Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities


Abstract
1. Age as an Important Factor in Preclinical Studies of Stroke
2. Comorbidities as Important Factors in Preclinical Studies of Stroke
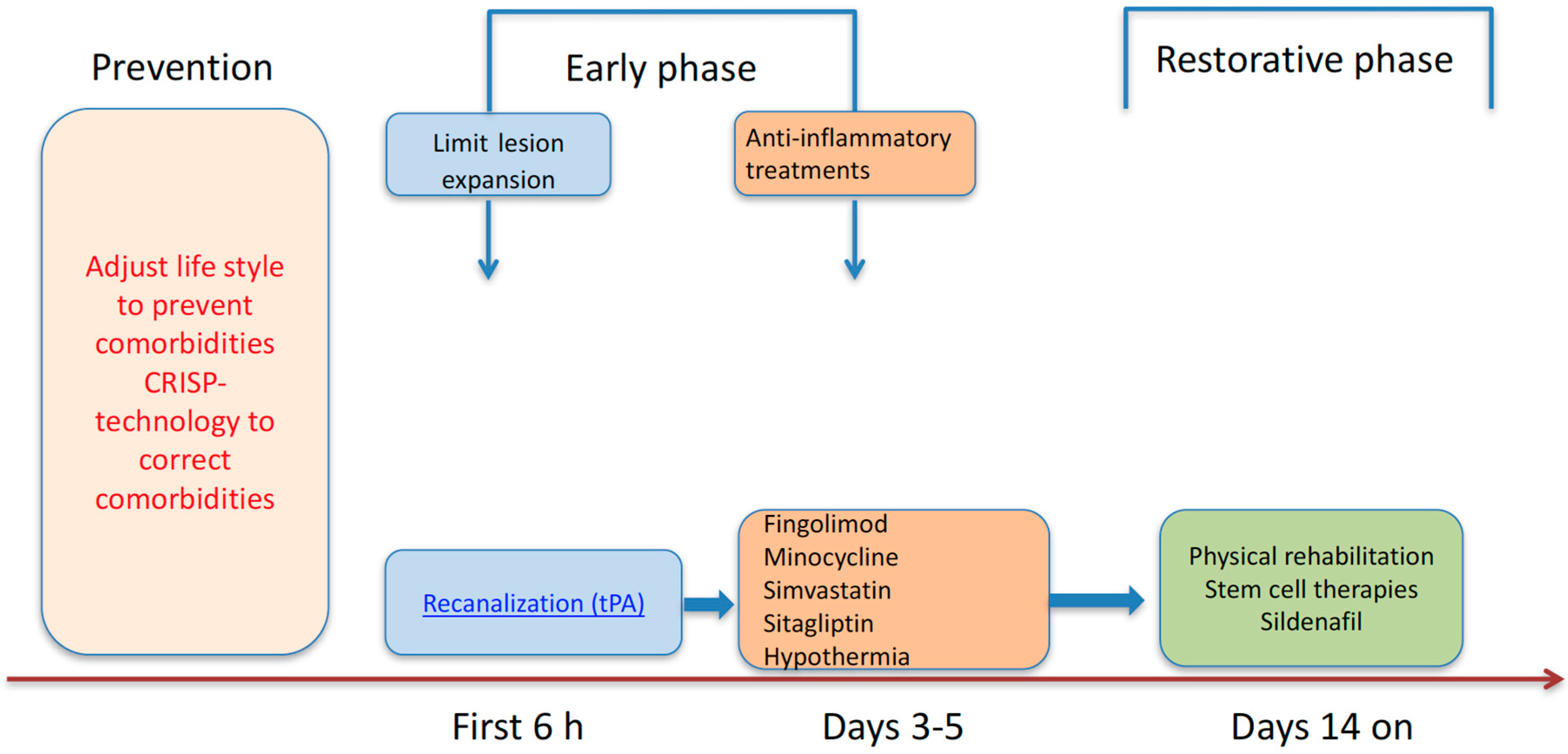
3. Promising New Therapeutic Strategies of Stroke
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Salvioli, S.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Bacalini, M.G.; Garagnani, P.; Giulian, C.; Fontanesi, E.; Ostan, R.; et al. Immune System, Cell Senescence, Aging and Longevity—Inflamm-Aging Reappraised. Curr. Pharm. Des. 2013, 19. [Google Scholar] [CrossRef]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Simen, A.; Bordner, K.; Martin, M.; Moy, L.; Barry, L. Cognitive dysfunction with aging and the role of inflammation. Ther. Adv. Chronic Dis. 2011, 2, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.B.; Zimmer, J.; Sams-Dodd, F. Specific behavioral effects related to age and cerebral ischemia in rats. Pharmacol. Biochem. Behav. 1999, 62, 673–682. [Google Scholar] [CrossRef]
- Davis, M.; Mendelow, A.D.; Perry, R.H.; Chambers, I.R.; James, O.F. Experimental stroke and neuroprotection in the aging rat brain. Stroke 1995, 26, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R.; Dix, G.A.; Auer, R.N. Effect of age in rodent models of focal and forebrain ischemia. Stroke 1996, 27, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.W.; Marlowe, K.J.; Bjelke, B. Age effect on motor recovery in a post-acute animal stroke model. Neurobiol. Aging 2003, 24, 607–614. [Google Scholar] [CrossRef]
- Wang, R.-Y.; Wang, P.; Yang, Y.R. Effect of age in rats following middle cerebral artery occlusion. Gerontology 2003, 49, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, R.L.; Wang, Y.; Zhang, C.; Zhang, Z.G.; Meng, H.; Chopp, M. Functional recovery in aged and young rats after embolic stroke: Treatment with a phosphodiesterase type 5 inhibitor. Stroke 2005, 36, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Xie, L.; Kim, S.H.; Tang, H.; Wang, Y.; Mao, X. Influence of age on the response to fibroblast growth factor-2 treatment in a rat model of stroke. Brain Res. 2006, 1123, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, F.Y. Age-related decrease of striatal neurogenesis is associated with apoptosis of neural precursors and newborn neurons in rat brain after ischemia. Brain Res. 2007, 166, 9–19. [Google Scholar] [CrossRef] [PubMed]
- DiNapoli, V.A.; Huber, J.D.; Houser, K.; Li, X.; Rosen, C.L. Early disruptions of the blood–brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol. Aging 2008, 29, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Wise, P.M.; Dubal, D.B. Estradiol protects against ischemic brain injury in middle-aged rats. Biol. Reprod. 2000, 63, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B.; Wise, P.M. Neuroprotective effects of estradiol in middle-aged female rats. Endocrinology 2001, 142, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.D.; Gribkoff, V.K.; Donlan, N.A.; Jones, T.A. Long-lasting functional disabilities in middle-aged rats with small cerebral infarcts. J. Neurosci. 2003, 23, 10913–10922. [Google Scholar] [PubMed]
- Karki, K.; Knight, R.A.; Shen, L.H.; Kapke, A.; Lu, M.; Li, Y.; Chopp, M. Chronic brain tissue remodeling after stroke in rat: A 1-year multiparametric magnetic resonance imaging study. Brain Res. 2010, 1360, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Brenneman, M.; Sharma, S.; Harting, M.; Strong, R.; Cocs, C.S.; Aronowski, J.; Grotta, J.C.; Savitz, S.I. Autologous bone marrow mononuclear sells enhance recovery after acute ischemic stroke in young and middle-aged rats. J. Cereb. Blood Flow Metab. 2010, 30, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.G.; Mitreva, R.G.; Kortenska, L.V.; Nicoletti, F.; Ngomba, R.T. Age-dependence of sensorimotor and cerebral EEG asymmetry in rats subjected to unilateral cerebrovascular stroke. J. Cereb. Blood Flow Metab. 2013, 5, 13. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Doeppner, T.R.; Hermann, D.M. Stem cell therapies in preclinical models of stroke associated with aging. Front. Cell Neurosci. 2014, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Yanev, P.; Seevinck, P.R.; Rudrapatna, U.S.; Bouts, M.J.; van der Toorn, A.; Gertz, K.; Kronenberg, G.; Endres, M.; van Tilborg, G.A.; Dijkhuizen, R.M. Magnetic resonance imaging of local and remote vascular remodelling after experimental stroke. J. Cereb. Blood Flow Metab. 2017, 37, 2768–2779. [Google Scholar] [CrossRef] [PubMed]
- Canese, R.; Fortuna, S.; Lorenzini, P.; Podo, F.; Michalek, H. Transient global brain ischemia in young and aged rats: Differences in severity and progression, but not localization, of lesions evaluated by magnetic resonance imaging. Magn. Res. Mater. Phys. Biol. Med. 1998, 7, 28–34. [Google Scholar] [CrossRef]
- Canese, R.; Lorenzini, P.; Fortuna, S.; Volpe, M.T.; Giannini, M.; Podo, F.; Michalek, H. Age-dependent MRI-detected lesions at early stages of transient global ischemia in rat brain. MAGMA 2004, 17, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Markus, M.T.; Tsai, S.Y.; Bollnow, M.R.; Farrer, R.G.; O’Brien, T.E.; Kindler-Baumann, D.R.; Rausch, M.; Rudin, M.; Wiessner, C.; Mir, A.K.; et al. Recovery and brain reorganization after stroke in adult and aged rats. Ann. Neurol. 2005, 58, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Macri, M.A.; D’Alessandro, N.; Di Giulio, C.; Di Iorio, P.; Di Luzio, S.; Giuliani, P.; Esposito, E.; Pokoski, M. Region-specific effects on brain metabolites of hypoxia and hyperoxia overlaid on cerebral ischemia in young and old rats: A quantitative proton magnetic resonance spectroscopy study. J. Biomed. Sci. 2010, 17. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Badan, I.; Walker, L.; Groppa, S.; Patrana, N.; Kessler, C. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol. 2007, 113, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Titova, E.M.; Ghosh, N.; Valadez, Z.G.; Zhang, J.H.; Bellinger, D.L.; Obenaus, A. The late phase of post-stroke neurorepair in aged rats is reflected by MRI-based measures. Neuroscience 2014, 283, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Wayman, C.; Duricki, D.A.; Roy, L.A.; Haenzi, B.; Tsai, S.Y.; Kartje, G.; Beech, J.S.; Cash, D.; Moon, L. Performing Permanent Distal Middle Cerebral with Common Carotid Artery Occlusion in Aged Rats to Study Cortical Ischemia with Sustained Disability. J. Vis. Exp. 2016, 53106. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Kim, S.T.; Jung, W.B.; Han, Y.H.; Im, G.H.; Lee, J.H. Altered white matter integrity and functional connectivity of hyperacute-stage cerebral ischemia in a rat model. Magn. Reson. Imaging 2016, 34, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Vintilescu, R.; Balseanu, A.T.; Moldovan, M.; Junker, H.; Walker, L.; Lotze, M.; Popa-Wagner, A. Prolonged gaseous hypothermia prevents the upregulation of phagocytosis-specific protein Annexin 1 and causes low-amplitude EEG activity in the aged rat brain after cerebral ischemia. J. Cereb. Blood Flow Metab. 2012, 32, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.G.; Dijkhuizen, R.M. Present status and future challenges of electroencephalography- and magnetic resonance imaging-based monitoring in preclinical models of focal cerebral ischemia. Brain Res. Bull. 2014, 102, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Moyanova, S.G.; Kortenska, L.V.; Mitreva, R.G.; Pashova, V.D.; Ngomba, R.T.; Nicoletti, F. Multimodal assessment of neuroprotection applied to the use of MK-801 in the endothelin-1 model of transient focal brain ischemia. Brain Res. 2007, 1153, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Sakowitz, O.W.; Fabricius, M.; Bosche, B.; Reithmeier, T.; Ernestus, R.I.; Brinker, G.; Dreier, J.P.; Woitzik, J.; Strong, A.J.; et al. Co-Operative Study of Brain Injury Depolarisations (COSBID). Spreading depolarizations occur in human ischemic stroke with high incidence. Ann. Neurol. 2008, 63, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, Á.; Makra, P.; Szepes, B.É.; Tóth, O.M.; Hertelendy, P.; Bari, F.; Farkas, E. High incidence of adverse cerebral blood flow responses to spreading depolarization in the aged ischemic rat brain. Neurobiol. Aging 2015, 36, 3269–3277. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, M.; Bittner, T.; Jung, C.; Burgold, S.; Page, R.; Mitteregger, G.; Haass, C.; LaFerla, F.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Zhao, J.W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Truman, L.A.; Ford, C.A.; Pasikowska, M.; Pound, J.D.; Wilkinson, S.J.; Dumitriu, I.E.; Melville, L.; Melrose, L.A.; Ogden, C.A.; Nibbs, R.; et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 2008, 112, 5026–5036. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Doi, Y.; Liang, J.; Kawanokuchi, J.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 2011, 286, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Badan, I.; Buchhold, B.; Hamm, A.; Gratz, M.; Walker, L.C.; Platt, D.; Kessler, C.; Popa-Wagner, A. Accelerated glial reactivity to stroke in aged rats correlates with reduced functional recovery. J. Cereb. Blood Flow Metab. 2003, 23, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, J.; Zhang, C.; Liu, M.; Geng, X.; Ji, X.; Du, H.; Zhao, H. Analysis of long non-coding RNA expression profiles following focal cerebral ischemia in mice. Neurosci. Lett. 2017, 665, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pecoraro, R.; Casuccio, A.; Di Raimondo, D.; Buttà, C.; Clemente, G.; Della Corte, V.; Guggino, G.; Arnao, V.; Maida, C.; et al. Peripheral Frequency of CD4+ CD28- Cells in Acute Ischemic Stroke: Relationship With Stroke Subtype and Severity Markers. Medicine (Baltimore) 2015, 94, e813. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pedone, C.; Pinto, A.; Di Raimondo, D.; Fernandez, P.; Di Sciacca, R.; Licata, G. Predictors of outcome in acute ischemiccerebrovascular syndromes: The GIFA study. Int. J. Cardiol. 2008, 125, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Buga, A.M.; Sascau, M.; Pisoschi, C.; Herndon, J.G.; Kessler, C.; Popa-Wagner, A. The genomic response of the ipsilateral and contralateral cortex to stroke in aged rats. J. Cell Mol. Med. 2008, 12, 2731–2753. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Hayes, K.C.; Shi, R. Recombinant human TNFalpha induces concentration-dependent and reversible alterations in the electrophysiological properties of axons in mammalian spinal cord. J. Neurotrauma 2006, 23, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Buga, A.M.; Turner, R.C.; Rosen, C.L.; Toescu, E. Cerebrovascular disorders: Role of aging. J. Aging Res. 2012, 128146. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Herndon, J.G. Mosaic aging. Med. Hypotheses 2010, 74, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Phys. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Marcker, M.L.; Singh, K.K.; Rasmussen, L.J. The importance of mitochondrial DNA in aging and cancer. J. Aging Res. 2001, 407536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Han, Z.; Ji, X.; Luo, Y. Epigenetic Regulation of Oxidative Stress in Ischemic Stroke. Aging Dis. 2016, 7, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Brendan, P.; Patrick, F.C. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1347–1353. [Google Scholar] [CrossRef]
- Buga, A.M.; Scholz, C.J.; Kumar, S.; Herndon, J.G.; Alexandru, D.; Cojocaru, G.R.; Dandekar, T.; Popa-Wagner, A. Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PLoS ONE 2012, 7, e50985. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Xing, P.F.; Zhang, C.Y.; Deng, B.Q. Association of CYP2J2 gene polymorphisms with ischemic stroke and stroke subtypes in Chinese population. Medicine (Baltimore) 2017, 96, e6266. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Chen, M.; Yang, K.; Shao, J.; Fu, Y.; Zhou, W. Association of CD147 genetic polymorphisms with carotid atherosclerotic plaques in a Han Chinese population with cerebral infarction. Thromb. Res. 2017, 156, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zaw, K.T.T.; Sato, N.; Ikeda, S.; Thu, K.S.; Mieno, M.N.; Arai, T.; Mori, S.; Furukawa, T.; Sasano, T.; Sawabe, M.; et al. Association of ZFHX3 gene variation with atrial fibrillation, cerebral infarction, and lung thromboembolism: An autopsy study. J. Cardiol. 2017, 70, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Rigoldi, M.; Concolino, D.; Morrone, A.; Pieruzzi, F.; Ravaglia, R.; Furlan, F.; Santus, F.; Strisciuglio, P.; Torti, G.; Parini, R. Inter-familial and intra familial phenotypic variability in three Sicilian families with Anderson-Fabry disease. Clin. Genet. 2014, 86, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, F.; Yang, Y.; Fu, F.; Xu, C.; Shi, L.; Li, S.; Xia, Y.; Wu, G.; Cheng, X.; et al. Significant association of SNP rs2106261 in the ZFHX3 gene with atrial fibrillation in a Chinese Han GeneID population. Hum. Genet. 2011, 129, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Tortorella, G.; Concolino, D.; Spanò, M.; D’Amico, A.; Bruno, C.; Santorelli, F.M.; Mercuri, E.; Bertini, E. Congenital muscular dystrophy with defective α-dystroglycan, cerebellar hypoplasia, and epilepsy. Neurology 2009, 10, 1599–1601. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Stetler, R.A.; Leak, R.K.; Shi, Y.; Li, Y.; Yu, W.; Bennett, M.V.L.; Chen, J. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hu, X.; Gan, Y.; Gao, Y.; Liang, W.; Chen, J. Mechanistic insight into DNA damage and repair in ischemic stroke: Exploiting the base excision repair pathway as a model of neuroprotection. Antioxid. Redox Signal. 2011, 14, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, C.; Raffoul, J.J.; Ge, Y.; Van Remmen, H.; Matherly, L.H.; Heydari, A.R. Age-related loss of the DNA repair re-sponse following exposure to oxidative stress. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Andressoo, J.O.; de Wit, J.; Huijmans, J.; Beems, R.B.; van Steeg, H.; Weeda, G.; van der Horst, G.T.; van Leeuwen, W.; Themmen, A.P.; et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002, 296, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004, 36, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Ueda, H.; Seiyama, A.; Seki, J.; Konaka, K.; Yanagida, T.; Sakoda, S.; Yanagihara, T. Ischemic vasoconstriction and tissue energy metabolism during global cerebral ischemia in gerbils. J. Neurotrauma 2007, 24, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.K.Y.; Rosenblatt, Y.; Brock, G.B.; O’Gorman, D.B.; Gan, B.S. Phosphodiesterase inhibitors in vascular ischemia: A case report and review of their use in ischemic conditions. Can. J. Plast. Surg. 2010, 18, e5–e9. [Google Scholar] [PubMed]
- Farkas, E.; de Vos, R.A.; Donka, G.; Jansen Steur, E.N.; Mihály, A.; Luiten, P.G. Age-related microvascular degeneration in the human cerebral periventricular white matter. Acta Neuropathol. 2006, 111, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Navaratna, D.; Guo, S.; Arai, K.; Lo, E.H. Mechanisms and targets for angiogenic therapy after stroke. Cell Adhes. Migr. 2009, 3, 216–223. [Google Scholar] [CrossRef]
- Font, M.A.; Arboix, A.; Krupinski, J. Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr. Cardiol. Rev. 2010, 6, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Sandu, R.E.; Uzoni, A.; Ciobanu, O.; Moldovan, M.; Anghel, A.; Radu, E.; Coogan, A.N.; Popa-Wagner, A. Post-stroke gaseous hypothermia increases vascular density but not neurogenesis in the ischemic penumbra of aged rats. Restor. Neurol. Neurosci. 2016, 34, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Kumar, P.; Gaffney, J.; Kumar, S.; Krupinsky, J. Can angiogenesis be exploited to improve stroke outcome? Mechanisms and therapeutic potential. Clin. Sci. 2006, 111, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Persichetti, E.; Chuzhanova, N.A.; Dardis, A.; Tappino, B.; Pohl, S.; Thomas, N.S.; Rosano, C.; Balducci, C.; Paciotti, S.; Dominissini, S.; et al. Identification and molecular characterization of six novel mutations in the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTG) gene in patients with mucolipidosis III gamma. Hum. Mutat. 2009, 30, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Lanterna, L.A.; Rigoldi, M.; Tredici, G.; Biroli, F.; Cesana, C.; Gaini, S.M.; Dalprà, L. APOE influences vasospasm and cognition of noncomatose patients with subarachnoid hemorrhage. Neurology. 2005, 64, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Apple, D.M.; Kokovay, E. Vascular niche contribution to age-associated neural stem cell dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H896–H902. [Google Scholar] [CrossRef] [PubMed]
- Darsalia, V.; Heldmann, U.; Lindvall, O.; Kokaia, Z. Stroke-Induced Neurogenesis in Aged Brain. Stroke 2005, 36, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Badan, I.; Dinca, I.; Buchhold, B.; Suofu, Y.; Walker, L.; Gratz, M.; Platt, D.; Kessler, C.H.; Popa-Wagner, A. Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur. J. Neurosci. 2004, 19, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, J.; Aswendt, M.; Kreutzer, C.; Rotheneichner, P.; Riou, A.; Selt, M.; Beyrau, A.; Uhlenküken, U.; Diedenhofen, M.; Nelles, M.; et al. Neurogenesis upregulation on the healthy hemisphere after stroke enhances compensation for age-dependent decrease of basal neurogenesis. Neurobiol. Dis. 2017, 99, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Martín, S.; Rojas, A.; Chamorro, A.; Falcón, C.; Bargalló, N.; Planas, A.M. Why does acute hyperglycemia worsen the outcome of transient focal cerebral ischemia? Role of corticosteroids, inflammation, and protein O-glycosylation. Stroke 2006, 37, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Rewell, S.; Fernandez, J.; Cox, S.; Spratt, N.; Hogan, L.; Aleksoska, E.; van Raay, L.; Liberatore, G.; Batchelor, P.; Howells, D. Inducing stroke in aged, hypertensive, diabetic rats. J. Cereb. Blood Flow Metab. 2010, 30, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Liu, T. Inflammator y cause of metabolic syndrome via brain stress and NF-κB. Aging 2012, 4, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhan, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, A.M.; Lenighan, Y.M.; O’Reilly, M.E.; McGillicuddy, F.C.; Roche, H.M. Nutritional modulation of metabolic inflammation. Biochem. Soc. Trans. 2017, 45, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, D.M.; Han, Y.; Yang, D.; Ding, J.; Savant-Bhonsale, S. Mannitol enhances delivery of marrow stromal cells to the brain after experimental intracerebral hemorrhage. Brain Res. 2008, 11, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Ayala-Grosso, C.A.; Ganguly, M.; Jordão, J.F.; Aubert, I.; Hynynen, K. Targeted delivery of neural stem cells to the brain using MRI-guided focused ultrasound to disrupt the blood-brain barrier. PLoS ONE 2011, 6, e27877. [Google Scholar] [CrossRef] [PubMed]
- Bible, E.; Dell’Acqua, F.; Solanky, B.; Balducci, A.; Crapo, P.M.; Badylak, S.F.; Ahrens, E.T.; Modo, M. Non-invasive imaging of transplanted human neural stem cells and ECM scaffold remodeling in the stroke-damaged rat brain by (19)F- and diffusion-MRI. Biomaterials 2012, 33, 2858–2871. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, H.J.; Görgens, A.; Schlechter, J.; Ludwig, A.K.; Radtke, S.; de Miroschedji, K.; Horn, P.A.; Giebel, B.; Hermann, D.M. Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl. Med. 2015, 4, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Manzanero, S.; Gelderblom, M.; Magnus, T.; Arumugam, T. Calorie restriction and stroke. Exp. Transl. Stroke Med. 2011, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Bendlin, B.B.; McLaren, D.G.; Canu, E.; Kastman, E.K.; Kosmatka, K.J.; Xu, G.; Field, A.S.; Alexander, A.L.; Colman, R.J.; et al. Age-related changes in neural volume and microstructure associated with interleukin-6 are ameliorated by a calorie-restricted diet in old rhesus monkeys. Neuroimage 2010, 51, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, O.; Sandu, R.E.; Balseanu, A.T.; Zavaleanu, A.; Gresita, A.; Petcu, E.B.; Uzoni, A.; Popa-Wagner, A. Caloric restriction stabilizes body weight and accelerates behavioral recovery in aged rats after focal ischemia. Aging Cell. 2017, 16, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Elsaesser, R.; Hungerhuber, E.; Zausinger, S.; Baethmann, A.; Reulen, H.J. Combination drug therapy and mild hypothermia. A promising treatment strategy for reversible, focal cerebral ischemia. Stroke 1999, 30, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Sandu, R.E.; Balseanu, A.T.; Bogdan, C.; Slevin, M.; Petcu, E.; Popa-Wagner, A. Stem cell therapies in preclinical models of stroke. Is the aged brain microenvironment refractory to cell therapy? Exp. Gerontol. 2017, 94, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Amico, L.; Cappellini, M.D.; Cassinerio, E.; Conti, M.; Donati, M.A.; Falvo, F.; Fiumara, A.; Maccarone, M.; Manna, R.; et al. Home infusion program with enzyme replacement therapy for Fabry disease: The experience of a large Italian collaborative group. Mol. Genet. Metab. Rep. 2017, 12, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lee, J.; Zhang, M. Magnetic nanoparticles in MR imaging and drug delivery. Atherosclerosis 2008, 209, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Deddens, L.H.; Van Tilborg, G.A.; Mulder, W.J.; De Vries, H.E.; Dijkhuizen, R.M. Imaging neuroinflammation after stroke: Current status of cellular and molecular MRI strategies. Cereb. Dis. 2012, 33, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Musunuru, K. Genome Editing for the Study of Cardiovascular Diseases. Curr. Cardiol. Rep. 2017, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Musunuru, K. CRISPR-Cas9 Genome Editing for Treatment of Atherogenic Dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Carmichael, S.T.; Kokaia, Z.; Kessler, C.; Walker, L.C. The response of the aged brain to stroke: Too much, too soon? Curr. Neurovasc. Res. 2007, 4, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Dinca, I.; Yalikun, S.; Walker, L.; Kroemer, H.; Kessler, C. Accelerated delimitation of the infarct zone by capillary-derived nestin-positive cells in aged rats. Curr. Neurovasc. Res. 2006, 3, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Buga, A.M.; Margaritescu, C.; Scholz, C.J.; Radu, E.; Zelenak, C.; Popa-Wagner, A. Transcriptomics of post-stroke angiogenesis in the aged brain. Front. Aging Neurosci. 2014, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, Z.; Liu, T.; Zhao, M.; Zhao, S.; Xiao, T.; Jolkkonen, J.; Zhao, C. Fluoxetine Enhances Neurogenesis in Aged Rats with Cortical Infarcts, but This is not Reflected in a Behavioral Recovery. J. Mol. Neurosci. 2016, 58, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.L.; Zhao, M.; Zhao, S.S.; Xiao, T.; Song, C.G.; Cao, Y.P.; Jolkkonen, J.; Zhao, C.S. Forced limb-use enhanced neurogenesis and behavioral recovery after stroke in the aged rats. Neuroscience 2015, 286, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.S.; Puurunen, K.; Schallert, T.; Sivenius, J.; Jolkkonen, J. Behavioral effects of photothrombotic ischemic cortical injury in aged rats treated with the sedative-hypnotic GABAergic drug zopiclone. Behav. Brain Res. 2005, 160, 260–266. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Event | Main Findings in the Aged Rat Brains | References |
|---|---|---|
| Neuroinflammation | Fulminant development of astroglial response; increased BBB permeability | [12,43,49] |
| Cell death | Accelerated apoptosis; Microglial engulfment of dying neurons | [26,38,39] |
| Early gene expression | Decreased number of transiently upregulated genes; Early upregulation of genes associated with DNA damage and down-regulation of anti-apoptosis related genes; Increased number of genes associated with phagocytosis; Increased number of genes coding for pro-inflammatory mediators; Persistent upregulation of genes encoding for extracellular matrix degradation | [47,55] |
| Infarct development | Precipitous development of the infarct | [26,100,101] |
| Late gene expression | Increased number of downregulated genes Delayed expression of angiogenesis-related genes Increased no of genes involved in fibrosis and scar build-up Disregulation of gene expression for brain development and CNS remodeling | [47,55,72,102] |
| Angiogenesis | Delayed sprouting angiogenesis and basal lamina build-up, decreased vascular density in the periinfarcted area but increased vascular density beyond the scar region | [72,102] |
| Neurogenesis | Impaired neurogenesis and decreased expression of the neuronal precursor marker, doublecortin | [71,72,77,79] |
| Behavioral recovery | Limited behavioral (motor, sensory, working memory) recovery | [26,78,92,103,104,105] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa-Wagner, A.; Glavan, D.-G.; Olaru, A.; Olaru, D.-G.; Margaritescu, O.; Tica, O.; Surugiu, R.; Sandu, R.E. Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities. Int. J. Mol. Sci. 2018, 19, 356. https://doi.org/10.3390/ijms19020356
Popa-Wagner A, Glavan D-G, Olaru A, Olaru D-G, Margaritescu O, Tica O, Surugiu R, Sandu RE. Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities. International Journal of Molecular Sciences. 2018; 19(2):356. https://doi.org/10.3390/ijms19020356
Chicago/Turabian StylePopa-Wagner, Aurel, Daniela-Gabriela Glavan, Andrei Olaru, Denissa-Greta Olaru, Otilia Margaritescu, Oana Tica, Roxana Surugiu, and Raluca Elena Sandu. 2018. "Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities" International Journal of Molecular Sciences 19, no. 2: 356. https://doi.org/10.3390/ijms19020356
APA StylePopa-Wagner, A., Glavan, D.-G., Olaru, A., Olaru, D.-G., Margaritescu, O., Tica, O., Surugiu, R., & Sandu, R. E. (2018). Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities. International Journal of Molecular Sciences, 19(2), 356. https://doi.org/10.3390/ijms19020356