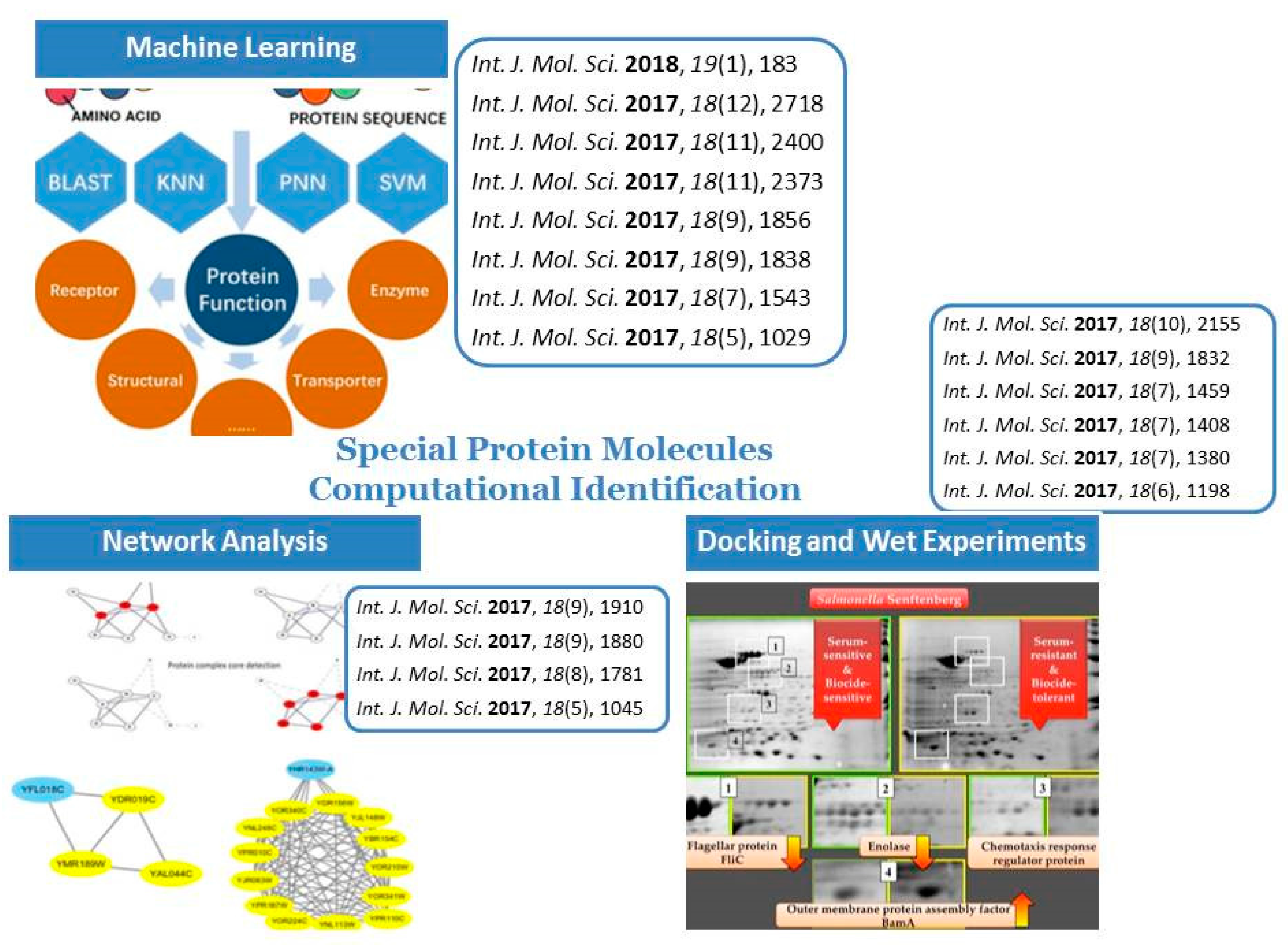
Special Protein Molecules Computational Identification



{kind=link}
Abstract
:1. Introduction
2. Machine Learning Related Researches
2.1. Protein–Protein Interaction Prediction
2.2. Special Proteins Identification
2.3. Protein Subcellular Localization and Function Analysis
3. Network Techniques Related Researches
4. Docking and Wet Experiments Researches
Acknowledgements
Conflicts of Interest
References
- Zeng, J.; Zou, Q.; Wu, Y.; Li, D.; Liu, X. An Empirical Study of Features Fusion Techniques for Protein–protein Interaction Prediction. Curr. Bioinform. 2016, 11, 4–12. [Google Scholar] [CrossRef]
- Garzón, J.I.; Deng, L.; Murray, D.; Shapira, S.; Petrey, D.; Honig, B. A computational interactome and functional annotation for the human proteome. eLife 2016, 5, e18715. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Jia, L.; Ren, Y.; Yu, G. Protein–protein Interactions Prediction Using a Novel Local Conjoint Triad Descriptor of Amino Acid Sequences. Int. J. Mol. Sci. 2017, 18, 2373. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, Z.; Li, X.; Chen, X.; Jiang, T.; Zhang, J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. Int. J. Mol. Sci. 2017, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Predicting protein–protein interactions via multivariate mutual information of protein sequences. BMC Bioinform. 2016, 17, 398. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of Protein–Protein Interactions via a Novel Matrix-Based Sequence Representation Model with Amino Acid Contact Information. Int. J. Mol. Sci. 2016, 17, 1623. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ding, Y.; Tang, J.; Xu, X.; Guo, F. An Ameliorated Prediction of Drug-Target Interactions Based on Multi-Scale Discrete Wavelet Transform and Network Features. Int. J. Mol. Sci. 2017, 18, 1781. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of Drug-Target Interactions via Multiple Information Integration. Inf. Sci. 2017, 418–419, 546–560. [Google Scholar] [CrossRef]
- Chen, P.; Li, J. Sequence-based identification of interface residues by an integrative profile combining hydrophobic and evolutionary information. BMC Bioinform. 2010, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, J.; Wong, L.; Kuwahara, H.; Huang, J.Z.; Gao, X. Accurate prediction of hot spot residues through physicochemical characteristics of amino acid sequences. Proteins-Struct. Funct. Bioinform. 2013, 81, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, Z.; Zhan, W.; Deng, L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hu, S.; Zhang, J.; Gao, X.; Li, J.; Xia, J.; Wang, B. A sequence-based dynamic ensemble learning system for protein ligand-binding site prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.; Peng, C.; Bing, W.; Li, J. Protein binding hot spots prediction from sequence only by a new ensemble learning method. Amino Acids 2017, 49, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, N.; Chen, P.; Zheng, C.; Wang, B. Prediction of Protein Hotspots from Whole Protein Sequences by a Random Projection Ensemble System. Int. J. Mol. Sci. 2017, 18, 1543. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Dong, Q.; Li, S.; Liu, X. Identification of DNA-binding proteins by combining auto-cross covariance transformation and ensemble learning. IEEE Trans. NanoBiosci. 2016, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou’s PseAAC and Physicochemical Distance Transformation. Mol. Inform. 2015, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Wang, X. DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation. Sci. Rep. 2015, 5, 15479. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Lan, X.; Xu, R.; Zhou, J.; Wang, X.; Chou, K.-C. iDNA-Prot|dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. PLoS ONE 2014, 9, e106691. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, D.; Zeng, X.; Wu, Y.; Guo, L.; Zou, Q. nDNA-Prot: Identification of DNA-binding Proteins Based on Unbalanced Classification. BMC Bioinform. 2014, 15, 298. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An Improved DNA-binding Protein Prediction Method by Exploring Local Evolutionary Information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Qu, K.; Han, K.; Wu, S.; Wang, G.; Wei, L. Identification of DNA-Binding Proteins Using Mixed Feature Representation Methods. Molecules 2017, 22, 1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, B. PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation. Int. J. Mol. Sci. 2017, 18, 1856. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ding, Y.; Tang, J.; Song, J.; Guo, F. Identification of DNA-protein Binding Sites through Multi-Scale Local Average Blocks on Sequence Information. Molecules 2017, 22, 2079. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Deng, E.Z.; Chen, W.; Lin, H. Identifying the subfamilies of voltage-gated potassium channels using feature selection technique. Int. J. Mol. Sci. 2014, 15, 12940–12951. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ding, H. Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J. Theor. Biol. 2011, 269, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, H. Identification of voltage-gated potassium channel subfamilies from sequence information using support vector machine. Comput. Biol. Med. 2012, 42, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.W.; Su, Z.D.; Yang, W.; Lin, H.; Chen, W.; Tang, H. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. Int. J. Mol. Sci. 2017, 18, 1838. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, B.; Sun, P.P.; Ma, Z.Q. A Topology Structure Based Outer Membrane Proteins Segment Alignment Method. Math. Probl. Eng. 2013, 2013, 541359. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Yan, L.; Su, L.; Xu, D. OMPcontact: An Outer Membrane Protein Inter-Barrel Residue Contact Prediction Method. J. Comput. Biol. 2017, 24, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, Z.; Zhang, C.; Zhang, L.; Xu, D. Transmembrane protein alignment and fold recognition based on predicted topology. PLoS ONE 2013, 8, e69744. [Google Scholar] [CrossRef] [PubMed]
- Antonets, K.S.; Nizhnikov, A.A. Predicting Amyloidogenic Proteins in the Proteomes of Plants. Int. J. Mol. Sci. 2017, 18, 2155. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S. Protein Sub-Nuclear Localization Based on Effective Fusion Representations and Dimension Reduction Algorithm LDA. Int. J. Mol. Sci. 2015, 16, 30343–30361. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Nie, B.; Yue, K.; Fei, Y.; Li, W.; Xu, D. Protein Subcellular Localization with Gaussian Kernel Discriminant Analysis and Its Kernel Parameter Selection. Int. J. Mol. Sci. 2017, 18, 2718. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, P. A New Feature Extraction Method Based on the Information Fusion of Entropy Matrix and Covariance Matrix and Its Application in Face Recognition. Entropy 2015, 17, 4664–4683. [Google Scholar] [CrossRef]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2017, 46, D1121–D1127. [Google Scholar]
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.; Chen, Y.; Xue, W.; Li, X.; Zhu, F. NOREVA: Normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, W162–W170. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Xu, J.Y.; Tao, L.; Li, X.F.; Li, S.; Zeng, X.; Chen, S.Y.; Zhang, P.; Qin, C.; Zhang, C. SVM-Prot 2016: A Web-Server for Machine Learning Prediction of Protein Functional Families from Sequence Irrespective of Similarity. PLoS ONE 2016, 11, e0155290. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, X.; Yang, H.; Li, Y.H.; Xue, W.; Chen, Y.; Tao, L.; Zhu, F. Assessing the Performances of Protein Function Prediction Algorithms from the Perspectives of Identification Accuracy and False Discovery Rate. Int. J. Mol. Sci. 2018, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Du, P.F.; Zhao, W.; Miao, Y.Y.; Wei, L.Y.; Wang, L. UltraPse: A Universal and Extensible Software Platform for Representing Biological Sequences. Int. J. Mol. Sci. 2017, 18, 2400. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Analyt. Biochem. 2012, 425, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Gu, S.; Jiao, Y. PseAAC-General: Fast Building Various Modes of General Form of Chou’s Pseudo-Amino Acid Composition for Large-Scale Protein Datasets. Int. J. Mol. Sci. 2014, 15, 3495–3506. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.S.; Du, P.F. Predicting Golgi-resident protein types using pseudo amino acid compositions: Approaches with positional specific physicochemical properties. J. Theor. Biol. 2015, 391, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Oti, M.; Snel, B.; Huynen, M.A.; Brunner, H.G. Predicting disease genes using protein–protein interactions. J. Med. Genet. 2006, 43, 691. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hao Xing, Z.; Huang, T.; Shu, Y.; Huang, G.; Li, H.-P. Application of the Shortest Path Algorithm for the Discovery of Breast Cancer-Related Genes. Curr. Bioinform. 2016, 11, 51–58. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Huang, T.; Shu, Y.; Chen, L. Identification of novel proliferative diabetic retinopathy related genes on protein–protein interaction network. Neurocomputing 2016, 217, 63–72. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Xing, Z.; Yuan, F.; Shu, Y.; Zhang, Y.; Kong, X.; Huang, T.; Li, H.; Cai, Y.D. An integrated method for the identification of novel genes related to oral cancer. PLoS ONE 2017, 12, e0175185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Chen, Z.; Lei, D. Integrating Multiple Heterogeneous Networks for Novel LncRNA-disease Association Inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Su, F.; Xu, Y.; Zou, Q. Network-based method for mining novel HPV infection related genes using random walk with restart algorithm. BBA-Mol. Basis Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Yan, Y.; Li, Z.; Chen, L.; Yang, J.; Zhang, Y.; Wang, S.; Liu, L. Determination of Genes Related to Uveitis by Utilization of the Random Walk with Restart Algorithm on a Protein–Protein Interaction Network. Int. J. Mol. Sci. 2017, 18, 1045. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liao, B. Protein Complexes Prediction Method Based on Core-Attachment Structure and Functional Annotations. Int. J. Mol. Sci. 2017, 18, 1910. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Meng, X.; Zheng, R.; Wu, F.X.; Li, Y.; Pan, Y.; Wang, J. Identification of protein complexes by using a spatial and temporal active protein interaction network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. BioSystems 2015, 127, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, G.; Chen, G.; Li, M.; Wu, F.X.; Pan, Y. ClusterViz: A cytoscape APP for cluster analysis of biological network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, J.; Wu, F.X.; Pan, Y.; Wang, J. DyNetViewer: A Cytoscape app for dynamic network construction, analysis and visualization. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Min, L.; Wang, J.; Wu, F.X. CytoCtrlAnalyser: A Cytoscape app for biomolecular network controllability analysis. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, D.; Tang, Y.; Wu, F.; Wang, J. CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks. Int. J. Mol. Sci. 2017, 18, 1880. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Sun, J.; Zhou, S.; Gao, J.; Liu, Y. Identification of Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by Structure-Based Virtual Screening and Molecular Docking Approach. Int. J. Mol. Sci. 2017, 18, 1408. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.L.; Luo, J.X.; Zhang, B.C.; Jiang, G.F.; Ding, W.; Zhang, Y.Q. 3D-QSAR and Molecular Docking Studies on the TcPMCA1-Mediated Detoxification of Scopoletin and Coumarin Derivatives. Int. J. Mol. Sci. 2017, 18, 1380. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Gao, F.; Yu, Y.; Chen, B. Biochemical and Computational Insights on a Novel Acid-Resistant and Thermal-Stable Glucose 1-Dehydrogenase. Int. J. Mol. Sci. 2017, 18, 1198. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.C.; Gandhi, N.S.; Mancera, R.L.; Smith, G.; Elizur, A.; Ventura, T. Understanding Insulin Endocrinology in Decapod Crustacea: Molecular Modelling Characterization of an Insulin-Binding Protein and Insulin-Like Peptides in the Eastern Spiny Lobster, Sagmariasus verreauxi. Int. J. Mol. Sci. 2017, 18, 1832. [Google Scholar] [CrossRef] [PubMed]
- FutomaKołoch, B.; Dudek, B.; Kapczyńska, K.; Krzyżewska, E.; Wańczyk, M.; Korzekwa, K.; Rybka, J.; Klausa, E. Relationship of Triamine-Biocide Tolerance of Salmonella enterica Serovar Senftenberg to Antimicrobial Susceptibility, Serum Resistance and Outer Membrane Proteins. Int. J. Mol. Sci. 2017, 18, 1459. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. NanoBiosci. 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, Q.; He, W. Special Protein Molecules Computational Identification. Int. J. Mol. Sci. 2018, 19, 536. https://doi.org/10.3390/ijms19020536
Zou Q, He W. Special Protein Molecules Computational Identification. International Journal of Molecular Sciences. 2018; 19(2):536. https://doi.org/10.3390/ijms19020536
Chicago/Turabian StyleZou, Quan, and Wenying He. 2018. "Special Protein Molecules Computational Identification" International Journal of Molecular Sciences 19, no. 2: 536. https://doi.org/10.3390/ijms19020536
APA StyleZou, Q., & He, W. (2018). Special Protein Molecules Computational Identification. International Journal of Molecular Sciences, 19(2), 536. https://doi.org/10.3390/ijms19020536