The Stress-Inducible Protein DRR1 Exerts Distinct Effects on Actin Dynamics

 ,
, 
Abstract
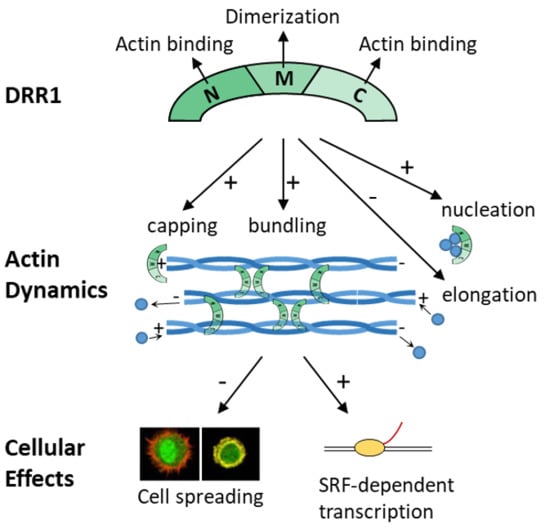
1. Introduction
2. Results
2.1. DRR1 Features an Actin Binding Site at Each Terminus
2.2. DRR1 Enhances Actin Bundling Via Its Two Actin Binding Regions and Potentially through Homo-Dimerization
2.3. DRR1 Bundling Diminishes Cellular Actin Treadmilling
2.4. DRR1 Reduces Actin Filament Elongation but Increases Nucleation
2.5. In the Presence of Profilin, DRR1, and the Mutants dM and dC Block Elongation More Effectively, Suggesting DRR1 as a Novel Barbed End Capping Factor
2.6. DRR1 Modulates Actin-Dependent Processes in Cells
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Culture and Transfection
4.3. SDS-PAGE, Colloidal Coomassie Staining, and Immunoblot
4.4. Protein Expression and Purification
4.5. Co-Immunoprecipitation
4.6. Dual Luciferase Reporter Assay
4.7. Chemical Crosslinking
4.8. Subcellular Fractionation
4.9. HeLa Cell Spreading
4.10. Fluorescence Recovery after Photobleaching (FRAP)
4.11. Cellular Stainings
4.12. Colocalization Analysis
4.13. Quantification of Mean Cellular F-actin Content
4.14. Actin Preparation
4.15. Pyrene-Actin Polymerization Assay
4.16. Actin-Filament Elongation and Nucleation Assay
4.17. Reconstituted Actin Networks
4.18. F-Actin Co-Sedimentation Assay
4.19. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variants |
| ATP | Adenosine triphosphate |
| BCA | Bicinchoninic acid |
| BMB | 1,4-Bismaleimidobutane |
| BSA | Bovine serum albumin |
| CA3 | Cornu Ammonis region 3 (hippocampal region) |
| CaMKIIb | Ca2+/calmodulin-dependent protein kinase IIb |
| CMV | Cytomegalovirus |
| CoIP | Co-immunoprecipitation |
| DRR1 | Down-regulated in renal cell carcinoma 1 (also known as FAM107A or TU3A) |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
| EGFP | Enhanced green fluorescent protein |
| F-actin | Filamentous actin |
| FAM107A | Family with sequence similarity 107, member A |
| G-actin | Globular actin |
| GFP | Green fluorescent protein |
| HEK-293 | Human embryonic kidney 293 cells |
| MBP | Maltose binding protein |
| PCC | Pearson’s correlation coefficient |
| PMSF | Phenylmethane sulfonyl fluoride |
| R | Molar ratio DRR1 (wt or mutants):actin |
| RT | Room temperature |
| SDS | Sodium dodecyl sulfate |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SRF | Serum response factor |
| TIRF | Total internal reflection fluorescence |
| TU3A | Tohoku University cDNA clone A on chromosome 3 |
| wt | Wild-type |
Appendix A





References
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Fenster, R.J.; Lebois, L.A.M.; Ressler, K.J.; Suh, J. Brain circuit dysfunction in post-traumatic stress disorder: From mouse to man. Nat. Rev. Neurosci. 2018, 19, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, L.A.; Goda, Y. Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008, 9, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooij, M.A.; Masana, M.; Rust, M.B.; Müller, M.B. The stressed cytoskeleton: How actin dynamics can shape stress-related consequences on synaptic plasticity and complex behavior. Neurosci. Biobehav. Rev. 2016, 62, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Flügge, G. Stress, glucocorticoids and structural plasticity of the hippocampus. Neurosci. Biobehav. Rev. 1998, 23, 295–300. [Google Scholar] [CrossRef]
- Kasai, H.; Fukuda, M.; Watanabe, S.; Hayashi-Takagi, A.; Noguchi, J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010, 33, 121–129. [Google Scholar] [CrossRef]
- Yan, Z.; Kim, E.; Datta, D.; Lewis, D.A.; Soderling, S.H. Synaptic Actin Dysregulation, a Convergent Mechanism of Mental Disorders? J. Neurosci. 2016, 36, 11411–11417. [Google Scholar] [CrossRef]
- Davidson, A.J.; Wood, W. Unravelling the Actin Cytoskeleton: A New Competitive Edge? Trends Cell Biol. 2016, 26, 569–576. [Google Scholar] [CrossRef]
- Kuhn, J.R.; Pollard, T.D. Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophys. J. 2005, 88, 1387–1402. [Google Scholar] [CrossRef]
- Harris, E.S.; Higgs, H.N. Biochemical analysis of mammalian formin effects on actin dynamics. Methods Enzymol. 2006, 406, 190–214. [Google Scholar] [PubMed]
- Dominguez, R. Structural insights into de novo actin polymerization. Curr. Opin. Struct. Biol. 2010, 20, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.D.; Heuser, J.A.; Pollard, T.D. The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA 1998, 95, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Pollard, T.D. Effect of capping protein on the kinetics of actin polymerization. Biochemistry 1985, 24, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.; Zwolak, A.; Schafer, D.A.; Sept, D.; Dominguez, R.; Cooper, J.A. Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Jayo, A.; Parsons, M. Fascin: A key regulator of cytoskeletal dynamics. Int. J. Biochem. Cell Biol. 2010, 42, 1614–1617. [Google Scholar] [CrossRef]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adh. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef]
- Sjöblom, B.; Salmazo, A.; Djinovic-Carugo, K. Alpha-actinin structure and regulation. Cell Mol. Life Sci. 2008, 65, 2688–2701. [Google Scholar] [CrossRef]
- Posern, G.; Treisman, R. Actin’ together: Serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 2006, 16, 588–596. [Google Scholar] [CrossRef]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef]
- Liebl, C.; Panhuysen, M.; Putz, B.; Trumbach, D.; Wurst, W.; Deussing, J.M.; Muller, M.B.; Schmidt, M.V. Gene expression profiling following maternal deprivation: Involvement of the brain Renin-Angiotensin system. Front. Mol. Neurosci. 2009, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Masana, M.; Su, Y.A.; Liebl, C.; Wang, X.D.; Jansen, L.; Westerholz, S.; Wagner, K.V.; Labermaier, C.; Scharf, S.H.; Santarelli, S.; et al. The stress-inducible actin-interacting protein DRR1 shapes social behavior. Psychoneuroendocrinology 2014, 48, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.V.; Schülke, J.P.; Liebl, C.; Stiess, M.; Avrabos, C.; Bock, J.; Wochnik, G.M.; Davies, H.A.; Zimmermann, N.; Scharf, S.H.; et al. Tumor suppressor down-regulated in renal cell carcinoma 1 (DRR1) is a stress-induced actin bundling factor that modulates synaptic efficacy and cognition. Proc. Natl. Acad. Sci. USA 2011, 108, 17213–17218. [Google Scholar] [CrossRef] [PubMed]
- Yamato, T.; Orikasa, K.; Fukushige, S.; Orikasa, S.; Horii, A. Isolation and characterization of the novel gene, TU3A, in a commonly deleted region on 3p14.3-->p14.2 in renal cell carcinoma. Cytogenet. Cell Genet. 1999, 87, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Darling, J.; Zhang, J.S.; Liu, W.; Qian, J.; Bostwick, D.; Hartmann, L.; Jenkins, R.; Bardenhauer, W.; Schutte, J.; et al. Loss of expression of the DRR 1 gene at chromosomal segment 3p21.1 in renal cell carcinoma. Genes Chromosom. Cancer 2000, 27, 1–10. [Google Scholar] [CrossRef]
- Udali, S.; Guarini, P.; Ruzzenente, A.; Ferrarini, A.; Guglielmi, A.; Lotto, V.; Tononi, P.; Pattini, P.; Moruzzi, S.; Campagnaro, T.; et al. DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clin. Epigenetics 2015, 7, 43. [Google Scholar] [CrossRef]
- Kiwerska, K.; Szaumkessel, M.; Paczkowska, J.; Bodnar, M.; Byzia, E.; Kowal, E.; Kostrzewska-Poczekaj, M.; Janiszewska, J.; Bednarek, K.; Jarmuz-Szymczak, M.; et al. Combined deletion and DNA methylation result in silencing of FAM107A gene in laryngeal tumors. Sci. Rep. 2017, 7, 5386. [Google Scholar] [CrossRef]
- Lawrie, A.; Han, S.; Sud, A.; Hosking, F.; Cezard, T.; Turner, D.; Clark, C.; Murray, G.I.; Culligan, D.J.; Houlston, R.S.; et al. Combined linkage and association analysis of classical Hodgkin lymphoma. Oncotarget 2018, 9, 20377–20385. [Google Scholar] [CrossRef]
- Pastuszak-Lewandoska, D.; Czarnecka, K.H.; Migdalska-Sek, M.; Nawrot, E.; Domanska, D.; Kiszalkiewicz, J.; Kordiak, J.; Antczak, A.; Gorski, P.; Brzezianska-Lasota, E. Decreased FAM107A Expression in Patients with Non-small Cell Lung Cancer. Adv. Exp. Med. Biol. 2015, 852, 39–48. [Google Scholar]
- Mu, P.; Akashi, T.; Lu, F.; Kishida, S.; Kadomatsu, K. A novel nuclear complex of DRR1, F-actin and COMMD1 involved in NF-kappaB degradation and cell growth suppression in neuroblastoma. Oncogene 2017, 36, 5745–5756. [Google Scholar] [CrossRef]
- Asano, Y.; Kishida, S.; Mu, P.; Sakamoto, K.; Murohara, T.; Kadomatsu, K. DRR1 is expressed in the developing nervous system and downregulated during neuroblastoma carcinogenesis. Biochem. Biophys. Res. Commun. 2010, 394, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Fèvre-Montange, M.; Champier, J.; Durand, A.; Wierinckx, A.; Honnorat, J.; Guyotat, J.; Jouvet, A. Microarray gene expression profiling in meningiomas: Differential expression according to grade or histopathological subtype. Int. J. Oncol. 2009, 35, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Kholodnyuk, I.D.; Kozireva, S.; Kost-Alimova, M.; Kashuba, V.; Klein, G.; Imreh, S. Down regulation of 3p genes, LTF, SLC38A3 and DRR1, upon growth of human chromosome 3-mouse fibrosarcoma hybrids in severe combined immunodeficiency mice. Int. J. Cancer 2006, 119, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, X.Y.; Bai, R.Z.; Liang, S.F.; Nie, C.L.; Yuan, Z.; Wang, C.T.; Wu, Y.; Chen, L.J.; Wei, Y.Q. Induction of tumor inhibition and apoptosis by a candidate tumor suppressor gene DRR1 on 3p21.1. Oncol. Rep. 2009, 22, 1069–1075. [Google Scholar] [PubMed]
- Vanaja, D.K.; Ballman, K.V.; Morlan, B.W.; Cheville, J.C.; Neumann, R.M.; Lieber, M.M.; Tindall, D.J.; Young, C.Y. PDLIM4 repression by hypermethylation as a potential biomarker for prostate cancer. Clin. Cancer Res. 2006, 12, 1128–1136. [Google Scholar] [CrossRef]
- Van den, B.J.; Wolter, M.; Blaschke, B.; Knobbe, C.B.; Reifenberger, G. Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int. J. Cancer 2006, 119, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular identity of human outer radial glia during cortical development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef]
- Ma, Y.S.; Wu, Z.J.; Bai, R.Z.; Dong, H.; Xie, B.X.; Wu, X.H.; Hang, X.S.; Liu, A.N.; Jiang, X.H.; Wang, G.R.; et al. DRR1 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via regulating AKT activation. Cancer Lett. 2018, 423, 86–94. [Google Scholar] [CrossRef]
- Le, P.U.; Angers-Loustau, A.; de Oliveira, R.M.; Ajlan, A.; Brassard, C.L.; Dudley, A.; Brent, H.; Siu, V.; Trinh, G.; Molenkamp, G.; et al. DRR drives brain cancer invasion by regulating cytoskeletal-focal adhesion dynamics. Oncogene 2010, 29, 4636–4647. [Google Scholar] [CrossRef]
- Quartier, A.; Chatrousse, L.; Redin, C.; Keime, C.; Haumesser, N.; Maglott-Roth, A.; Brino, L.; Le, G.S.; Benchoua, A.; Mandel, J.L.; et al. Genes and Pathways Regulated by Androgens in Human Neural Cells, Potential Candidates for the Male Excess in Autism Spectrum Disorder. Biol. Psychiatry 2018, 84, 239–252. [Google Scholar] [CrossRef]
- Wan, B.; Feng, P.; Guan, Z.; Sheng, L.; Liu, Z.; Hua, Y. A severe mouse model of spinal muscular atrophy develops early systemic inflammation. Hum. Mol. Genet. 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, M.D.; Burns, T.C.; Morgan, A.A.; Khatri, P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol. Commun. 2014, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Vawter, M.P. Shared gene expression alterations in schizophrenia and bipolar disorder. Biol. Psychiatry 2008, 64, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ranno, E.; D’Antoni, S.; Spatuzza, M.; Berretta, A.; Laureanti, F.; Bonaccorso, C.M.; Pellitteri, R.; Longone, P.; Spalloni, A.; Iyer, A.M.; et al. Endothelin-1 is over-expressed in amyotrophic lateral sclerosis and induces motor neuron cell death. Neurobiol. Dis. 2014, 65, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.M.; Lee, S.; Baumer, D.; Parson, S.H.; Talbot, K.; Gillingwater, T.H. Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet. 2010, 19, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Salameh, J.S.; Richter, J.D. Impaired neurodevelopment by the low complexity domain of CPEB4 reveals a convergent pathway with neurodegeneration. Sci. Rep. 2016, 6, 29395. [Google Scholar] [CrossRef] [PubMed]
- Masana, M.; Westerholz, S.; Kretzschmar, A.; Treccani, G.; Liebl, C.; Santarelli, S.; Dournes, C.; Popoli, M.; Schmidt, M.V.; Rein, T.; et al. Expression and glucocorticoid-dependent regulation of the stress-inducible protein DRR1 in the mouse adult brain. Brain Struct. Funct. 2018, 223, 4039–4052. [Google Scholar] [CrossRef]
- Masana, M.; Jukic, M.M.; Kretzschmar, A.; Wagner, K.V.; Westerholz, S.; Schmidt, M.V.; Rein, T.; Brodski, C.; Muller, M.B. Deciphering the spatio-temporal expression and stress regulation of Fam107B, the paralog of the resilience-promoting protein DRR1 in the mouse brain. Neuroscience 2015, 290, 147–158. [Google Scholar] [CrossRef]
- Stankiewicz, A.M.; Goscik, J.; Swiergiel, A.H.; Majewska, A.; Wieczorek, M.; Juszczak, G.R.; Lisowski, P. Social stress increases expression of hemoglobin genes in mouse prefrontal cortex. BMC Neurosci. 2014, 15, 130. [Google Scholar] [CrossRef]
- Jene, T.; Gassen, N.C.; Opitz, V.; Endres, K.; Muller, M.B.; van der Kooij, M.A. Temporal profiling of an acute stress-induced behavioral phenotype in mice and role of hippocampal DRR1. Psychoneuroendocrinology 2018, 91, 149–158. [Google Scholar] [CrossRef]
- Lupas, A.N.; Gruber, M. The structure of alpha-helical coiled coils. Adv. Protein Chem. 2005, 70, 37–78. [Google Scholar] [PubMed]
- Liu, J.; Rost, B. Comparing function and structure between entire proteomes. Protein Sci. 2001, 10, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Falahzadeh, K.; Banaei-Esfahani, A.; Shahhoseini, M. The potential roles of actin in the nucleus. Cell J. 2015, 17, 7–14. [Google Scholar] [PubMed]
- Zhao, X.Y.; Liang, S.F.; Yao, S.H.; Ma, F.X.; Hu, Z.G.; Yan, F.; Yuan, Z.; Ruan, X.Z.; Yang, H.S.; Zhou, Q.; et al. Identification and preliminary function study of Xenopus laevis DRR1 gene. Biochem. Biophys. Res. Commun. 2007, 361, 74–78. [Google Scholar] [CrossRef]
- Siton-Mendelson, O.; Bernheim-Groswasser, A. Functional Actin Networks under Construction: The Cooperative Action of Actin Nucleation and Elongation Factors. Trends Biochem. Sci. 2017, 42, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Higgs, H.N. The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr. Biol. 2003, 13, 1335–1340. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Hertzog, M.; Milanesi, F.; Hazelwood, L.; Disanza, A.; Liu, H.; Perlade, E.; Malabarba, M.G.; Pasqualato, S.; Maiolica, A.; Confalonieri, S.; et al. Molecular basis for the dual function of Eps8 on actin dynamics: Bundling and capping. PLoS Biol. 2010, 8, e1000387. [Google Scholar] [CrossRef]
- Gremm, D.; Wegner, A. Gelsolin as a calcium-regulated actin filament-capping protein. Eur. J. Biochem. 2000, 267, 4339–4345. [Google Scholar] [CrossRef]
- Schirenbeck, A.; Bretschneider, T.; Arasada, R.; Schleicher, M.; Faix, J. The Diaphanous-related formin dDia2 is required for the formation and maintenance of filopodia. Nat. Cell Biol. 2005, 7, 619–625. [Google Scholar] [CrossRef]
- Paavilainen, V.O.; Oksanen, E.; Goldman, A.; Lappalainen, P. Structure of the actin-depolymerizing factor homology domain in complex with actin. J. Cell Biol. 2008, 182, 51–59. [Google Scholar] [CrossRef]
- Paavilainen, V.O.; Hellman, M.; Helfer, E.; Bovellan, M.; Annila, A.; Carlier, M.F.; Permi, P.; Lappalainen, P. Structural basis and evolutionary origin of actin filament capping by twinfilin. Proc. Natl. Acad. Sci. USA 2007, 104, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Way, M.; Pope, B.; Weeds, A.G. Evidence for functional homology in the F-actin binding domains of gelsolin and alpha-actinin: Implications for the requirements of severing and capping. J. Cell Biol. 1992, 119, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.C.; Mejillano, M.; Le, V.P.; Burtnick, L.D.; Yin, H.L.; Choe, S. Domain movement in gelsolin: A calcium-activated switch. Science 1999, 286, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- McGough, A.; Chiu, W.; Way, M. Determination of the gelsolin binding site on F-actin: Implications for severing and capping. Biophys. J. 1998, 74, 764–772. [Google Scholar] [CrossRef]
- Falzone, T.T.; Lenz, M.; Kovar, D.R.; Gardel, M.L. Assembly kinetics determine the architecture of alpha-actinin crosslinked F-actin networks. Nat. Commun. 2012, 3, 861. [Google Scholar] [CrossRef] [PubMed]
- Schmoller, K.M.; Semmrich, C.; Bausch, A.R. Slow down of actin depolymerization by cross-linking molecules. J. Struct. Biol. 2011, 173, 350–357. [Google Scholar] [CrossRef]
- Chamaraux, F.; Fache, S.; Bruckert, F.; Fourcade, B. Kinetics of cell spreading. Phys. Rev. Lett. 2005, 94, 158102. [Google Scholar] [CrossRef]
- Li, J.; Han, D.; Zhao, Y.P. Kinetic behaviour of the cells touching substrate: The interfacial stiffness guides cell spreading. Sci. Rep. 2014, 4, 3910. [Google Scholar] [CrossRef]
- Yauch, R.L.; Felsenfeld, D.P.; Kraeft, S.K.; Chen, L.B.; Sheetz, M.P.; Hemler, M.E. Mutational evidence for control of cell adhesion through integrin diffusion/clustering, independent of ligand binding. J. Exp. Med. 1997, 186, 1347–1355. [Google Scholar] [CrossRef]
- Cuvelier, D.; Théry, M.; Chu, Y.S.; Dufour, S.; Thiery, J.P.; Bornens, M.; Nassoy, P.; Mahadevan, L. The universal dynamics of cell spreading. Curr. Biol. 2007, 17, 694–699. [Google Scholar] [CrossRef]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabro, V.; La, M.G. p14ARF interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef] [PubMed]
- Frost, N.A.; Shroff, H.; Kong, H.; Betzig, E.; Blanpied, T.A. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron 2010, 67, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Honkura, N.; Matsuzaki, M.; Noguchi, J.; Ellis-Davies, G.C.; Kasai, H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 2008, 57, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Star, E.N.; Kwiatkowski, D.J.; Murthy, V.N. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat. Neurosci. 2002, 5, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Narayanan, R.; Lee, S.H.; Murata, K.; Hayashi, Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. USA 2007, 104, 6418–6423. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci. 2003, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rüegg, J.; Holsboer, F.; Turck, C.; Rein, T. Cofilin 1 is revealed as an inhibitor of glucocorticoid receptor by analysis of hormone-resistant cells 2195. Mol. Cell Biol. 2004, 24, 9371–9382. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Durand, C.M.; Perroy, J.; Loll, F.; Perrais, D.; Fagni, L.; Bourgeron, T.; Montcouquiol, M.; Sans, N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 2012, 17, 71–84. [Google Scholar] [CrossRef]
- Nakatani, N.; Ohnishi, T.; Iwamoto, K.; Watanabe, A.; Iwayama, Y.; Yamashita, S.; Ishitsuka, Y.; Moriyama, K.; Nakajima, M.; Tatebayashi, Y.; et al. Expression analysis of actin-related genes as an underlying mechanism for mood disorders. Biochem. Biophys. Res. Commun. 2007, 352, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, N.; Aburatani, H.; Nishimura, K.; Semba, J.; Yoshikawa, T. Comprehensive expression analysis of a rat depression model. Pharmacogenomics J. 2004, 4, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Bucan, M. Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007, 81, 1278–1283. [Google Scholar] [CrossRef]
- Huang, T.L.; Sung, M.L.; Chen, T.Y. 2D-DIGE proteome analysis on the platelet proteins of patients with major depression. Proteome Sci. 2014, 12. [Google Scholar] [CrossRef]
- Calabrese, B.; Halpain, S. Lithium prevents aberrant NMDA-induced F-actin reorganization in neurons. Neuroreport 2014, 25, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Piubelli, C.; Vighini, M.; Mathe, A.A.; Domenici, E.; Carboni, L. Escitalopram affects cytoskeleton and synaptic plasticity pathways in a rat gene-environment interaction model of depression as revealed by proteomics. Part II: Environmental challenge. Int. J. Neuropsychopharmacol. 2011, 14, 834–855. [Google Scholar] [CrossRef]
- O’Dushlaine, C.; Ripke, S.; Ruderfer, D.M.; Hamilton, S.P.; Fava, M.; Iosifescu, D.V.; Kohane, I.S.; Churchill, S.E.; Castro, V.M.; Clements, C.C.; et al. Rare copy number variation in treatment-resistant major depressive disorder. Biol. Psychiatry 2014, 76, 536–541. [Google Scholar] [CrossRef]
- Frijters, R.; Fleuren, W.; Toonen, E.J.; Tuckermann, J.P.; Reichardt, H.M.; van der, M.H.; van, E.A.; van Lierop, M.J.; Dokter, W.; de, V.J.; et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics 2010, 11, 359. [Google Scholar] [CrossRef]
- Kretzschmar, A. The Modulation of Actin Dynamics by the Stress-Induced Protein DRR1 and Antidepressants. Ph.D. Thesis, Ludmig Maximilians University Munich, Munich, Germany, 2015. [Google Scholar]
- Zhao, X.Y.; Li, H.X.; Liang, S.F.; Yuan, Z.; Yan, F.; Ruan, X.Z.; You, J.; Xiong, S.Q.; Tang, M.H.; Wei, Y.Q. Soluble expression of human DRR1 (down-regulated in renal cell carcinoma 1) in Escherichia coli and preparation of its polyclonal antibodies. Biotechnol. Appl. Biochem. 2008, 49, 17–23. [Google Scholar] [CrossRef]
- Schülke, J.P.; Wochnik, G.M.; Lang-Rollin, I.; Gassen, N.C.; Knapp, R.T.; Berning, B.; Yassouridis, A.; Rein, T. Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS ONE 2010, 5, e11717. [Google Scholar] [CrossRef] [PubMed]
- Rino, J.; Martin, R.M.; Carvalho, T.; Carmo-Fonseca, M. Imaging dynamic interactions between spliceosomal proteins and pre-mRNA in living cells. Methods 2014, 65, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Walker, S.B.; Pollard, T.D. Pyrene actin: Documentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil. 1983, 4, 253–262. [Google Scholar] [CrossRef]
- Liu, X.; Pollack, G.H. Stepwise sliding of single actin and Myosin filaments. Biophys. J. 2004, 86, 353–358. [Google Scholar] [CrossRef]
- Pollard, T.D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J. Cell Biol. 1986, 103, 2747–2754. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F-Actin Binding | Bundling | Filament Elongation | Nucleation | Capping | |
|---|---|---|---|---|---|
| DRR1 wt | + + + | + + + | − − − | + + | + + + |
| dN | + | + + | 0 | 0 | 0 |
| dC | + + | + | − (n.s.) | + (n.s.) | + |
| dM | + + | + | − − − | + (n.s.) | + + + |
| M | 0 | 0 | 0 | 0 | 0 |
| DRR1 | Colocalization F-Actin | Cellular F-Actin | Actin Treadmillling | Cell Spreading | SRF Activation |
|---|---|---|---|---|---|
| wt | +++ | +++ | − − − | − − − | + + + |
| dN | ++ | + | − − − | − | + + |
| dC | + | 0 | 0 | − − − | 0 |
| dM | ++ | ++ | 0 | − | + |
| M | 0 | 0 | 0 | 0 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretzschmar, A.; Schülke, J.-P.; Masana, M.; Dürre, K.; Müller, M.B.; Bausch, A.R.; Rein, T. The Stress-Inducible Protein DRR1 Exerts Distinct Effects on Actin Dynamics. Int. J. Mol. Sci. 2018, 19, 3993. https://doi.org/10.3390/ijms19123993
Kretzschmar A, Schülke J-P, Masana M, Dürre K, Müller MB, Bausch AR, Rein T. The Stress-Inducible Protein DRR1 Exerts Distinct Effects on Actin Dynamics. International Journal of Molecular Sciences. 2018; 19(12):3993. https://doi.org/10.3390/ijms19123993
Chicago/Turabian StyleKretzschmar, Anja, Jan-Philip Schülke, Mercè Masana, Katharina Dürre, Marianne B. Müller, Andreas R. Bausch, and Theo Rein. 2018. "The Stress-Inducible Protein DRR1 Exerts Distinct Effects on Actin Dynamics" International Journal of Molecular Sciences 19, no. 12: 3993. https://doi.org/10.3390/ijms19123993
APA StyleKretzschmar, A., Schülke, J.-P., Masana, M., Dürre, K., Müller, M. B., Bausch, A. R., & Rein, T. (2018). The Stress-Inducible Protein DRR1 Exerts Distinct Effects on Actin Dynamics. International Journal of Molecular Sciences, 19(12), 3993. https://doi.org/10.3390/ijms19123993