Potential Role of Humoral IL-6 Cytokine in Mediating Pro-Inflammatory Endothelial Cell Response in Amyotrophic Lateral Sclerosis

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Humoral Effectors in ALS Patients
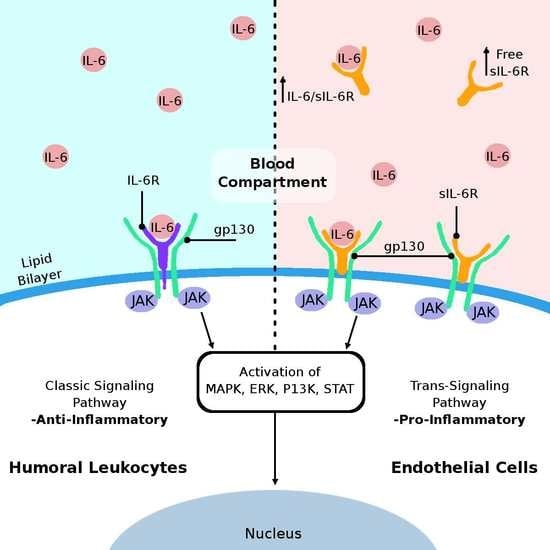
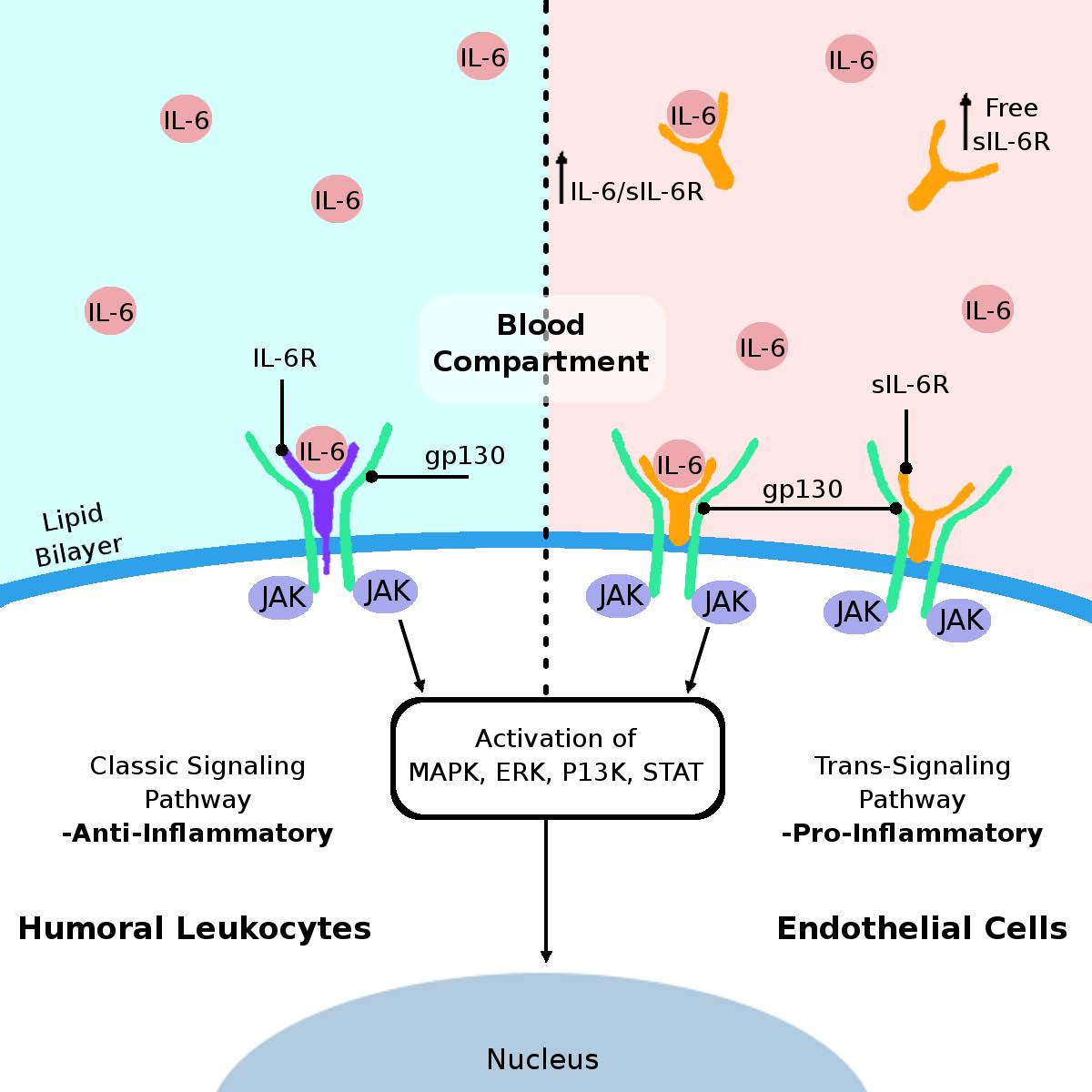
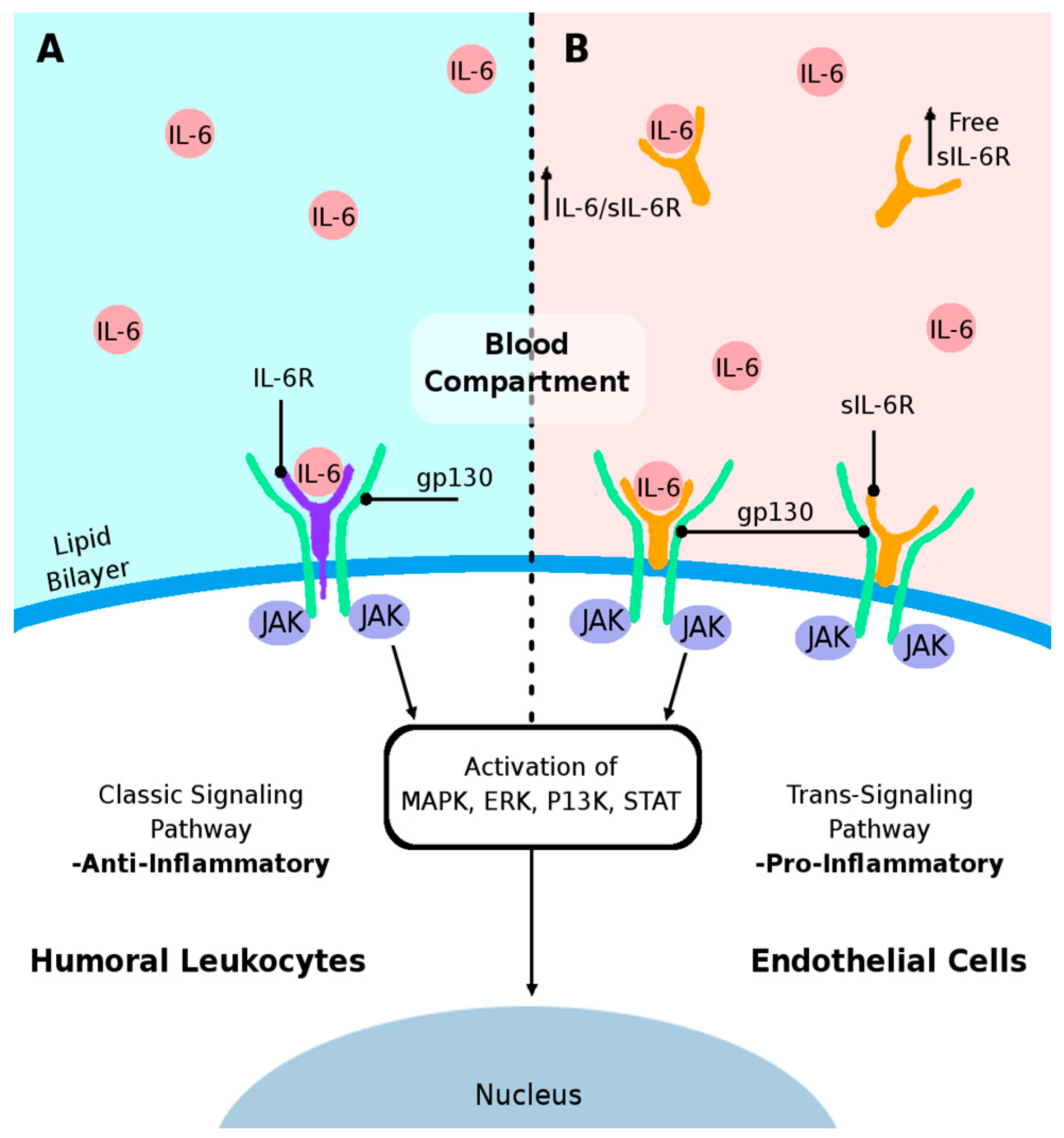
3. IL-6 Cytokine and Its Receptors
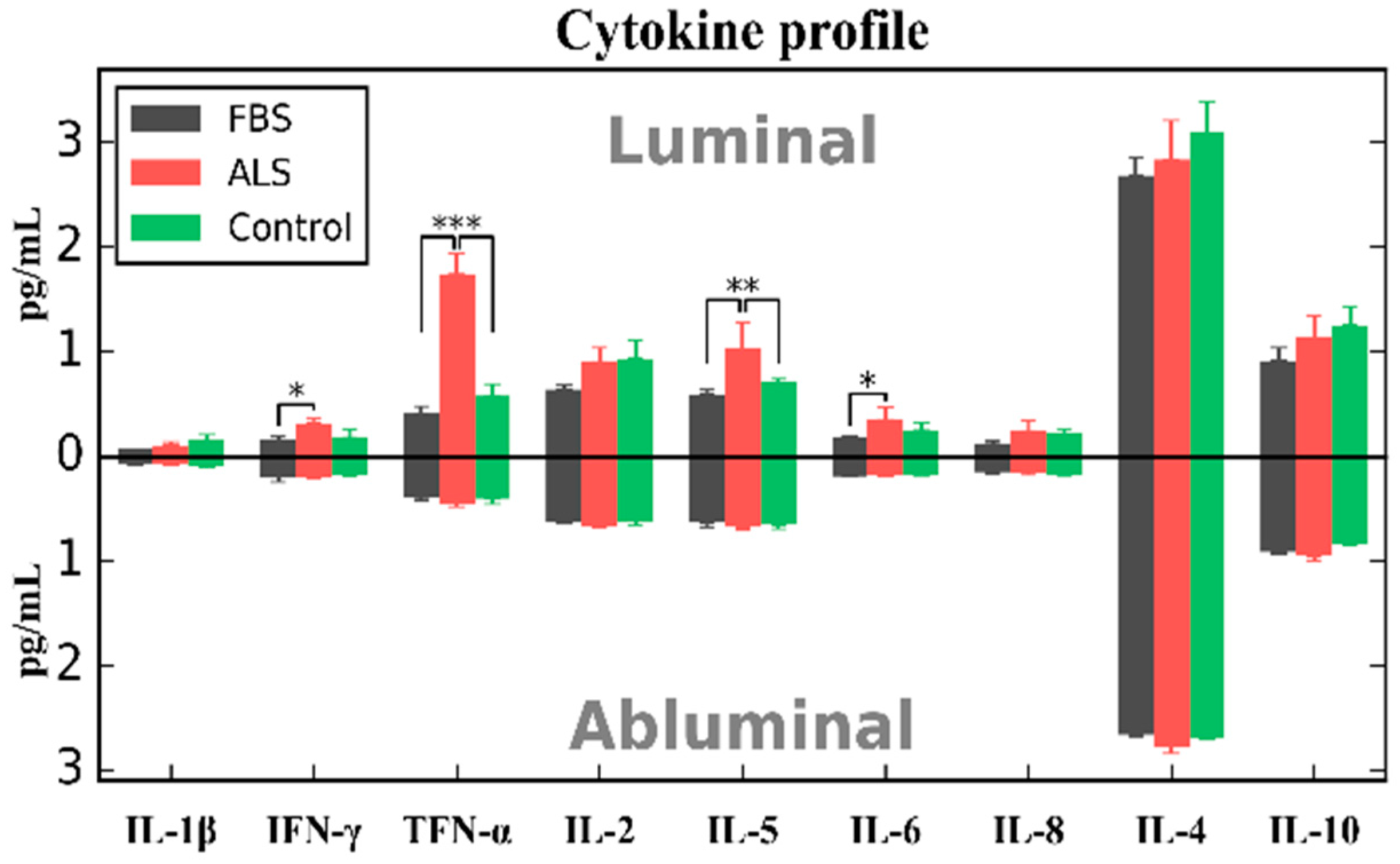
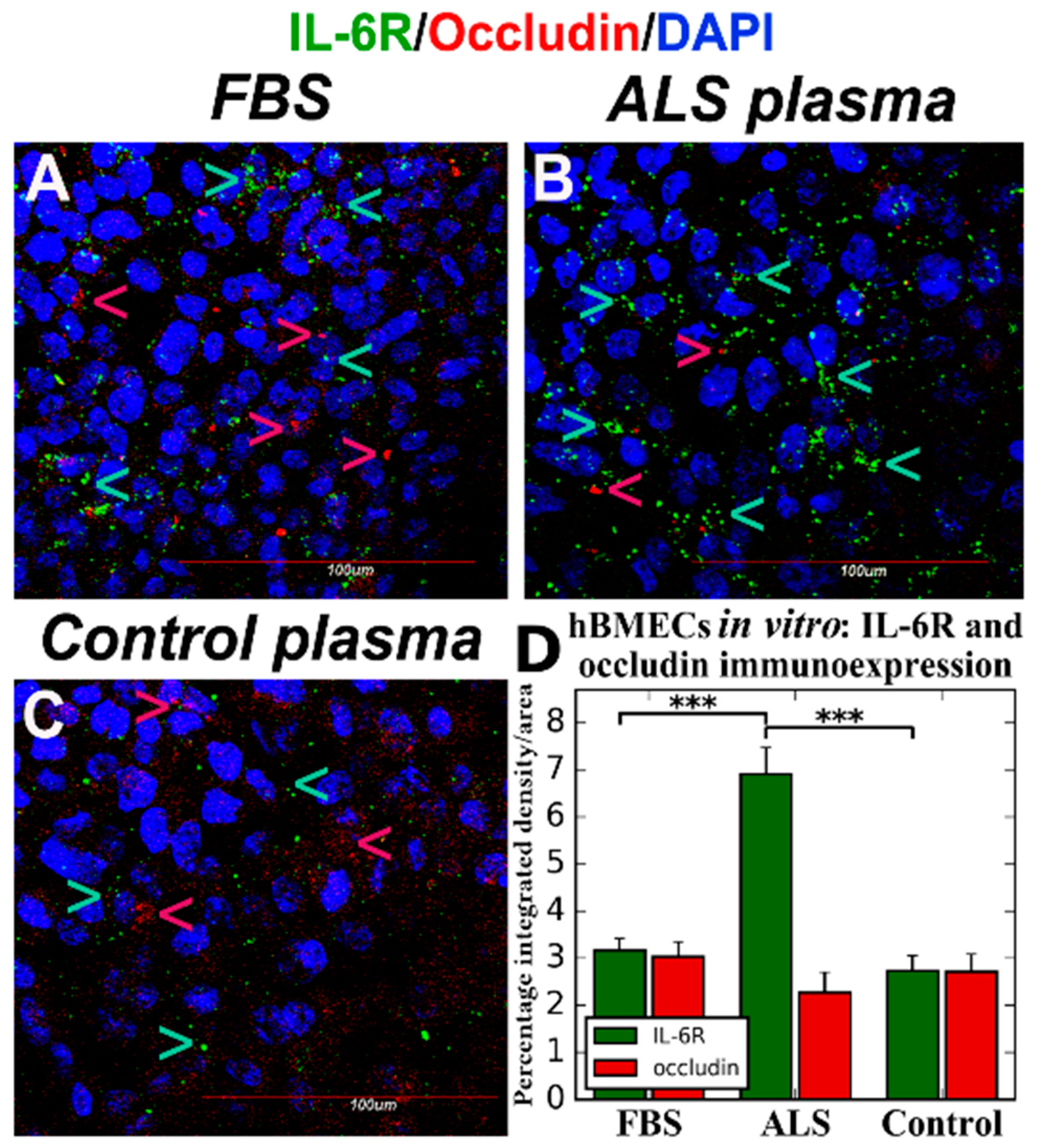
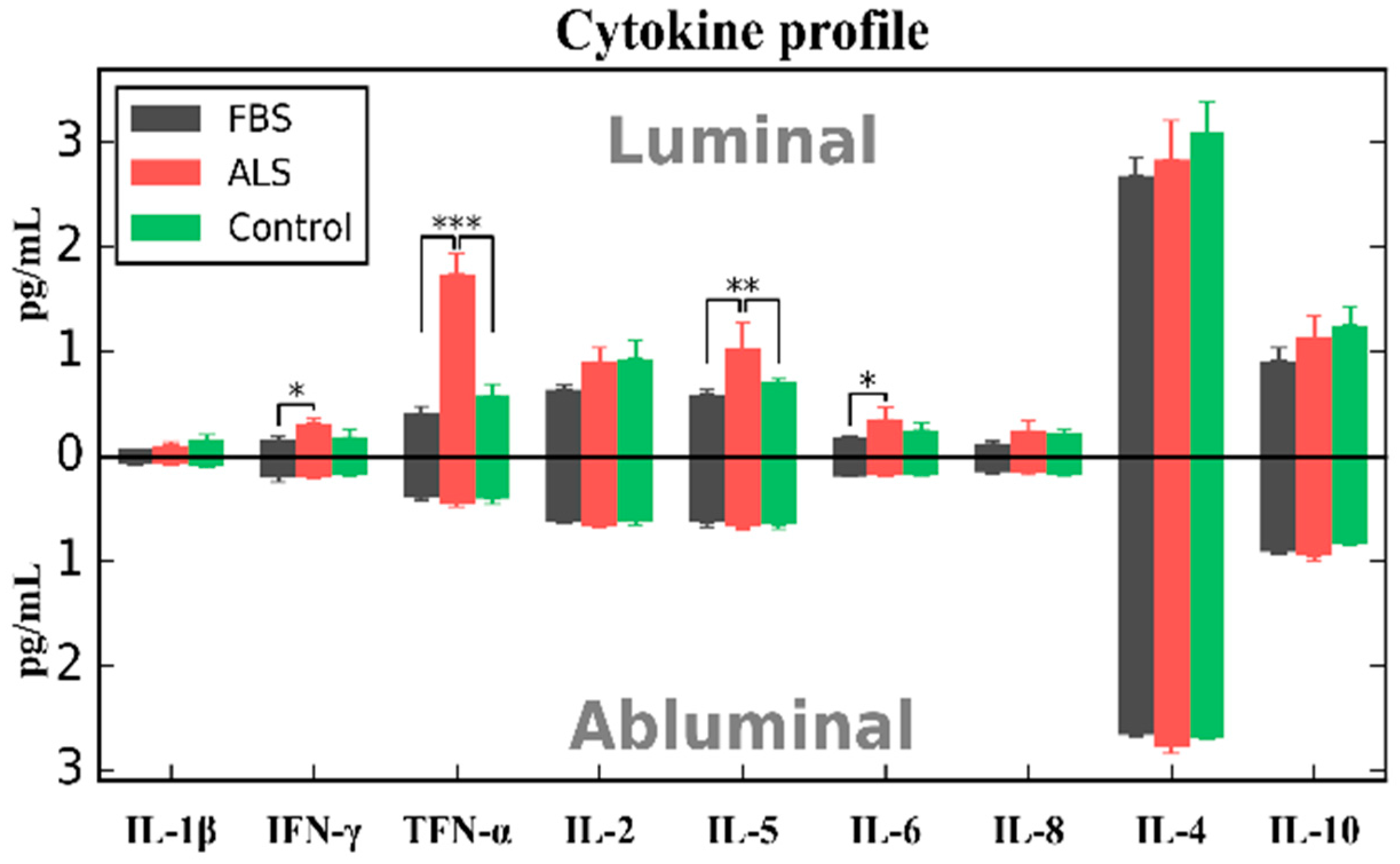
4. Effects of ALS Plasma Proteins on Human Bone Marrow Derived Endothelial Cells In Vitro
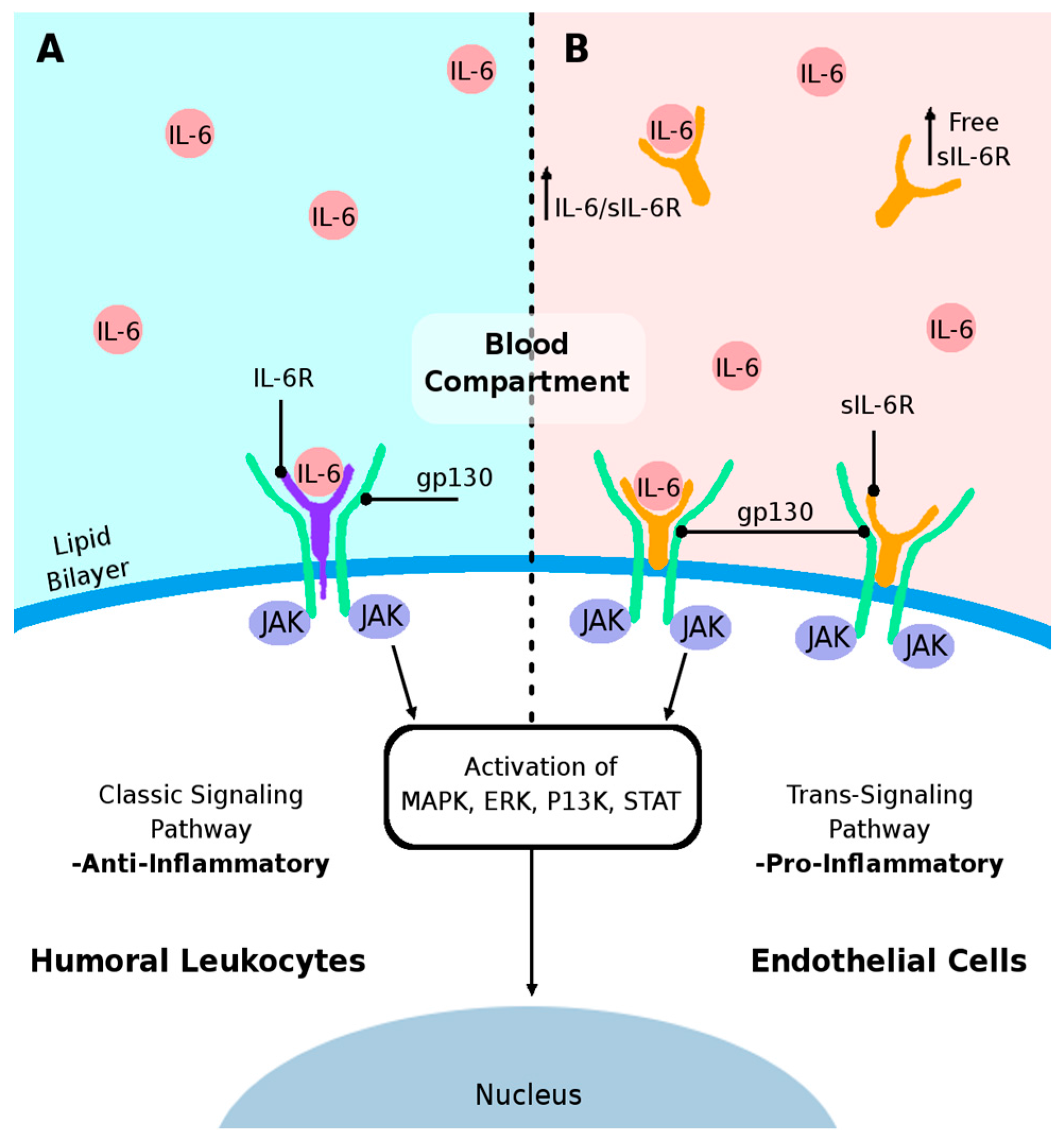
5. Proposed Mechanisms of Humoral IL-6-Mediated Inflammation Triggering Endothelial Cell Damage
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ALSRS-R | amyotrophic lateral sclerosis functional rating scale-revised |
| BBB | blood-brain barrier |
| B-CNS-B | blood-CNS barrier |
| BSCB | blood-spinal cord barrier |
| CCL2 | C-C motif chemokine ligand 2 |
| CNS | central nervous system |
| CX3CL1 | C-X3-C motif chemokine ligand 1 |
| CXC | cysteine-X-cysteine |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DIV | days in vitro |
| EC | endothelial cell |
| ERK | extracellular signal-regulated kinase |
| FALS | familial ALS |
| FBS | fetal bovine serum |
| FITC | fluorescein Isothiocyanate |
| G-CSF | granulocyte colony-stimulating factor |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| gp130 | glycoprotein 130 |
| GSH | glutathione |
| hBMEC | human bone marrow derived endothelial cell |
| hUVEC | human umbilical vein endothelial cell |
| IFN-β | interferon-beta |
| IFN-γ | interferon-gamma |
| IgG | immunoglobulin G |
| IL | interleukin |
| IL-6R | interleukin-6 receptor |
| JAK | janus kinase |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemotactic protein-1 |
| MHC | major histocompatibility complex |
| MMP | matrix metalloproteinase |
| mRNA | messenger RNA |
| PI3K | phosphatidyl-inositol-3-kinase |
| SALS | sporadic ALS |
| sgp130 | soluble form of gp130 |
| sIL-6R | soluble form of IL-6R |
| SOD-1 | superoxide dismutase 1 |
| STAT | signal transducer and activator of transcription |
| TGF- β | transforming growth factor-beta |
| TNF-α | tumor necrosis factor-alpha |
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Sorarù, G.; Ermani, M.; Logroscino, G.; Palmieri, A.; D’Ascenzo, C.; Orsetti, V.; Volpe, M.; Cima, V.; Zara, G.; Pegoraro, E.; et al. Natural history of upper motor neuron-dominant ALS. Amyotroph. Lateral Scler. 2010, 11, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K. Motor neuron disease: The bare essentials. Pract. Neurol. 2009, 9, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H. Amyotrophic lateral sclerosis: Pathophysiology, diagnosis and management. CNS Drugs 2011, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.A.; Reis, C.; Simões, F.; Fonseca, J.; Mendes Ribeiro, J. Diagnostic investigation and multidisciplinary management in motor neuron disease. J. Neurol. 2005, 252, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A. Diagnosis and Clinical Management of Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. Contin. Lifelong Learn. Neurol. 2017, 23, 1332–1359. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.C.; Manzano, R.; Mendonça, D.M.F.; Muñoz, M.J.; Zaragoza, P.; Osta, R. Amyotrophic lateral sclerosis: A focus on disease progression. BioMed Res. Int. 2014, 2014, 925101. [Google Scholar] [CrossRef] [PubMed]
- Tomik, J.; Tomik, B.; Gajec, S.; Ceranowicz, P.; Pihut, M.; Olszanecki, R.; Stręk, P.; Składzień, J. The balloon-based manometry evaluation of swallowing in patients with amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- McConnel, F.M.; Cerenko, D.; Hersh, T.; Weil, L.J. Evaluation of pharyngeal dysphagia with manofluorography. Dysphagia 1988, 2, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The pathobiology of amyotrophic lateral sclerosis: A proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Price, A.C.; Kaiser, A.; Shaikh, A.Y.; Liu, Z. Mechanisms for neuronal degeneration in amyotrophic lateral sclerosis and in models of motor neuron death (Review). Int. J. Mol. Med. 2000, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.J.; Getzoff, E.D.; Perry, J.J.P. Amyotrophic lateral sclerosis: Update and new developments. Degener. Neurol. Neuromuscul. Dis. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Hugon, J. Riluzole and ALS therapy. Wien. Med. Wochenschr. 1996, 146, 185–187. [Google Scholar] [PubMed]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2003, 4, 191–206. [Google Scholar] [PubMed]
- Garbuzova-Davis, S.; Saporta, S.; Sanberg, P.R. Implications of blood-brain barrier disruption in ALS. Amyotroph. Lateral Scler. 2008, 9, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Haller, E.; Saporta, S.; Kolomey, I.; Nicosia, S.V.; Sanberg, P.R. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 2007, 1157, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.O.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS barrier impairment in ALS patients versus an animal model. Front. Cell. Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, C.; Mitrecic, D.; Demetter, P.; De Decker, R.; Authelet, M.; Boom, A.; Pochet, R. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009, 1301, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Alterations of the blood-spinal cord barrier in sporadic amyotrophic lateral sclerosis. Neuropathology 2015, 35, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Rodrigues, M.C.O.; Hernandez-Ontiveros, D.G.; Louis, M.K.; Willing, A.E.; Borlongan, C.V.; Sanberg, P.R. Amyotrophic lateral sclerosis: A neurovascular disease. Brain Res. 2011, 1398, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.C.O.; Hernandez-Ontiveros, D.G.; Louis, M.K.; Willing, A.E.; Borlongan, C.V.; Sanberg, P.R.; Voltarelli, J.C.; Garbuzova-Davis, S. Neurovascular aspects of amyotrophic lateral sclerosis. Int. Rev. Neurobiol. 2012, 102, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Chattopadhay, M.; La Cava, A.; Tse, E.; Liu, G.; Lourenco, E.; Eskin, A.; Liu, P.T.; Magpantay, L.; Tse, S.; et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflamm. 2010, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossù, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflamm. 2014, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.M.; Simmons, Z.; Beard, J.L.; Stephens, H.E.; Connor, J.R. Plasma biomarkers associated with ALS and their relationship to iron homeostasis. Muscle Nerve 2010, 42, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Hu, J.; Shimizu, N.; Imai, T.; Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 187, 27–34. [Google Scholar] [CrossRef]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.C.; Couch, Y.; Sibson, N.; Turner, M.R. Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 2013, 53, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Simmons, Z.; Mitchell, R.M.; Kong, L.; Stephens, H.E.; Connor, J.R. Biomarker-based predictive models for prognosis in amyotrophic lateral sclerosis. JAMA Neurol. 2013, 70, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncology 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Fattori, E.; Cappelletti, M.; Costa, P.; Sellitto, C.; Cantoni, L.; Carelli, M.; Faggioni, R.; Fantuzzi, G.; Ghezzi, P.; Poli, V. Defective inflammatory response in interleukin 6-deficient mice. J. Exp. Med. 1994, 180, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Dinarello, C.A.; Mier, J.W. IL-6 and APPs: Anti-inflammatory and immunosuppressive mediators. Immunol. Today 1997, 18, 428–432. [Google Scholar] [CrossRef]
- Xing, Z.; Gauldie, J.; Cox, G.; Baumann, H.; Jordana, M.; Lei, X.F.; Achong, M.K. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Investig. 1998, 101, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.M.; Spehlmann, M.E.; Hammond, D.C.; Iimura, M.; Hase, K.; Choi, L.J.; Hanson, E.; Eckmann, L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 2008, 180, 6816–6826. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier biology and methodology. J. Neurovirol. 1999, 5, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cao, C.; Qin, X.-Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: A meta-analysis study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef] [PubMed]
- Robelin, L.; Gonzalez De Aguilar, J.L. Blood biomarkers for amyotrophic lateral sclerosis: Myth or reality? BioMed Res. Int. 2014, 2014, 525097. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.C.O.; Voltarelli, J.C.; Sanberg, P.R.; Borlongan, C.V.; Garbuzova-Davis, S. Immunological aspects in amyotrophic lateral sclerosis. Transl. Stroke Res. 2012, 3, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.C.O.; Sanberg, P.R.; Cruz, L.E.; Garbuzova-Davis, S. The innate and adaptive immunological aspects in neurodegenerative diseases. J. Neuroimmunol. 2014, 269, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, S.; Garbelli, S.; Pasini, A.; Alimonti, D.; Perotti, C.; Melazzini, M.; Bendotti, C.; Mora, G. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J. Neuroimmunol. 2009, 210, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain J. Neurol. 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Evangelopoulos, E.; Sereti, E.; Zouvelou, V.; Marmara, S.; Alexakis, T.; Evdokimidis, I. Alterations of T cell subsets in ALS: A systemic immune activation? Acta Neurol. Scand. 2012, 125, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Gascon, R.; Miller, R.G.; Gelinas, D.F.; Mass, J.; Hadlock, K.; Jin, X.; Reis, J.; Narvaez, A.; McGrath, M.S. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2005, 159, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Schubert, W.; Schwan, H. Detection by 4-parameter microscopic imaging and increase of rare mononuclear blood leukocyte types expressing the Fc gamma RIII receptor (CD16) for immunoglobulin G in human sporadic amyotrophic lateral sclerosis (ALS). Neurosci. Lett. 1995, 198, 29–32. [Google Scholar] [CrossRef]
- Keizman, D.; Rogowski, O.; Berliner, S.; Ish-Shalom, M.; Maimon, N.; Nefussy, B.; Artamonov, I.; Drory, V.E. Low-grade systemic inflammation in patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 2009, 119, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Murdock, B.J.; Bender, D.E.; Kashlan, S.R.; Figueroa-Romero, C.; Backus, C.; Callaghan, B.C.; Goutman, S.A.; Feldman, E.L. Increased ratio of circulating neutrophils to monocytes in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e242. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.; Chin, L.; Halder, R.C.; Sagong, B.; Famenini, S.; Sayre, J.; Montoya, D.; Rubbi, L.; Pellegrini, M.; Fiala, M. Epigenetic changes in T-cell and monocyte signatures and production of neurotoxic cytokines in ALS patients. FASEB J. 2016, 30, 3461–3473. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Sekizawa, T.; Openshaw, H.; Ohbo, K.; Sugamura, K.; Itoyama, Y.; Niland, J.C. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: Immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J. Neurol. Sci. 1998, 154, 194–199. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Gutierrez, E.G. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci. Lett. 1994, 179, 53–56. [Google Scholar] [CrossRef]
- Ringheim, G.E.; Burgher, K.L.; Heroux, J.A. Interleukin-6 mRNA expression by cortical neurons in culture: Evidence for neuronal sources of interleukin-6 production in the brain. J. Neuroimmunol. 1995, 63, 113–123. [Google Scholar] [CrossRef]
- Moreau, C.; Devos, D.; Brunaud-Danel, V.; Defebvre, L.; Perez, T.; Destée, A.; Tonnel, A.B.; Lassalle, P.; Just, N. Elevated IL-6 and TNF-α levels in patients with ALS: Inflammation or hypoxia? Neurology 2005, 65, 1958–1960. [Google Scholar] [CrossRef] [PubMed]
- Utgaard, J.O.; Jahnsen, F.L.; Bakka, A.; Brandtzaeg, P.; Haraldsen, G. Rapid secretion of prestored interleukin 8 from Weibel-Palade bodies of microvascular endothelial cells. J. Exp. Med. 1998, 188, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef]
- Yu, H.; Huang, X.; Ma, Y.; Gao, M.; Wang, O.; Gao, T.; Shen, Y.; Liu, X. Interleukin-8 regulates endothelial permeability by down-regulation of tight junction but not dependent on integrins induced focal adhesions. Int. J. Biol. Sci. 2013, 9, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Melani, C.; Mattia, G.F.; Silvani, A.; Carè, A.; Rivoltini, L.; Parmiani, G.; Colombo, M.P. Interleukin-6 expression in human neutrophil and eosinophil peripheral blood granulocytes. Blood 1993, 81, 2744–2749. [Google Scholar] [PubMed]
- Ericson, S.G.; Zhao, Y.; Gao, H.; Miller, K.L.; Gibson, L.F.; Lynch, J.P.; Landreth, K.S. Interleukin-6 production by human neutrophils after Fc-receptor cross-linking or exposure to granulocyte colony-stimulating factor. Blood 1998, 91, 2099–2107. [Google Scholar] [PubMed]
- Cicco, N.A.; Lindemann, A.; Content, J.; Vandenbussche, P.; Lübbert, M.; Gauss, J.; Mertelsmann, R.; Herrmann, F. Inducible production of interleukin-6 by human polymorphonuclear neutrophils: Role of granulocyte-macrophage colony-stimulating factor and tumor necrosis factor-α. Blood 1990, 75, 2049–2052. [Google Scholar] [PubMed]
- Horii, Y.; Muraguchi, A.; Suematsu, S.; Matsuda, T.; Yoshizaki, K.; Hirano, T.; Kishimoto, T. Regulation of BSF-2/IL-6 production by human mononuclear cells. Macrophage-dependent synthesis of BSF-2/IL-6 by T cells. J. Immunol. 1988, 141, 1529–1535. [Google Scholar] [PubMed]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Jirik, F.; Kabouridis, P.; Tsoukas, C.; Hirano, T.; Kishimoto, T.; Carson, D.A. B cell stimulating factor 2/interleukin 6 is a costimulant for human thymocytes and T lymphocytes. J. Exp. Med. 1988, 167, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, P.; Tuscano, J.M.; Kehrl, J.H. Tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in B-lymphocyte function. Methods 1997, 11, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Brown, B.D.; Merad, M. Studying the mononuclear phagocyte system in the molecular age. Nat. Rev. Immunol. 2011, 11, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L. The CD14+ CD16+ blood monocytes: Their role in infection and inflammation. J. Leukoc. Biol. 2007, 81, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Clanchy, F.I.L.; Holloway, A.C.; Lari, R.; Cameron, P.U.; Hamilton, J.A. Detection and properties of the human proliferative monocyte subpopulation. J. Leukoc. Biol. 2006, 79, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L. Monocyte subsets in man and other species. Cell. Immunol. 2014, 289, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Grage-Griebenow, E.; Flad, H.D.; Ernst, M. Heterogeneity of human peripheral blood monocyte subsets. J. Leukoc. Biol. 2001, 69, 11–20. [Google Scholar] [PubMed]
- Mukherjee, R.; Kanti Barman, P.; Kumar Thatoi, P.; Tripathy, R.; Kumar Das, B.; Ravindran, B. Non-Classical monocytes display inflammatory features: Validation in sepsis and systemic lupus erythematous. Sci. Rep. 2015, 5, 13886. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F.; Steeber, D.A.; Chen, A.; Engel, P. The selectins: Vascular adhesion molecules. FASEB J. 1995, 9, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Hamada, E.; Shodai, T.; Kamoshida, G.; Kudo, S.; Itoh, S.; Koike, J.; Nagata, K.; Irimura, T.; Tsuji, T. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int. Arch. Allergy Immunol. 2013, 160, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Makó, V.; Czúcz, J.; Weiszhár, Z.; Herczenik, E.; Matkó, J.; Prohászka, Z.; Cervenak, L. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1β, TNF-α, and LPS. Cytom. Part A 2010, 77, 962–970. [Google Scholar] [CrossRef] [PubMed]
- May, L.T.; Torcia, G.; Cozzolino, F.; Ray, A.; Tatter, S.B.; Santhanam, U.; Sehgal, P.B.; Stern, D. Interleukin-6 gene expression in human endothelial cells: RNA start sites, multiple IL-6 proteins and inhibition of proliferation. Biochem. Biophys. Res. Commun. 1989, 159, 991–998. [Google Scholar] [CrossRef]
- Ancuta, P.; Wang, J.; Gabuzda, D. CD16+ monocytes produce IL-6, CCL2, and matrix metalloproteinase-9 upon interaction with CX3CL1-expressing endothelial cells. J. Leukoc. Biol. 2006, 80, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Cai, Y.; Matsumoto, K.; Agui, T. Endothelin-induced interleukin-6 production by rat aortic endothelial cells. Endocrinology 1995, 136, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Pedersen, B.K. The role of exercise-induced myokines in muscle homeostasis and the defense against chronic diseases. J. Biomed. Biotechnol. 2010, 2010, 520258. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Wolsk-Petersen, E.; Febbraio, M. The metabolic role of IL-6 produced during exercise: Is IL-6 an exercise factor? Proc. Nutr. Soc. 2004, 63, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Fischer, C.P. Beneficial health effects of exercise—The role of IL-6 as a myokine. Trends Pharmacol. Sci. 2007, 28, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.W.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Fischer, C.P.; Keller, C.; Møller, K.; Pedersen, B.K. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E433–E437. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Febbraio, M.A.; Whitham, M. From cytokine to myokine: The emerging role of interleukin-6 in metabolic regulation. Immunol. Cell Biol. 2014, 92, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cánoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; An, J. Cytokines, inflammation and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Rose-John, S. Interleukin-6 and its receptor: From bench to bedside. Med. Microbiol. Immunol. 2006, 195, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Van Snick, J. Interleukin-6: An overview. Annu. Rev. Immunol. 1990, 8, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.C.; Anderson, M.E.; Moots, R.J. The many faces of interleukin-6: The role of IL-6 in inflammation, vasculopathy, and fibrosis in systemic sclerosis. Int. J. Rheumatol. 2011, 2011, 721608. [Google Scholar] [CrossRef] [PubMed]
- Müllberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Koyanagi, Y.; Zhou, Y.; Miyamoto, H.; Tanaka, Y.; Waki, M.; Matsumoto, A.; Yamamoto, M.; Yamamoto, N. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur. J. Immunol. 1994, 24, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Jacobs, S.; Ehlers, M.; Vollmer, P.; Müllberg, J.; Wolf, E.; Brem, G.; Meyer zum Büschenfelde, K.H.; Rose-John, S. The function of the soluble interleukin 6 (IL-6) receptor in vivo: Sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J. Exp. Med. 1996, 183, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Leggate, M.; Nowell, M.A.; Jones, S.A.; Nimmo, M.A. The response of interleukin-6 and soluble interleukin-6 receptor isoforms following intermittent high intensity and continuous moderate intensity cycling. Cell Stress Chaperones 2010, 15, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Jones, S.A.; Richards, P.J.; Scheller, J.; Rose-John, S. IL-6 transsignaling: The in vivo consequences. J. Interferon Cytokine Res. 2005, 25, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Waetzig, G.H.; Scheller, J.; Grötzinger, J.; Seegert, D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin. Ther. Targets 2007, 11, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S3. [Google Scholar] [CrossRef] [PubMed]
- Modur, V.; Li, Y.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Retrograde inflammatory signaling from neutrophils to endothelial cells by soluble interleukin-6 receptor α. J. Clin. Investig. 1997, 100, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Montero-Julian, F.A.; Grès, S.; Boulay, V.; Bongrand, P.; Farnarier, C.; Kaplanski, G. The IL-6-soluble IL-6Rα autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: An experimental model involving thrombin. J. Immunol. 2001, 167, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Maruo, N.; Morita, I.; Shirao, M.; Murota, S. IL-6 increases endothelial permeability in vitro. Endocrinology 1992, 131, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Erta, M.; Lim, S.L.; Frausto, R.; May, U.; Rose-John, S.; Scheller, J.; Hidalgo, J. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J. Neurosci. 2014, 34, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Keystone, E.; Tony, H.P.; Cantagrel, A.; van Vollenhoven, R.; Sanchez, A.; Alecock, E.; Lee, J.; Kremer, J. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: Results from a 24-week multicentre randomised placebo-controlled trial. Ann. Rheum. Dis. 2008, 67, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ripley, B.; Serada, S.; Naka, T.; Fujimoto, M. Interleukin-6 deficiency does not affect motor neuron disease caused by superoxide dismutase 1 mutation. PLoS ONE 2016, 11, e0153399. [Google Scholar] [CrossRef] [PubMed]
- Patin, F.; Baranek, T.; Vourc’h, P.; Nadal-Desbarats, L.; Goossens, J.-F.; Marouillat, S.; Dessein, A.-F.; Descat, A.; Hounoum, B.M.; Bruno, C.; et al. Combined metabolomics and transcriptomics approaches to assess the IL-6 blockade as a therapeutic of ALS: Deleterious alteration of lipid metabolism. Neurother. J. Am. Soc. Exp. Neurother. 2016, 13, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Saleh, I.A.; Zesiewicz, T.; Xie, Y.; Sullivan, K.L.; Miller, A.M.; Kuzmin-Nichols, N.; Sanberg, P.R.; Garbuzova-Davis, S. Evaluation of humoral immune response in adaptive immunity in ALS patients during disease progression. J. Neuroimmunol. 2009, 215, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Paradis, A.; Leblanc, D.; Dumais, N. Optimization of an in vitro human blood-brain barrier model: Application to blood monocyte transmigration assays. MethodsX 2016, 3, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Müllberg, J.; Dittrich, E.; Graeve, L.; Gerhartz, C.; Yasukawa, K.; Taga, T.; Kishimoto, T.; Heinrich, P.C.; Rose-John, S. Differential shedding of the two subunits of the interleukin-6 receptor. FEBS Lett. 1993, 332, 174–178. [Google Scholar] [CrossRef]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [PubMed]
- Zhang, J.G.; Zhang, Y.; Owczarek, C.M.; Ward, L.D.; Moritz, R.L.; Simpson, R.J.; Yasukawa, K.; Nicola, N.A. Identification and characterization of two distinct truncated forms of gp130 and a soluble form of leukemia inhibitory factor receptor α-chain in normal human urine and plasma. J. Biol. Chem. 1998, 273, 10798–10805. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbuzova-Davis, S.; Ehrhart, J.; Sanberg, P.R.; Borlongan, C.V. Potential Role of Humoral IL-6 Cytokine in Mediating Pro-Inflammatory Endothelial Cell Response in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 423. https://doi.org/10.3390/ijms19020423
Garbuzova-Davis S, Ehrhart J, Sanberg PR, Borlongan CV. Potential Role of Humoral IL-6 Cytokine in Mediating Pro-Inflammatory Endothelial Cell Response in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2018; 19(2):423. https://doi.org/10.3390/ijms19020423
Chicago/Turabian StyleGarbuzova-Davis, Svitlana, Jared Ehrhart, Paul R. Sanberg, and Cesario V. Borlongan. 2018. "Potential Role of Humoral IL-6 Cytokine in Mediating Pro-Inflammatory Endothelial Cell Response in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 19, no. 2: 423. https://doi.org/10.3390/ijms19020423
APA StyleGarbuzova-Davis, S., Ehrhart, J., Sanberg, P. R., & Borlongan, C. V. (2018). Potential Role of Humoral IL-6 Cytokine in Mediating Pro-Inflammatory Endothelial Cell Response in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 19(2), 423. https://doi.org/10.3390/ijms19020423