| 134F | 5′-CTGGTGAAAGAGGATGCACTGGCCCTGCTG-3′ |
| 134R | 5′-CCAGCAGGGCCAGTGCATCCTCTTTCACC-3′ |
| 704F | 5′-GGAGCTGCTGGGTGCGTTGGACCCCC-3′ |
| 704R | 5′-GGGGGTCCAACGCACCCAGCAGCTCC-3′ |
| 740F | 5′-CCCCCTGGCTGCGCTGGTGAACCCGG-3′ |
| 740R | 5′-GGGTTCACCAGCGCAGCCAGGGGGG-3′ |
| 789F | 5′-GGTCTGCCTGGGCAATGTGGTGAGAGAGGATTCC-3′ |
| 789R | 5′-GGAATCCTCTCTCACCACATTGCCCAGGCAGACC-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakkalakal, S.A.; Heilig, J.; Baumann, U.; Paulsson, M.; Zaucke, F. Impact of Arginine to Cysteine Mutations in Collagen II on Protein Secretion and Cell Survival. Int. J. Mol. Sci. 2018, 19, 541. https://doi.org/10.3390/ijms19020541
Chakkalakal SA, Heilig J, Baumann U, Paulsson M, Zaucke F. Impact of Arginine to Cysteine Mutations in Collagen II on Protein Secretion and Cell Survival. International Journal of Molecular Sciences. 2018; 19(2):541. https://doi.org/10.3390/ijms19020541
Chicago/Turabian StyleChakkalakal, Salin A., Juliane Heilig, Ulrich Baumann, Mats Paulsson, and Frank Zaucke. 2018. "Impact of Arginine to Cysteine Mutations in Collagen II on Protein Secretion and Cell Survival" International Journal of Molecular Sciences 19, no. 2: 541. https://doi.org/10.3390/ijms19020541
APA StyleChakkalakal, S. A., Heilig, J., Baumann, U., Paulsson, M., & Zaucke, F. (2018). Impact of Arginine to Cysteine Mutations in Collagen II on Protein Secretion and Cell Survival. International Journal of Molecular Sciences, 19(2), 541. https://doi.org/10.3390/ijms19020541