2′-Fluoro-Pyrimidine-Modified RNA Aptamers Specific for Lipopolysaccharide Binding Protein (LBP)

Abstract

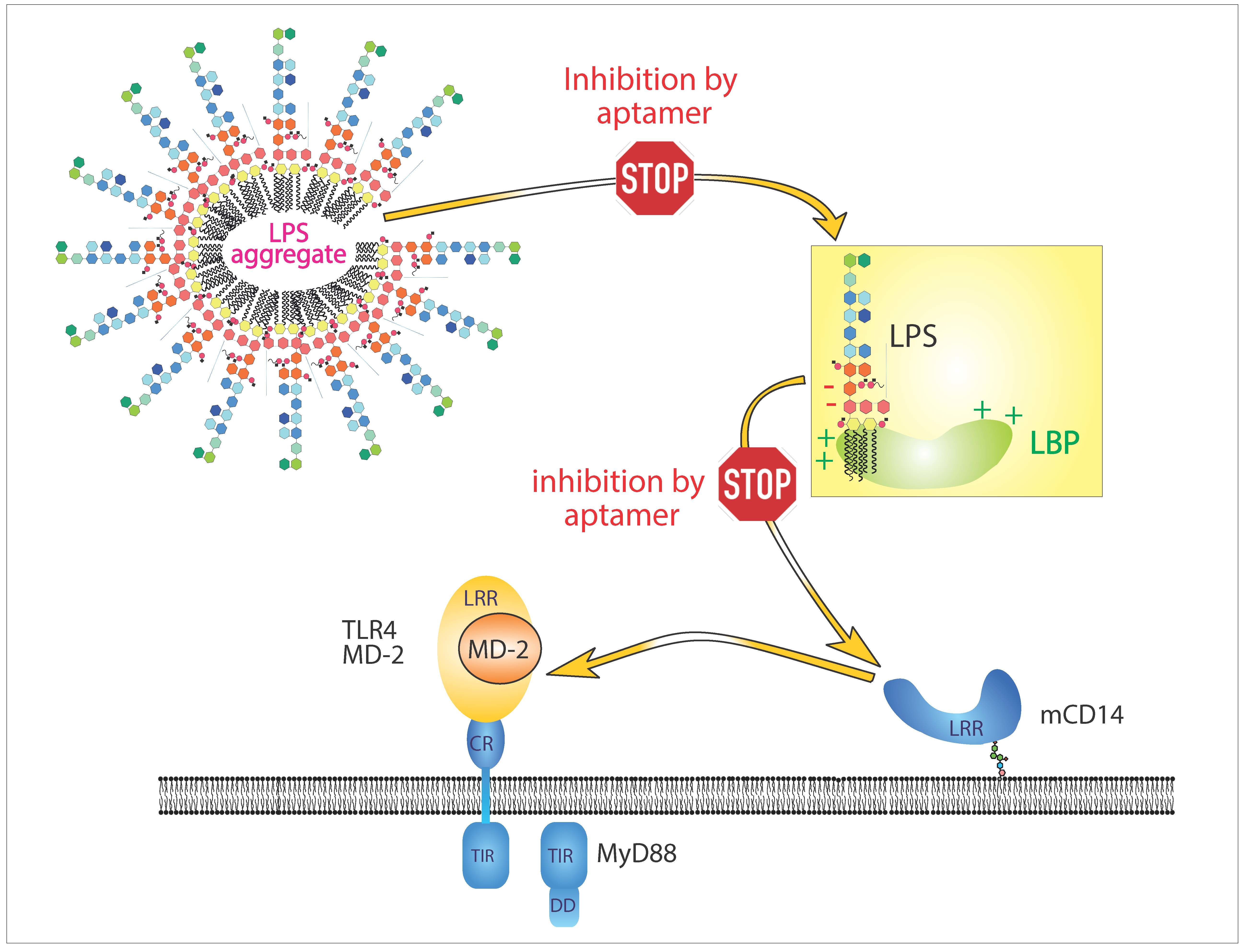
1. Introduction
2. Results and Discussion
2.1. In Vitro Selection of LBP-Binding Aptamers
2.2. Secondary Structures of Aptamers
2.3. Structural Probing
2.4. Boundary Experiment
2.5. Affinity and Selectivity of A011 Binding to mLBP
3. Materials and Methods
3.1. Materials
3.2. RNA Library
3.3. In Vitro Selection
3.4. 3′- and 5′-Endlabeling
3.5. Determination of Equilibrium Dissociation Constants
3.6. Enzymatic and Chemical Structure Probing
3.7. Boundary Experiments
4. Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Schmieg, R.E., Jr.; Hui, J.J.; Chang, K.C.; Osborne, D.F.; Freeman, B.D.; Cobb, J.P.; Buchman, T.G.; et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J. Immunol. 2001, 166, 6952–6963. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Schmieg, R.E., Jr.; Swanson, P.E.; Freeman, B.D.; Tinsley, K.W.; Cobb, J.P.; Karl, I.E.; Buchman, T.G. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit. Care Med. 2000, 28, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Glauser, M.P.; Zanetti, G.; Baumgartner, J.D.; Cohen, J. Septic shock: Pathogenesis. Lancet 1991, 338, 732–736. [Google Scholar] [CrossRef]
- Parillo, J.E. Pathogenic mechanisms of septic shock. N. Engl. J. Med. 1993, 328, 1471–1477. [Google Scholar]
- Schumann, R.R.; Kirschning, C.J.; Unbehaun, A.; Aberle, H.P.; Knope, H.P.; Lamping, N.; Ulevitch, R.J.; Herrmann, F. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol. Cell. Biol. 1996, 16, 3490–3503. [Google Scholar] [CrossRef] [PubMed]
- Schumann, R.R.; Zweigner, J. A novel acute-phase marker: Lipopolysaccharide binding protein (LBP). Clin. Chem. Lab. Med. 1999, 37, 271–274. [Google Scholar] [CrossRef]
- Schumann, R.R. Recognition of bacterial endotoxin and modulation of the inflammatory response: The LBP/CD14 pathway during the acute phase. Sepsis 1998, 149–155. [Google Scholar] [CrossRef]
- Ulevitch, R.J.; Tobias, P.S. Recognition of gram-negative bacteria and endotoxin by the innate immune system. Curr. Opin. Immunol. 1999, 11, 19–22. [Google Scholar] [CrossRef]
- Villar, J.; Pérez-Méndez, L.; Espinosa, E.; Flores, C.; Blanco, J.; Muriel, A.; Basaldúa, S.; Muros, M.; Blanch, L.; Artigas, A.; Kacmarek, R.M. GRECIA and GEN-SEP Groups. Serum lipopolysaccharide binding protein levels predict severity of lung injury and mortality in patients with severe sepsis. PLoS ONE 2009, 4, e6818. [Google Scholar] [CrossRef]
- Kim, H.I.; Park, S. Sepsis: Early Recognition and Optimized Treatment. Tuberc. Respir. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tobias, P.S.; Mathison, J.C.; Ulevitch, R.J. A family of lipopolysaccharide binding proteins involved in responses to gram-negative sepsis. J. Biol. Chem. 1988, 263, 13479–13481. [Google Scholar] [PubMed]
- Agellon, L.B.; Quinet, E.M.; Gillette, T.G.; Drayna, D.T.; Brown, M.L.; Tall, A.R. Organization of the human cholesteryl ester transfer protein gene. Biochemistry 1990, 29, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Au-Young, J.; Fielding, C.J. Synthesis and secretion of wild-type and mutant human plasma cholesteryl ester transfer protein in baculovirus-transfected insect cells: The carboxyl-terminal region is required for both lipoprotein binding and catalysis of transfer. Proc. Natl. Acad. Sci. USA 1992, 89, 4094–4098. [Google Scholar] [CrossRef] [PubMed]
- Day, J.R.; Albers, J.J.; Lofton-Day, C.E.; Gilbert, T.L.; Ching, A.F.; Grant, F.J.; O’Hara, P.J.; Marcovina, S.M.; Adolphson, J.L. Complete cDNA encoding human phospholipid transfer protein from human endothelial cells. J. Biol. Chem. 1994, 269, 9388–9391. [Google Scholar] [PubMed]
- Hubacek, J.A.; Buchler, C.; Aslanidis, C.; Schmitz, G. The genomic organization of the genes for human lipopolysaccharide binding protein (LBP) and bactericidal permeability increasing protein (BPI) is highly conserved. Biochem. Biophys. Res. Commun. 1997, 236, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Kirschning, C.J.; Au-Young, J.; Lamping, N.; Reuter, D.; Pfeil, D.; Seilhamer, J.J.; Schumann, R.R. Similar organization of the lipopolysaccharide-binding protein (LBP) and phospholipid transfer protein (PLTP) genes suggests a common gene family of lipid-binding proteins. Genomics 1997, 46, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zähringer, U.; Lindner, B.; Rietschel, E.T. Molecular structure of lipid A, the endotoxic center of bacterial lipopolysaccharides. Adv. Carbohydr. Chem. Biochem. 1994, 50, 211–276. [Google Scholar] [PubMed]
- Tobias, P.S.; Soldau, K.; Ulevitch, R.J. Identification of a lipid A binding site in the acute phase reactant lipopolysaccharide binding protein. J. Biol. Chem. 1989, 264, 10867–10871. [Google Scholar]
- Gazzano-Santoro, H.; Meszaros, K.; Birr, C.; Carroll, S.F.; Theofan, G.; Horwitz, A.H.; Lim, E.; Aberle, S.; Kasler, H.; Parent, J.B. Competition between rBPI23, a recombinant fragment of bactericidal/permeability-increasing protein, and lipopolysaccharide (LPS)-binding protein for binding to LPS and gram-negative bacteria. Infect. Immun. 1994, 62, 1185–1191. [Google Scholar]
- Lamping, N.; Hoess, A.; Yu, B.; Park, T.C.; Kirschning, C.J.; Pfeil, D.; Reuter, D.; Wright, S.D.; Herrmann, F.; Schumann, R.R. Effects of site-directed mutagenesis of basic residues (Arg 94, Lys 95, Lys 99) of lipopolysaccharide (LPS)-binding protein on binding and transfer of LPS and subsequent immune cell activation. J. Immunol. 1996, 157, 4648–4656. [Google Scholar] [PubMed]
- Taylor, A.H.; Heavner, G.; Nedelman, M.; Sherris, D.; Brunt, E.; Knight, D.; Ghrayeb, J. Lipopolysaccharide (LPS) neutralizing peptides reveal a lipid A binding site of LPS binding protein. J. Biol. Chem. 1995, 270, 17934–17938. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Mathison, J.C.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide (LPS) binding protein, truncated at Ile-197, binds LPS but does not transfer LPS to CD14. J. Biol. Chem. 1994, 269, 8172–8175. [Google Scholar]
- Theofan, G.; Horwitz, A.H.; Williams, R.E.; Liu, P.S.; Chan, I.; Birr, C.; Carroll, S.F.; Meszaros, K.; Parent, J.B.; Kasler, H.; et al. An amino-terminal fragment of human lipopolysaccharide-binding protein retains lipid A binding but not CD14-stimulatory activity. J. Immunol. 1994, 152, 3623–3629. [Google Scholar] [PubMed]
- Schumann, R.R.; Leong, S.R.; Flaggs, G.W.; Gray, P.W.; Wright, S.D.; Mathison, J.C.; Tobias, P.S.; Ulevitch, R.J. Structure and function of lipopolysaccharide binding protein. Science 1990, 249, 1429–1431. [Google Scholar] [CrossRef]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Triantafilou, K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends. Immunol. 2002, 23, 301–304. [Google Scholar] [CrossRef]
- Zhao, B.; Bowden, R.A.; Stavchansky, S.A.; Bowman, P.D. Human endothelial cell response to gram-negative lipopolysaccharide assessed with cDNA microarrays. Am. J. Physiol. Cell. Physiol. 2001, 281, C1587–C1595. [Google Scholar] [CrossRef]
- Fessler, M.B.; Malcolm, K.C.; Duncan, M.W.; Worthen, G.S. A genomic and proteomic analysis of activation of the human neutrophil by lipopolysaccharide and its mediation by p38 mitogen-activated protein kinase. J. Biol. Chem. 2002, 277, 31291–31302. [Google Scholar] [CrossRef]
- Riedemann, N.C.; Guo, R.F.; Ward, P.A. Novel strategies for the treatment of sepsis. Nat. Med. 2003, 9, 517–524. [Google Scholar] [CrossRef]
- Marshall, J.C. Such stuff as dreams are made on: Mediator-directed therapy in sepsis. Nat. Rev. Drug Discov. 2003, 2, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, N.C.; Guo, R.F.; Ward, P.A. The enigma of sepsis. J. Clin. Invest. 2003, 112, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Peters van Ton, A.M.; Kox, M.; Abdo, W.F.; Pickkers, P. Precision Immunotherapy for Sepsis. Front. Immunol. 2018, 9, 1926. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.L.; Gan, S.T.; Ho, B. Single-stranded DNA oligoaptamers: Molecular recognition and LPS antagonism are length- and secondary structure-dependent. J. Innate. Immun. 2009, 1, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Grünweller, A.; Hartmann, R.K.; Kjems, J. Using chemical modification to enhance siRNA performance. In Handbook of RNA Biochemistry, 2nd ed.; Hartmann, R.K., Bindereif, A., Schön, A., Westhof, E., Eds.; Wiley-VCH: Weinheim, Germany, 2014; Volume 2, pp. 1243–1277. ISBN 978-3-527-32764-5. [Google Scholar]
- Green, L.S.; Jellinek, D.; Bell, C.; Beebe, L.A.; Feistner, B.D.; Gill, S.C.; Jucker, F.M.; Janjic, N. Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem. Biol. 1995, 2, 683–695. [Google Scholar] [CrossRef]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.P.; Limmer, S.; Hornung, V.; Sprinzl, M. Ni2+-binding RNA motifs with an asymmetric purine-rich internal loop and a G-A base pair. RNA 1997, 3, 1289–1300. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Ciesiolka, J.; Hardt, W.D.; Schlegl, J.; Erdmann, V.A.; Hartmann, R.K. Lead-ion-induced cleavage of RNase P RNA. Eur. J. Biochem. 1994, 219, 49–56. [Google Scholar] [CrossRef]
- Ehresmann, C.; Baudin, F.; Mougel, M.; Romby, P.; Ebel, J.P.; Ehresmann, B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987, 15, 9109–9128. [Google Scholar] [CrossRef] [PubMed]
- Persson, T.; Cuzic, S.; Hartmann, R.K. Catalysis by RNase P RNA. J. Biol. Chem. 2003, 278, 43394–43401. [Google Scholar] [CrossRef]
- Wen, A.Q.; Yang, Q.W.; Li, J.C.; Lv, F.L.; Zhong, Q.; Chen, C.Y. A novel lipopolysaccharide-antagonizing aptamer protects mice against endotoxemia. Biochem. Biophys. Res. Commun. 2009, 382, 140–144. [Google Scholar] [CrossRef]
- Sousa, R.; Padilla, R. A mutant T7 RNA polymerase as a DNA polymerase. EMBO J. 1995, 14, 4609–4621. [Google Scholar] [CrossRef]
- Persson, T.; Hartmann, R.K.; Eckstein, F. Selection of hammerhead ribozyme variants with low Mg2+ requirement: Importance of stem-loop II. Chembiochem 2002, 3, 1066–1071. [Google Scholar] [CrossRef]
- Turunen, J.J.; Pavlova, L.V.; Hengesbach, M.; Helm, M.; Müller, S.; Hartmann, R.K.; Frilander, M.J. RNA Ligation. In Handbook of RNA Biochemistry, 2nd ed.; Hartmann, R.K., Bindereif, A., Schön, A., Westhof, E., Eds.; WILEY-VCH: Weinheim, Germany, 2014; pp. 45–88. [Google Scholar]
- Hardt, W.D.; Warnecke, J.M.; Erdmann, V.A.; Hartmann, R.K. Rp-phosphorothioate modifications in RNase P RNA that interfere with tRNA binding. EMBO J. 1995, 14, 2935–2944. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Round | 2′-F-Y-RNA (μM) | mLBP (μM) | Total Volume (μl) | RNA:mLBP ratio | Preselections |
|---|---|---|---|---|---|
| 1 | 1.37 | 1.6 | 400 | 1.0 : 1.2 | + |
| 2 | 0.85 | 0.9 | 300 | 1.0 : 1.3 | + |
| 3 | 0.45 | 0.8 | 200 | 1.0 : 1.8 | + |
| 4 | 0.5 | 0.8 | 200 | 1.0 : 1.5 | + |
| 5MB | 0.48 | 0.8 | 100 | 1.0 : 1.6 | + |
| 6MB | 0.7 | 0.8 | 200 | 1.0 : 1.1 | + |
| 7 | 0.79 | 0.8 | 200 | 1.0 : 1.0 | + |
| 8 | 1.1 | 0.8 | 200 | 1.4 : 1.0 | + |
| Ni-NTA Agarose Matrix | ||
|---|---|---|
| Round | 2′-F-Y-RNA Preselection (%) | 2′-F-Y-RNA Selection (%) |
| 1 | 10.70 | 1.28 |
| 2 | 17.00 | 0.96 |
| 3 | 21.00 | 2.50 |
| 4 | 18.60 | 1.83 |
| 5MB | 8.00 | 4.10 |
| 6MB | 7.53 | 4.92 |
| 7 | 31.00 | 2.64 |
| 8 | 11.00 | 2.72 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldag, J.; Persson, T.; Hartmann, R.K. 2′-Fluoro-Pyrimidine-Modified RNA Aptamers Specific for Lipopolysaccharide Binding Protein (LBP). Int. J. Mol. Sci. 2018, 19, 3883. https://doi.org/10.3390/ijms19123883
Aldag J, Persson T, Hartmann RK. 2′-Fluoro-Pyrimidine-Modified RNA Aptamers Specific for Lipopolysaccharide Binding Protein (LBP). International Journal of Molecular Sciences. 2018; 19(12):3883. https://doi.org/10.3390/ijms19123883
Chicago/Turabian StyleAldag, Jasmin, Tina Persson, and Roland K. Hartmann. 2018. "2′-Fluoro-Pyrimidine-Modified RNA Aptamers Specific for Lipopolysaccharide Binding Protein (LBP)" International Journal of Molecular Sciences 19, no. 12: 3883. https://doi.org/10.3390/ijms19123883
APA StyleAldag, J., Persson, T., & Hartmann, R. K. (2018). 2′-Fluoro-Pyrimidine-Modified RNA Aptamers Specific for Lipopolysaccharide Binding Protein (LBP). International Journal of Molecular Sciences, 19(12), 3883. https://doi.org/10.3390/ijms19123883