Nanobody Based Dual Specific CARs

 , ,
, , 
Abstract
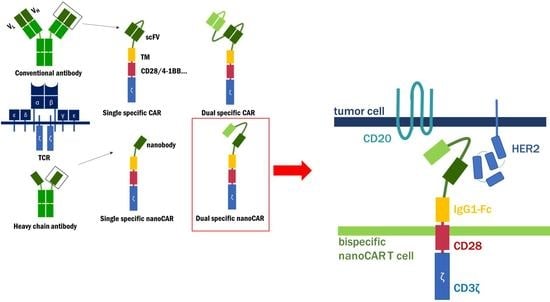
{kind=link}
{kind=link}
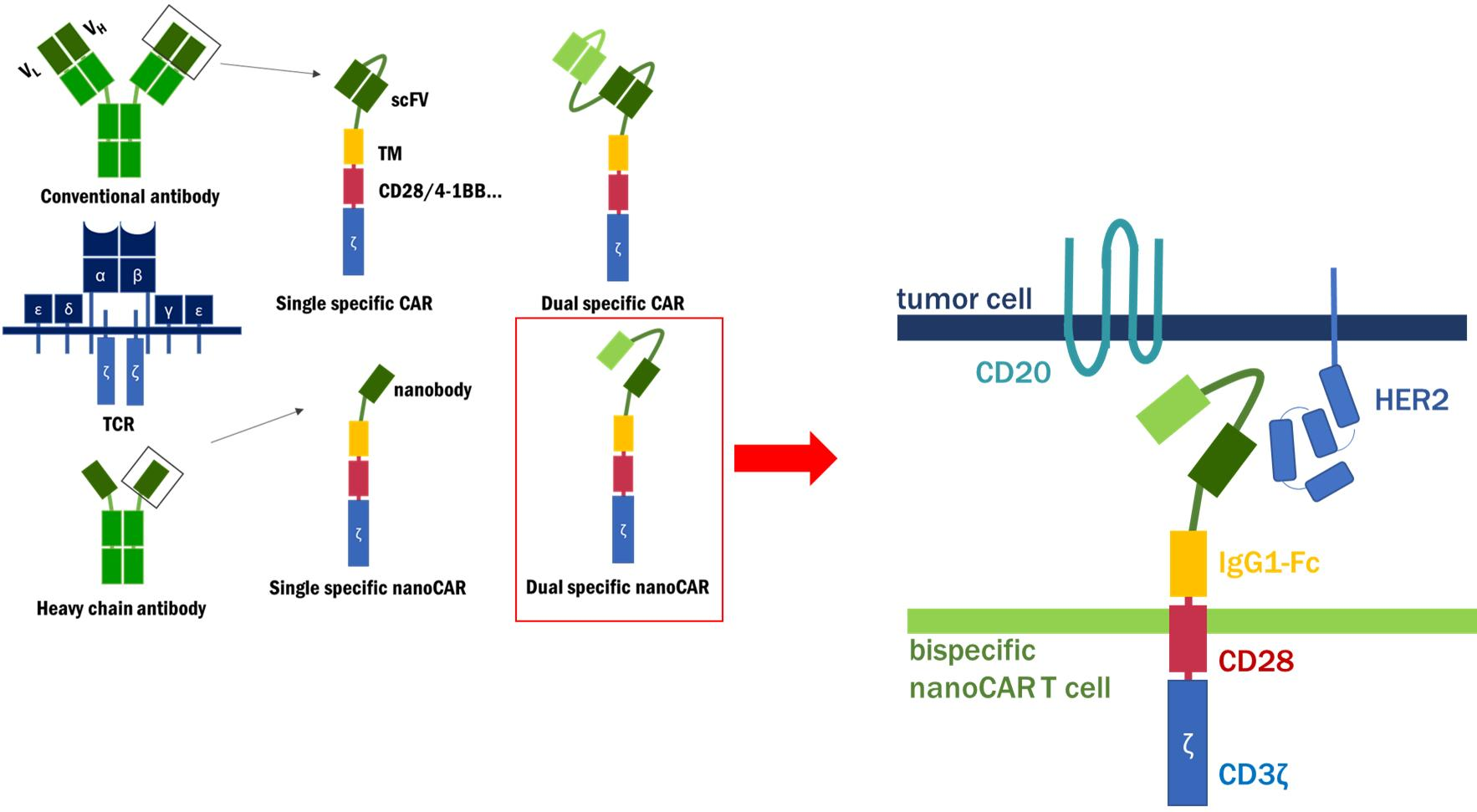
{kind=link}
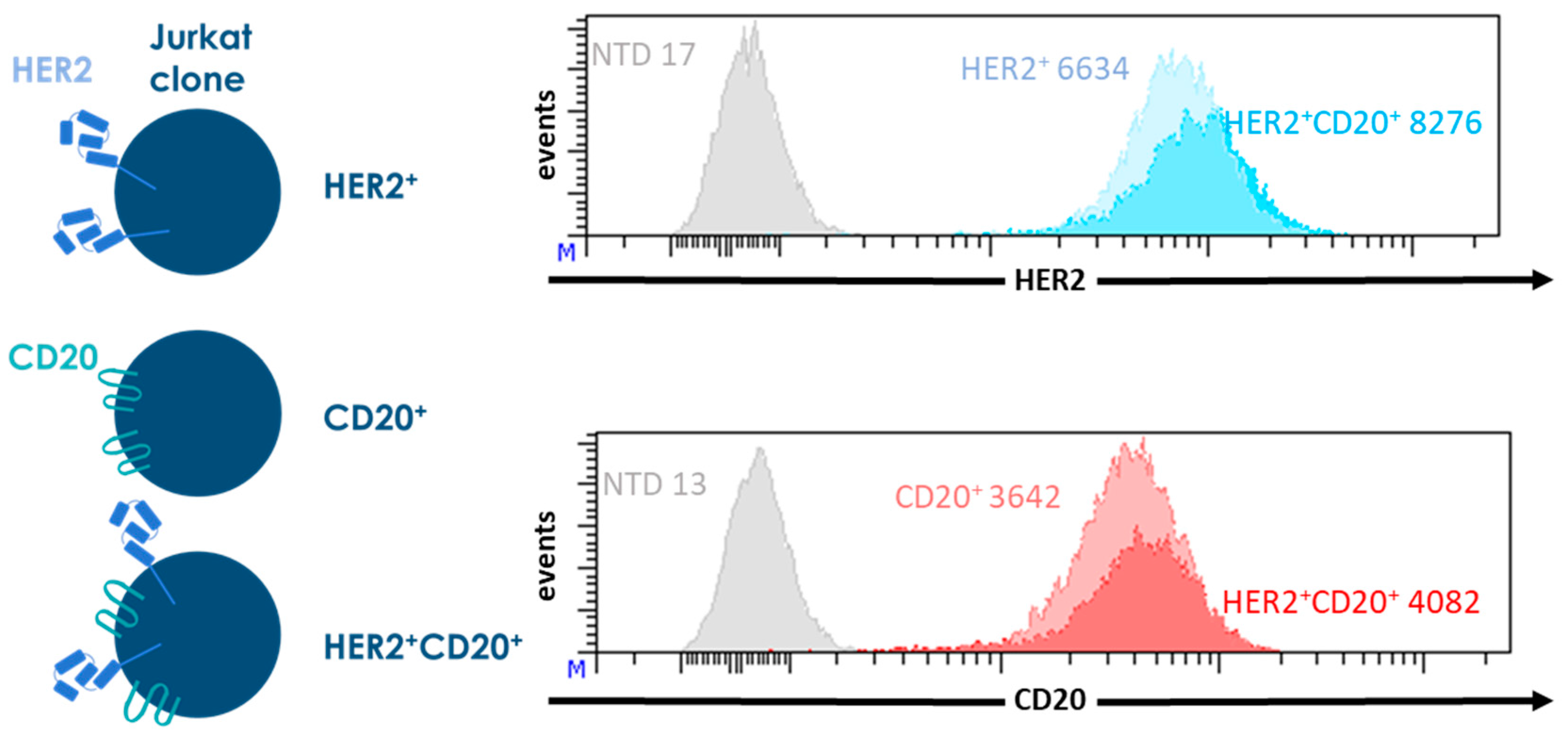
{kind=link}
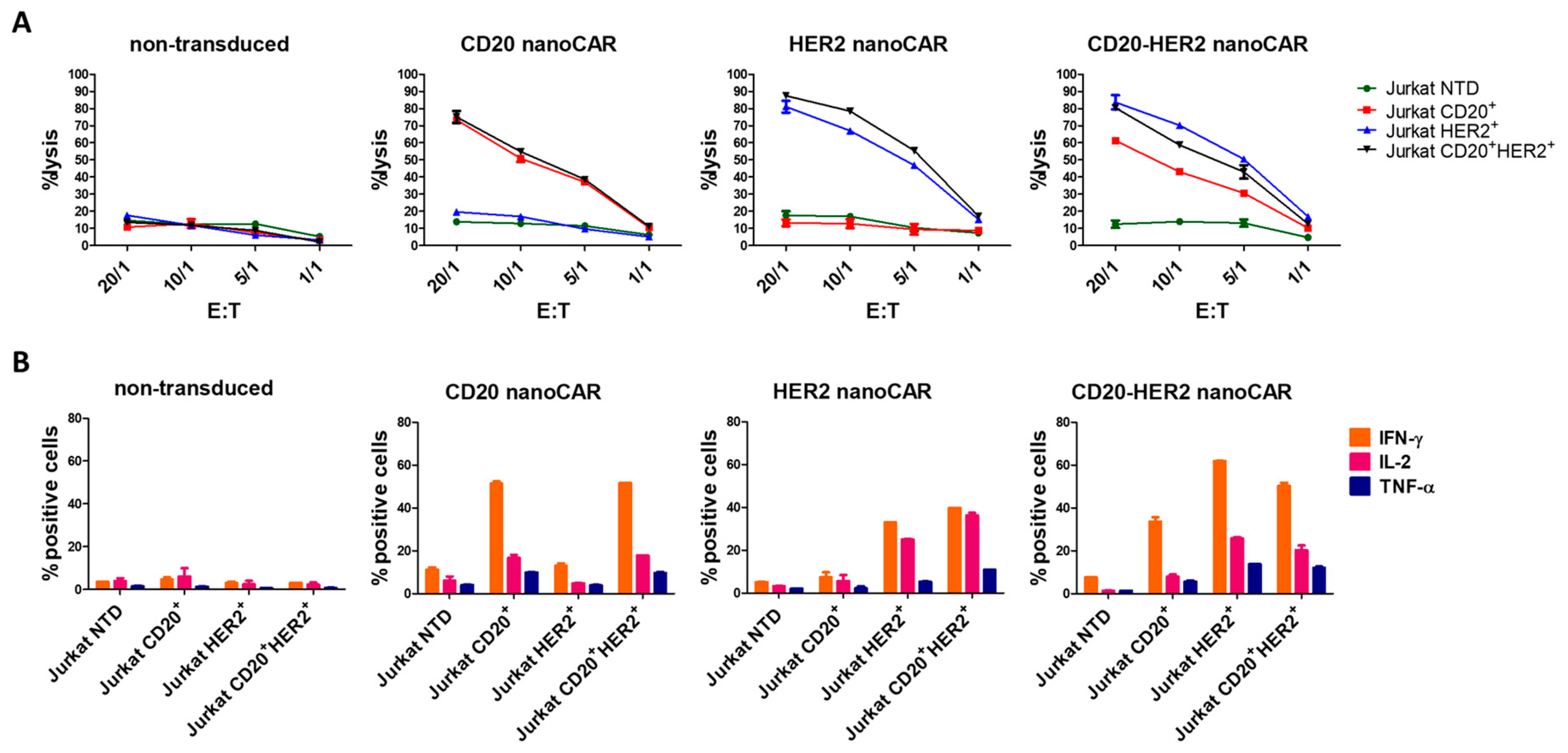
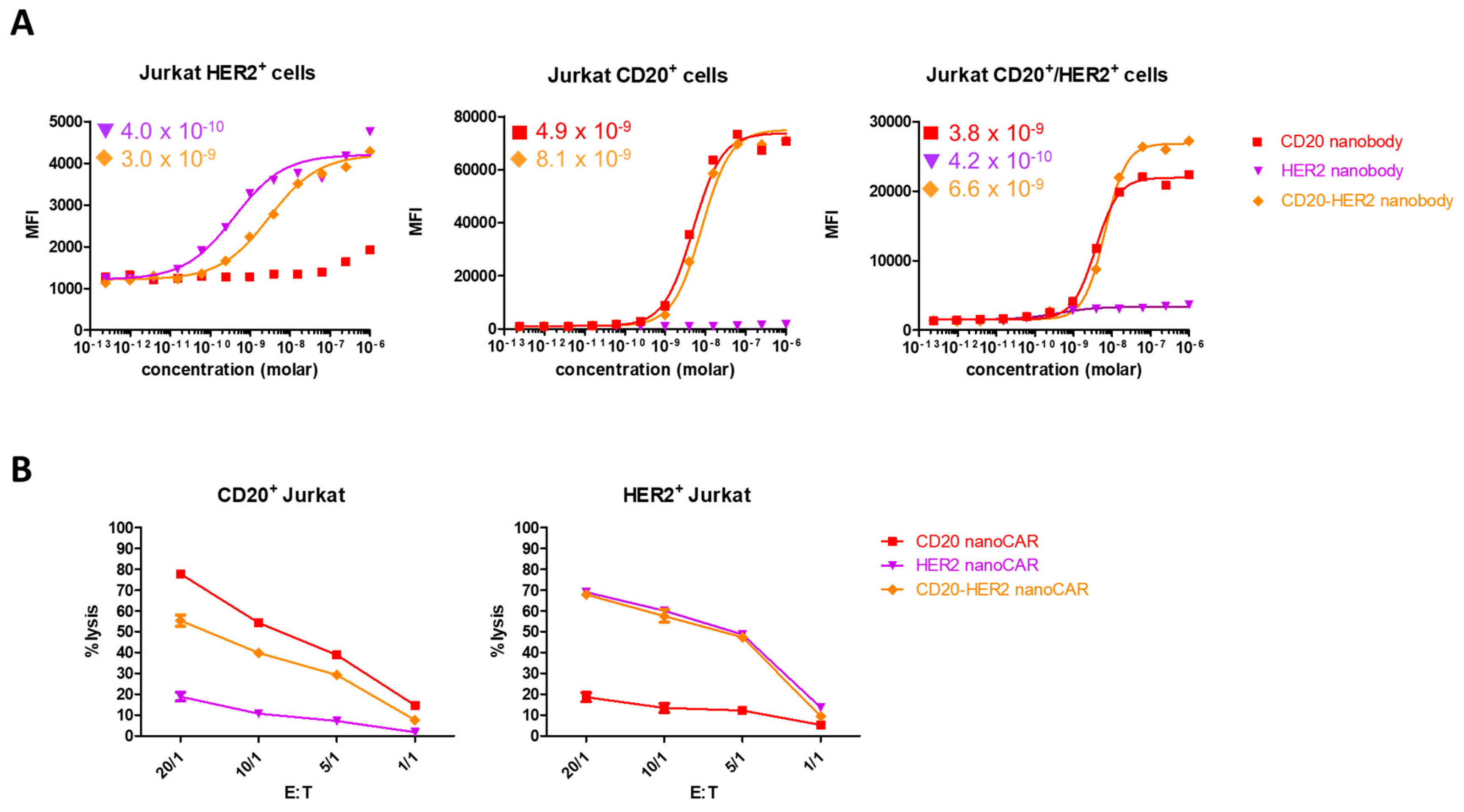{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Culture of Cell Lines
3.2. Production of Retroviral Vectors
3.3. Generation of NanoCAR Expressing Human T Cells
3.4. Flow Cytometry and Antibodies
3.5. 51Chromium Release Assay
3.6. Flowcytometric Determination of Cytokine Production
3.7. Affinity Determination
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR Immune Pressure Induces B-Precursor Acute Lymphoblastic Leukaemia Lineage Switch Exposing Inherent Leukaemic Plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19 Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T Cell Therapy. Blood 2016, 127, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Sadelain, M. Myeloid Leukemia Switch as Immune Escape from CD19 Chimeric Antigen Receptor (CAR) Therapy. Transl. Cancer Res. 2016, 5, S221–S225. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T Cells of Defined CD4+: CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New Development in CAR-T Cell Therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Herlyn, M. Resistance to BRAF Inhibitors: Unraveling Mechanisms and Future Treatment Options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Stong, R.C.; Uckun, F.; Youle, R.J.; Kersey, J.H.; Vallera, D.A. Use of Multiple T Cell-Directed Intact Ricin Immunotoxins for Autologous Bone Marrow Transplantation. Blood 1985, 66, 627–635. [Google Scholar] [PubMed]
- Ghetie, M.A.; Tucker, K.; Richardson, J.; Uhr, J.W.; Vitetta, E.S. The Antitumor Activity of an Anti-CD22 Immunotoxin in SCID Mice with Disseminated Daudi Lymphoma Is Enhanced by Either an Anti-CD19 Antibody or an Anti-CD19 Immunotoxin. Blood 1992, 80, 2315–2320. [Google Scholar] [PubMed]
- Flavell, D.J.; Noss, A.; Pulford, K.A.; Ling, N.; Flavell, S.U. Systemic Therapy with 3BIT, a Triple Combination Cocktail of Anti-CD19, -CD22, and -CD38-Saporin Immunotoxins, Is Curative of Human B-Cell Lymphoma in Severe Combined Immunodeficient Mice. Cancer Res. 1997, 57, 4824–4829. [Google Scholar] [PubMed]
- Jensen, M.C.; Riddell, S.R. Design and Implementation of Adoptive Therapy with Chimeric Antigen Receptor-Modified T Cells. Immunol. Rev. 2014, 257, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Navai, S.A.; Ahmed, N. Targeting the Tumour Profile Using Broad Spectrum Chimaeric Antigen Receptor T-Cells. Biochem. Soc. Trans. 2016, 44, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T Cells Targeting HER2 and IL13R α 2 Mitigate Tumor Antigen Escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.A.; MacKrell, A.J.; Anderson, W.F. Determination of the Binding Affinity of an Anti-CD34 Single-Chain Antibody Using a Novel, Flow Cytometry Based Assay. J. Immunol. Methods 1997, 201, 223–231. [Google Scholar] [CrossRef]
- Kumar, M.; Keller, B.; Makalou, N.; Sutton, R.E. Systematic Determination of the Packaging Limit of Lentiviral Vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Bos, T.J.; De Bruyne, E.; Van Lint, S.; Heirman, C.; Vanderkerken, K. Large Double Copy Vectors Are Functional but Show a Size-Dependent Decline in Transduction Efficiency. J. Biotechnol. 2010, 150, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally Occurring Antibodies Devoid of Light Chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S.; Lauwereys, M. Unique Single-Domain Antigen Binding Fragments Derived from Naturally Occurring Camel Heavy-Chain Antibodies. J. Mol. Recognit. 1999, 12, 131–140. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; De Bruyn, S.; Duby, C.; Verschueren, K.; Baumeister, J.; Sargentini-Maier, L.; Ververken, C.; Holz, J.-B. A Novel Individualized Treatment Approach in Open-Label Extension Study of Ozoralizumab (ATN-103) in Subjects with Rheumatoid Arthritis on a Background of Methotrexate. Arthritis Rheum. 2012, 64, S563. [Google Scholar]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knöbl, P.; Wu, H.; Artoni, A.; Westwood, J.-P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General Strategy to Humanize a Camelid Single-Domain Antibody and Identification of a Universal Humanized Nanobody Scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Nieba, L.; Honegger, A.; Krebber, C.; Pluckthun, A. Disrupting the Hydrophobic Patches at the Antibody Variable/constant Domain Interface: Improved in Vivo Folding and Physical Characterization of an Engineered scFv Fragment. Protein Eng. Des. Sel. 1997, 10, 435–444. [Google Scholar] [CrossRef]
- Sharifzadeh, Z.; Rahbarizadeh, F.; Shokrgozar, M.A.; Ahmadvand, D.; Mahboudi, F.; Jamnani, F.R.; Moghimi, S.M. Genetically Engineered T Cells Bearing Chimeric Nanoconstructed Receptors Harboring TAG-72-Specific Camelid Single Domain Antibodies as Targeting Agents. Cancer Lett. 2013, 334, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Iri-Sofla, F.J.; Rahbarizadeh, F.; Ahmadvand, D.; Rasaee, M.J. Nanobody-Based Chimeric Receptor Gene Integration in Jurkat Cells Mediated by PhiC31 Integrase. Exp. Cell Res. 2011, 317, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Jamnani, F.R.; Rahbarizadeh, F.; Shokrgozar, M.A.; Mahboudi, F.; Ahmadvand, D.; Sharifzadeh, Z.; Parhamifar, L.; Moghimi, S.M. T Cells Expressing VHH-Directed Oligoclonal Chimeric HER2 Antigen Receptors: Towards Tumor-Directed Oligoclonal T Cell Therapy. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, L.; Cui, H.; Wang, X.; Zhang, G.; Ma, J.; Han, H.; He, W.; Wang, W.; Zhao, Y.; et al. Anti-Melanoma Activity of T Cells Redirected with a TCR-like Chimeric Antigen Receptor. Sci. Rep. 2014, 4, 3571. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel Single-Domain Antibodies as Modular Building Units in Bispecific and Bivalent Antibody Constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; Fitzgerald, D.J.; Barrett, D.M.; et al. Anti-CD22—Chimeric Antigen Receptors Targeting B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2013, 121, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; van der Merwe, P.A. The Kinetic-Segregation Model: TCR Triggering and beyond. Nat. Immunol. 2006, 7, 803–809. [Google Scholar] [CrossRef] [PubMed]
- James, J.R.; Vale, R.D. Biophysical Mechanism of T-Cell Receptor Triggering in a Reconstituted System. Nature 2012, 487, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Chang, V.T.; Fernandes, R.A.; Ganzinger, K.A.; Lee, S.F.; Siebold, C.; McColl, J.; Jönsson, P.; Palayret, M.; Harlos, K.; Coles, C.H.; et al. Initiation of T Cell Signaling by CD45 Segregation at “Close Contacts”. Nat. Immunol. 2016, 17, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G. From the Bench to the Bedside: Ways to Improve Rituximab Efficacy. Blood 2004, 104, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-J.; Ord, D.; Omori, S.; Tedder, T. Structure of the Genes Encoding the CD19 Antigen of Human and Mouse B Lymphocytes. Immunogenetics 1992, 35, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Van Caeneghem, Y.; De Munter, S.; Tieppo, P.; Goetgeluk, G.; Weening, K.; Verstichel, G.; Bonte, S.; Taghon, T.; Leclercq, G.; Kerre, T.; et al. Antigen Receptor-Redirected T Cells Derived from Hematopoietic Precursor Cells Lack Expression of the Endogenous TCR/CD3 Receptor and Exhibit Specific Antitumor Capacities. Oncoimmunology 2017, 6, e1283460. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody Based Dual Specific CARs. Int. J. Mol. Sci. 2018, 19, 403. https://doi.org/10.3390/ijms19020403
De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, Kerre T, Abken H, Vandekerckhove B. Nanobody Based Dual Specific CARs. International Journal of Molecular Sciences. 2018; 19(2):403. https://doi.org/10.3390/ijms19020403
Chicago/Turabian StyleDe Munter, Stijn, Joline Ingels, Glenn Goetgeluk, Sarah Bonte, Melissa Pille, Karin Weening, Tessa Kerre, Hinrich Abken, and Bart Vandekerckhove. 2018. "Nanobody Based Dual Specific CARs" International Journal of Molecular Sciences 19, no. 2: 403. https://doi.org/10.3390/ijms19020403
APA StyleDe Munter, S., Ingels, J., Goetgeluk, G., Bonte, S., Pille, M., Weening, K., Kerre, T., Abken, H., & Vandekerckhove, B. (2018). Nanobody Based Dual Specific CARs. International Journal of Molecular Sciences, 19(2), 403. https://doi.org/10.3390/ijms19020403