The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases


Abstract
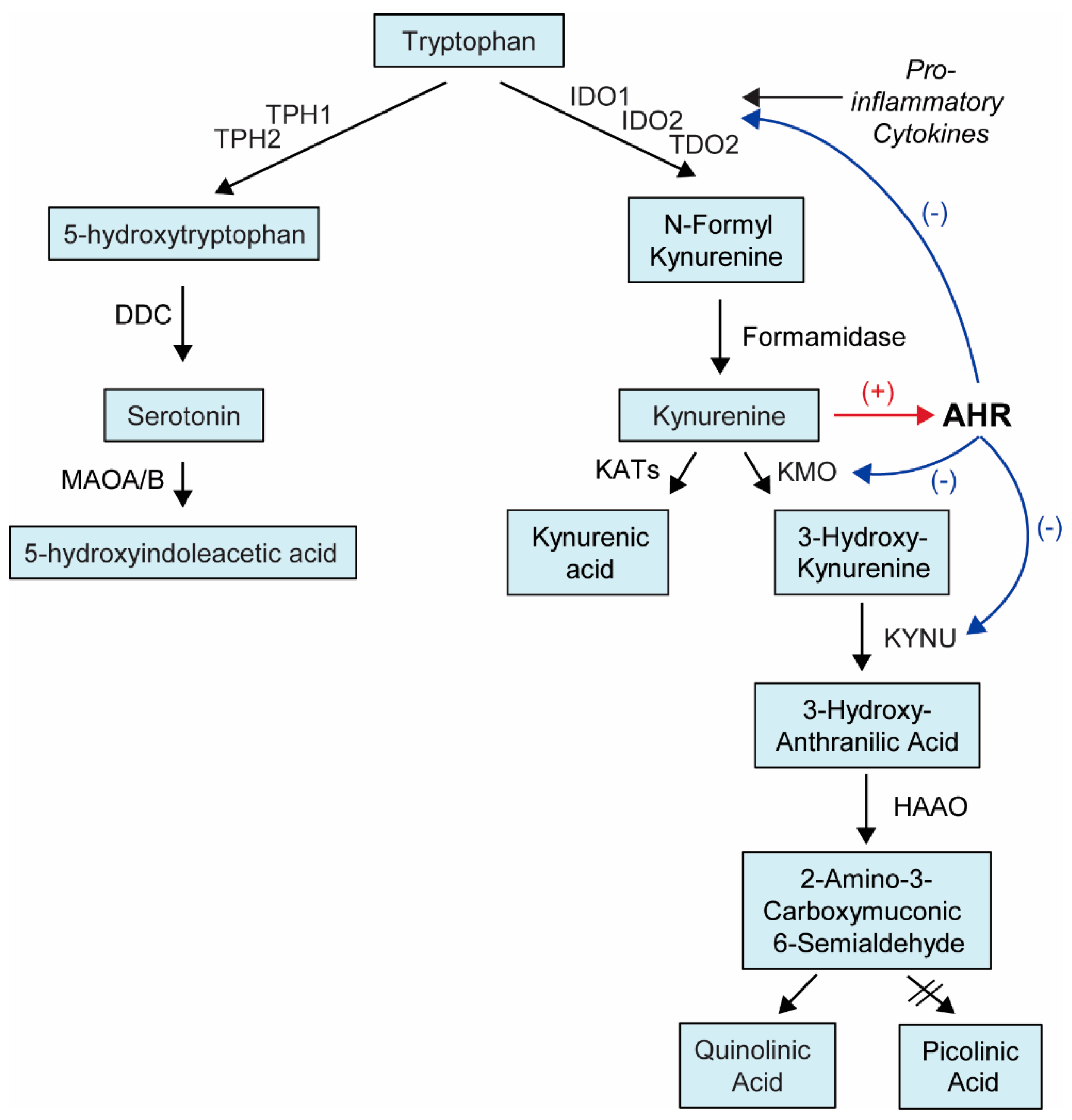
1. Introduction
1.1. AHR Mechanism of Action
1.2. AHR Ligands
2. AHR-Dependent Pathogen Response
2.1. Microbial Pathogens
2.2. Viral Pathogens
2.3. Parasitic Pathogens
3. AHR and the Central Nervous System
3.1. AHR and Major Depressive Disorder
3.2. AHR and Multiple Sclerosis
3.3. AHR and Congenital Nystagmus
4. AHR and the Gut Microbiome
5. AHR and Inflammatory Bowel Disease
6. AHR and Rheumatoid Arthritis
7. AHR and Psoriasis
8. AHR and Atherosclerosis
9. AHR and Single Nucleotide Polymorphisms
10. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-MC | 3-methylcholanthrene |
| AHR | aryl hydrocarbon receptor |
| AHRE | aryl hydrocarbon receptor response elements |
| AHRR | aryl hydrocarbon receptor repressor |
| ARNT | aryl hydrocarbon nuclear translocator |
| ANF | α-napthoflavone |
| BNF | β-napthoflavone |
| CD | Crohn’s disease |
| CNS | central nervous system |
| COPD | chronic obstructive pulmonary disease |
| CYP1A1 | cytochrome P450 Family 1 Subfamily A Member 1 |
| CYP1A2 | cytochrome P450 Family 1 Subfamily A Member 2 |
| CYP1B1 | cytochrome P450 Family 1 Subfamily B Member 1 |
| DC | dendritic cell |
| DIM | diindolylmethane |
| DSS | dextran sulfate sodium |
| EAE | experimental autoimmune encephalitis |
| eQTL | expression quantitative trait locus |
| FICZ | 6-formylindolo[3,2-b]carbazole |
| FOXP3 | forkhead Box P3 |
| I3C | indole-3-carbinol |
| IAA | indole-3-acetic acid |
| IBD | inflammatory bowel disease |
| ICZ | indolo[3,2-b]carbazole |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-g | interferon gamma |
| IL-12 | interleukin 12 |
| IL-17 | interleukin 17 |
| IL-18 | interleukin 18 |
| IL-1β | interleukin 1 beta |
| IL-22 | interleukin 22 |
| IL-6 | interleukin 6 |
| ILC | innate lymphoblastoid cell |
| KD | knock down |
| KMO | kynurenine 3-monooxygenase |
| KYN | kynurenine |
| KYNU | kynureninase |
| LM | Listeria Monocytogenes |
| LPS | lipopolysaccharide |
| MDD | major depressive disorder |
| MS | multiple sclerosis |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| NOR | Norisoboldine |
| PGx | pharmacogenomic |
| RA | rheumatoid arthritis |
| ROS | reactive oxygen species |
| SNP | single nucleotide polymorphism |
| TCDD | 2,3,7,8-tetraclorodibenzo-p-dioxina |
| TDO2 | tryptophan 2,3-dioxygenase |
| TIPARP | TCDD Inducible poly(ADP-Ribose) polymerase |
| TNF-a | tumor necrosis factor alpha |
| UC | ulcerative colitis |
| WT | wildtype |
References
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ramachandran, I.; Gabrilovich, D.I. Regulation of plasmacytoid dendritic cell development in mice by aryl hydrocarbon receptor. Immunol. Cell Biol. 2014, 92, 200–203. [Google Scholar] [CrossRef]
- Nakahama, T.; Kimura, A.; Nguyen, N.T.; Chinen, I.; Hanieh, H.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 14222–14227. [Google Scholar] [CrossRef]
- Quintana, F.J. LeA(H)Rning self-control. Cell Res. 2014, 24, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef]
- Colonna, M. AHR: Making the keratinocytes thick skinned. Immunity 2014, 40, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Rothhammer, V.; Quintana, F.J. Control of immune-mediated pathology via the aryl hydrocarbon receptor. J. Biol. Chem. 2017, 292, 12383–12389. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor κB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef]
- Korecka, A.; Dona, A.; Lahiri, S.; Tett, A.J.; Al-Asmakh, M.; Braniste, V.; D’Arienzo, R.; Abbaspour, A.; Reichardt, N.; Fujii-Kuriyama, Y.; et al. Bidirectional communication between the Aryl hydrocarbon Receptor (AhR) and the microbiome tunes host metabolism. NPJ Biofilms Microbiomes 2016, 2, 16014. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Nogueira, S.V.; Drongelen, V.V.; Coit, P.; Ling, S.; Rosloniec, E.F.; Sawalha, A.H.; Holoshitz, J. Shared epitope-aryl hydrocarbon receptor crosstalk underlies the mechanism of gene-environment interaction in autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2018, 115, 4755–4760. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R. RelB and the aryl hydrocarbon receptor: Dendritic cell tolerance at the epithelial interface. Immunol. Cell Biol. 2013, 91, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Casado, F.L.; Singh, K.P.; Gasiewicz, T.A. The aryl hydrocarbon receptor: Regulation of hematopoiesis and involvement in the progression of blood diseases. Blood Cells Mol. Dis. 2010, 44, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J. Regulation of central nervous system autoimmunity by the aryl hydrocarbon receptor. Semin. Immunopathol. 2013, 35, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Mimura, J.; Oshima, M.; Okawa, H.; Kanno, J.; Igarashi, K.; Gonzalez, F.J.; Ikuta, T.; Kawajiri, K.; Fujii-Kuriyama, Y. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 2009, 29, 6391–6400. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Seyedhosseini, F.S.; Behnampour, N.; Yazdani, Y. Indole-3-carbinol induces G1 cell cycle arrest and apoptosis through aryl hydrocarbon receptor in THP-1 monocytic cell line. J. Recept. Signal Transduct. Res. 2017, 37, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Hirota, K.; Duarte, J.; Veldhoen, M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin. Immunol. 2011, 23, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H. Toward understanding the role of aryl hydrocarbon receptor in the immune system: Current progress and future trends. BioMed Res. Int. 2014, 2014, 520763. [Google Scholar] [CrossRef]
- Kimura, A.; Abe, H.; Tsuruta, S.; Chiba, S.; Fujii-Kuriyama, Y.; Sekiya, T.; Morita, R.; Yoshimura, A. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen species production. Int. Immunol. 2014, 26, 209–220. [Google Scholar] [CrossRef]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ray, B.; Neavin, D.R.; Zhang, J.; Athreya, A.P.; Biernacka, J.M.; Bobo, W.V.; Hall-Flavin, D.K.; Skime, M.K.; Zhu, H.; et al. β-defensin 1, aryl hydrocarbon receptor and plasma kynurenine in major depressive disorder: Metabolomics-informed genomics. Transl. Psychiatry 2018, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sorrentino, C.; Denison, M.S.; Kolaja, K.; Fielden, M.R. Induction of CYP1A1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: Results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol. Pharmacol. 2007, 71, 1475–1486. [Google Scholar] [CrossRef]
- Bjeldanes, L.F.; Kim, J.Y.; Grose, K.R.; Bartholomew, J.C.; Bradfield, C.A. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: Comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc. Natl. Acad. Sci. USA 1991, 88, 9543–9547. [Google Scholar] [CrossRef]
- Nguyen, L.P.; Bradfield, C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.; Safe, S. PCB isomers and congeners: Induction of aryl hydrocarbon hydroxylase and ethoxyresorufin O-deethylase enzyme activities in rat hepatoma cells. Toxicol. Lett. 1982, 13, 87–93. [Google Scholar] [CrossRef]
- Soshilov, A.A.; Denison, M.S. Ligand Promiscuity of Aryl Hydrocarbon Receptor Agonists and Antagonists Revealed by Site-Directed Mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef]
- Seok, S.H.; Ma, Z.X.; Feltenberger, J.B.; Chen, H.; Chen, H.; Scarlett, C.; Lin, Z.; Satyshur, K.A.; Cortopassi, M.; Jefcoate, C.R.; et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J. Biol. Chem. 2018, 293, 1994–2005. [Google Scholar] [CrossRef]
- Hu, W.; Zhao, J.; Pei, G. Activation of aryl hydrocarbon receptor (AhR) by tranilast, an anti-allergy drug, promotes miR-302 expression and cell reprogramming. J. Biol. Chem. 2013, 288, 22972–22984. [Google Scholar] [CrossRef]
- Miller, C.A., 3rd. Expression of the human aryl hydrocarbon receptor complex in yeast. Activation of transcription by indole compounds. J. Biol. Chem. 1997, 272, 32824–32829. [Google Scholar] [CrossRef] [PubMed]
- Baricza, E.; Tamasi, V.; Marton, N.; Buzas, E.I.; Nagy, G. The emerging role of aryl hydrocarbon receptor in the activation and differentiation of Th17 cells. Cell. Mol. Life Sci. 2016, 73, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Barisione, C.; Garibaldi, S.; Furfaro, A.L.; Nitti, M.; Palmieri, D.; Passalacqua, M.; Garuti, A.; Verzola, D.; Parodi, A.; Ameri, P.; et al. Moderate Increase of Indoxyl Sulfate Promotes Monocyte Transition into Profibrotic Macrophages. PLoS ONE 2016, 11, e0149276. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Barroso, A.; Gualdron-Lopez, M.; Esper, L.; Brant, F.; Araujo, R.R.; Carneiro, M.B.; Avila, T.V.; Souza, D.G.; Vieira, L.Q.; Rachid, M.A.; et al. The Aryl Hydrocarbon Receptor Modulates Production of Cytokines and Reactive Oxygen Species and Development of Myocarditis during Trypanosoma cruzi Infection. Infect. Immun. 2016, 84, 3071–3082. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Huai, W.; Zhao, R.; Song, H.; Zhao, J.; Zhang, L.; Zhang, L.; Gao, C.; Han, L.; Zhao, W. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat. Commun. 2014, 5, 4738. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Parga, T.; Suryawanshi, A.; Rouse, B.T. Controlling viral immuno-inflammatory lesions by modulating aryl hydrocarbon receptor signaling. PLoS Pathog. 2011, 7, e1002427. [Google Scholar] [CrossRef] [PubMed]
- Wagage, S.; John, B.; Krock, B.L.; Hall, A.O.; Randall, L.M.; Karp, C.L.; Simon, M.C.; Hunter, C.A. The aryl hydrocarbon receptor promotes IL-10 production by NK cells. J. Immunol. 2014, 192, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- NIH. NINDS Multiple Sclerosis Information Page. Available online: https://www.ninds.nih.gov/disorders/all-disorders/multiple-sclerosis-information-page (accessed on 2 December 2018).
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS ONE 2013, 8, e79819. [Google Scholar] [CrossRef] [PubMed]
- Juricek, L.; Carcaud, J.; Pelhaitre, A.; Riday, T.T.; Chevallier, A.; Lanzini, J.; Auzeil, N.; Laprevote, O.; Dumont, F.; Jacques, S.; et al. AhR-deficiency as a cause of demyelinating disease and inflammation. Sci. Rep. 2017, 7, 9794. [Google Scholar] [CrossRef] [PubMed]
- Stange, J.; Veldhoen, M. The aryl hydrocarbon receptor in innate T cell immunity. Semin. Immunopathol. 2013, 35, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.U.; McPherson, Z.E.; Tan, B.; Korecka, A.; Pettersson, S. Host-microbiome interactions: The aryl hydrocarbon receptor and the central nervous system. J. Mol. Med. 2017, 95, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Goudot, C.; Coillard, A.; Villani, A.C.; Gueguen, P.; Cros, A.; Sarkizova, S.; Tang-Huau, T.L.; Bohec, M.; Baulande, S.; Hacohen, N.; et al. Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages. Immunity 2017, 47, 582–596.e6. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, L. Aryl hydrocarbon receptor promotes RORγt+ group 3 ILCs and controls intestinal immunity and inflammation. Semin. Immunopathol. 2013, 35, 657–670. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, K.; Han, B.; Sheng, B.; Yin, J.; Pu, A.; Li, L.; Sun, L.; Yu, M.; Qiu, Y.; et al. Aryl hydrocarbon receptor inhibits inflammation in DSS-induced colitis via the MK2/p-MK2/TTP pathway. Int. J. Mol. Med. 2018, 41, 868–876. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, K.; Qiao, S.M.; Dai, Y.; Wei, Z.F. Norisoboldine, a natural aryl hydrocarbon receptor agonist, alleviates TNBS-induced colitis in mice, by inhibiting the activation of NLRP3 inflammasome. Chin. J. Nat. Med. 2018, 16, 161–174. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, K.; Qiao, S.; Yang, L.; Xin, Y.; Dai, Y.; Wei, Z. Norisoboldine, a natural AhR agonist, promotes Treg differentiation and attenuates colitis via targeting glycolysis and subsequent NAD+/SIRT1/SUV39H1/H3K9me3 signaling pathway. Cell Death Dis. 2018, 9, 258. [Google Scholar] [CrossRef]
- Wei, Z.F.; Lv, Q.; Xia, Y.; Yue, M.F.; Shi, C.; Xia, Y.F.; Chou, G.X.; Wang, Z.T.; Dai, Y. Norisoboldine, an Anti-Arthritis Alkaloid Isolated from Radix Linderae, Attenuates Osteoclast Differentiation and Inflammatory Bone Erosion in an Aryl Hydrocarbon Receptor-Dependent Manner. Int. J. Biol. Sci. 2015, 11, 1113–1126. [Google Scholar] [CrossRef]
- Kazantseva, M.G.; Highton, J.; Stamp, L.K.; Hessian, P.A. Dendritic cells provide a potential link between smoking and inflammation in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R208. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a Natural AhR Agonist that Resolves Skin Inflammation in Mice and Humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef]
- Bridgman, A.C.; Kirchhof, M.G. Treatment of psoriasis vulgaris using low-dose naltrexone. JAAD Case Rep. 2018, 4, 827–829. [Google Scholar] [CrossRef]
- Xiao, L.; Zhang, Z.; Luo, X. Roles of xenobiotic receptors in vascular pathophysiology. Circ. J. 2014, 78, 1520–1530. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Wan, M.; Ding, J.; Taylor, J.R.; Lohman, K.; Su, D.; Bennett, B.D.; Porter, D.K.; Gimple, R.; Pittman, G.S.; et al. DNA Methylation of the Aryl Hydrocarbon Receptor Repressor Associations with Cigarette Smoking and Subclinical Atherosclerosis. Circ. Cardiovasc. Genet. 2015, 8, 707–716. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco Smoking Leads to Extensive Genome-Wide Changes in DNA Methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Kerley-Hamilton, J.S.; Trask, H.W.; Ridley, C.J.; Dufour, E.; Lesseur, C.; Ringelberg, C.S.; Moodie, K.L.; Shipman, S.L.; Korc, M.; Gui, J.; et al. Inherent and benzo[a]pyrene-induced differential aryl hydrocarbon receptor signaling greatly affects life span, atherosclerosis, cardiac gene expression, and body and heart growth in mice. Toxicol. Sci. 2012, 126, 391–404. [Google Scholar] [CrossRef]
- Marinkovic, N.; Pasalic, D.; Potocki, S. Polymorphisms of genes involved in polycyclic aromatic hydrocarbons’ biotransformation and atherosclerosis. Biochem. Medica 2013, 23, 255–265. [Google Scholar] [CrossRef]
- Pernomian, L.; da Silva, C.H. Current basis for discovery and development of aryl hydrocarbon receptor antagonists for experimental and therapeutic use in atherosclerosis. Eur. J. Pharmacol. 2015, 764, 118–123. [Google Scholar] [CrossRef]
- Kim, J.B.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Iyer, D.; Liu, B.; Wang, T.; Sazonova, O.; Carcamo-Orive, I.; Matic, L.P.; et al. TCF21 and the environmental sensor aryl-hydrocarbon receptor cooperate to activate a pro-inflammatory gene expression program in coronary artery smooth muscle cells. PLoS Genet. 2017, 13, e1006750. [Google Scholar] [CrossRef]
- Cornelis, M.C.; Byrne, E.M.; Esko, T.; Nalls, M.A.; Ganna, A.; Paynter, N.; Monda, K.L.; Amin, N.; Fischer, K.; Renstrom, F.; et al. Genome-wide meta-analysis identifies six novel loci associated with habitual coffee consumption. Mol. Psychiatry 2015, 20, 647–656. [Google Scholar] [CrossRef]
- Tsaprouni, L.G.; Yang, T.P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Vinuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef]
- Luo, Z.; Li, X.; Zhu, M.; Tang, J.; Li, Z.; Zhou, X.; Song, G.; Liu, Z.; Zhou, H.; Zhang, W. Identification of novel variants associated with warfarin stable dosage by use of a two-stage extreme phenotype strategy. J. Thromb. Haemost. 2017, 15, 28–37. [Google Scholar] [CrossRef]
- Li, R.; Shugart, Y.Y.; Zhou, W.; An, Y.; Yang, Y.; Zhou, Y.; Zhang, B.; Lu, D.; Wang, H.; Qian, J.; et al. Common genetic variations of the cytochrome P450 1A1 gene and risk of hepatocellular carcinoma in a Chinese population. Eur. J. Cancer 2009, 45, 1239–1247. [Google Scholar] [CrossRef]
- Zou, J.G.; Ma, Y.T.; Xie, X.; Yang, Y.N.; Pan, S.; Adi, D.; Liu, F.; Chen, B.D. The association between CYP1A1 genetic polymorphisms and coronary artery disease in the Uygur and Han of China. Lipids Health Dis. 2014, 13, 145. [Google Scholar] [CrossRef]
- Philibert, R.A.; Beach, S.R.; Lei, M.K.; Brody, G.H. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin. Epigenet. 2013, 5, 19. [Google Scholar] [CrossRef]
- Dogan, M.V.; Shields, B.; Cutrona, C.; Gao, L.; Gibbons, F.X.; Simons, R.; Monick, M.; Brody, G.H.; Tan, K.; Beach, S.R.; et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genom. 2014, 15, 151. [Google Scholar] [CrossRef]
- Liu, D.; Qin, S.; Ray, B.; Kalari, K.R.; Wang, L.; Weinshilboum, R.M. Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction. Drug Metab. Dispos. 2018, 46, 1372–1381. [Google Scholar] [CrossRef]
- Ho, M.F.; Bongartz, T.; Liu, M.; Kalari, K.R.; Goss, P.E.; Shepherd, L.E.; Goetz, M.P.; Kubo, M.; Ingle, J.N.; Wang, L.; et al. Estrogen, SNP-Dependent Chemokine Expression and Selective Estrogen Receptor Modulator Regulation. Mol. Endocrinol. 2016, 30, 382–398. [Google Scholar] [CrossRef]
- Ho, M.F.; Ingle, J.N.; Bongartz, T.; Kalari, K.R.; Goss, P.E.; Shepherd, L.E.; Mushiroda, T.; Kubo, M.; Wang, L.; Weinshilboum, R.M. TCL1A Single-Nucleotide Polymorphisms and Estrogen-Mediated Toll-Like Receptor-MYD88-Dependent Nuclear Factor-κB Activation: Single-Nucleotide Polymorphism- and Selective Estrogen Receptor Modulator-Dependent Modification of Inflammation and Immune Response. Mol. Pharmacol. 2017, 92, 175–184. [Google Scholar] [CrossRef]
- Liu, M.; Wang, L.; Bongartz, T.; Hawse, J.R.; Markovic, S.N.; Schaid, D.J.; Mushiroda, T.; Kubo, M.; Nakamura, Y.; Kamatani, N.; et al. Aromatase inhibitors, estrogens and musculoskeletal pain: Estrogen-dependent T-cell leukemia 1A (TCL1A) gene-mediated regulation of cytokine expression. Breast Cancer Res. 2012, 14, R41. [Google Scholar] [CrossRef]
- Qin, S.; Ingle, J.N.; Liu, M.; Yu, J.; Wickerham, D.L.; Kubo, M.; Weinshilboum, R.M.; Wang, L. Calmodulin-like protein 3 is an estrogen receptor alpha coregulator for gene expression and drug response in a SNP, estrogen, and SERM-dependent fashion. Breast Cancer Res. 2017, 19, 95. [Google Scholar] [CrossRef]
- Ingle, J.N.; Liu, M.; Wickerham, D.L.; Schaid, D.J.; Wang, L.; Mushiroda, T.; Kubo, M.; Costantino, J.P.; Vogel, V.G.; Paik, S.; et al. Selective estrogen receptor modulators and pharmacogenomic variation in ZNF423 regulation of BRCA1 expression: Individualized breast cancer prevention. Cancer Discov. 2013, 3, 812–825. [Google Scholar] [CrossRef]
- Ghigliotti, G.; Barisione, C.; Garibaldi, S.; Fabbi, P.; Brunelli, C.; Spallarossa, P.; Altieri, P.; Rosa, G.; Spinella, G.; Palombo, D.; et al. Adipose tissue immune response: Novel triggers and consequences for chronic inflammatory conditions. Inflammation 2014, 37, 1337–1353. [Google Scholar] [CrossRef]
- Jurado-Manzano, B.B.; Zavala-Reyes, D.; Turrubiartes-Martinez, E.A.; Portales-Perez, D.P.; Gonzalez-Amaro, R.; Layseca-Espinosa, E. FICZ generates human tDCs that induce CD4+ CD25(high) Foxp3+ Treg-like cell differentiation. Immunol. Lett. 2017, 190, 84–92. [Google Scholar] [CrossRef]
- Lawrence, B.P.; Denison, M.S.; Novak, H.; Vorderstrasse, B.A.; Harrer, N.; Neruda, W.; Reichel, C.; Woisetschlager, M. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low-molecular-weight compound. Blood 2008, 112, 1158–1165. [Google Scholar] [CrossRef]
- Zhang, S.; Patel, A.; Chu, C.; Jiang, W.; Wang, L.; Welty, S.E.; Moorthy, B.; Shivanna, B. Aryl hydrocarbon receptor is necessary to protect fetal human pulmonary microvascular endothelial cells against hyperoxic injury: Mechanistic roles of antioxidant enzymes and RelB. Toxicol. Appl. Pharmacol. 2015, 286, 92–101. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ligand | Abbreviation | EC50 (M) * | Exogenous/Endogenous | Synthetic/Natural | Source |
|---|---|---|---|---|---|
| 2,3,7,8-Tetrachlorodibenzo-p-dioxin | TCDD | 10−11–10−9 [24,25,26,27,28] | Exogenous | Synthetic | Chemical contaminant (i.e., Agent orange contaminant) |
| 6-Formylindolo[3,2-b]carbazole | FICZ | 10−11–10−10 [29,30] | Endogenous | Natural | Ultraviolet derivative of tryptophan |
| Benzo[a]pyrene | BaP | 10−9–10−8 [28,31] | Exogenous | Synthetic | Product of burning of organic compounds and cigarette smoke |
| 3-Methylcholanthrene | 3-MC | 10−9–10−6 [27,28,30] | Exogenous | Synthetic | Product of burning of organic compounds |
| Kynurenine | KYN | 10−9–10−5 [29] | Endogenous | Natural | Tryptophan metabolite |
| β-napthoflavone | BNF | 10−8 [31] | Exogenous | Synthetic | Flavone derivative |
| α-napthoflavone | ANF | 10−7 [28] | Exogenous | Synthetic | Flavone derivative |
| Indolo[3,2-b]carbazole | ICZ | 10−7 [25] | Endogenous | Natural | Indole-3-carbinol derivative |
| Diindolylmethane | DIM | 10−5 [30] | Exogenous | Natural | Indole-3-carbinol derivative |
| Indole-3-carbinol | I3C | 10−3–10−5 [31] | Exogenous | Natural | Cruciferous vegetables |
| Tryptophan | TRP | 10−4 [31] | Exogenous | Natural | Essential amino acid |
| Indole-3-acetic acid | IAA | 10−4 [26,31] | Exogenous & Endogenous | Natural | Microbiome product Tryptophan metabolite |
| Tryptamine | TRYP | 10−4 [26,31] | Endogenous | Natural | Tryptophan metabolite |
| Norisoboldine | NOR | NA | Exogenous | Natural | Alkaloid isolated from Radix Linderae |
| Disease | AHR Ligands | AHR Activation Phenotype | AHR Inactivation Method | AHR Inactivation Phenotype | Contributing Pathways | References |
|---|---|---|---|---|---|---|
| LPS-induced septic shock | 3-MC, TCDD, FICZ, KYN | Decreased death | AHR−/− | Increased death | IDO/TDO activation; TRP metabolism | [15,21,38] |
| Listeria Monocytogenes | TCCD, FICZ | Decreased death | AHR−/− | Increased death | ROS formation and cytokine expression | [19] |
| Herpes-simplex virus-induced ocular Infection | TCDD | Decrease herpes keratitis lesions | NA | NA | Unclear but decreased numbers of inflammatory IFN-γ+ secreting CD4+ T cells (Th1) and Th17 cells | [39] |
| FICZ | No effect | |||||
| Toxoplasma gondii infection | NA | NA | AHR−/− | Decreased anti-inflammatory response | IL-10 expression | [40] |
| Major depressive disorder | AHR SNP eQTL, 3-MC | Worse MDD symptoms, increased KYN | AHR SNP eQTL, AHR KD | Less MDD symptoms, decreased KYN | TRP metabolism; IDO/TDO, KMO, KYNU activation | [23] |
| Multiple Sclerosis | TCDD, I3C, DIM | Decreased disease scores | AHR KD | Increased disease scores | FOXP3 expression, Treg expansion, Th17 expansion | [6,8,14,18,41,42,43] |
| FICZ | Systemic exposure: decreased disease scores; Local administration: increased disease scores | |||||
| Congenital nystagmus | NA | NA | AHR−/− | Development of congenital nystagmus | Proinflammatory cytokine expression, STAT1 | [44] |
| Gut microbiome | TRP indoles | DC differentiation, ILC balance | AHR−/−, Remove diet TRP, antibiotics | Pathogen susceptibility, ILC balance, DC differentiation | Unclear | [17,18,45,46,47] |
| Inflammatory Bowel Disease | TCDD, NOR, FICZ | Relieve colitis symptoms | AHR−/− | More severe colitis symptoms | Pro-inflammatory cytokine expression, Th17 differentiation, Treg differentiation, NLRP3 inflammasome expression | [18,48,49,50,51] |
| Rheumatoid Arthritis | TCDD, FICZ | Increased disease severity | AHR−/− | Decreased disease severity | Pro-inflammatory cytokine expression, NF-κB | [3,9,11,43,52,53] |
| NOR | Decreased disease severity | |||||
| Psoriasis | FICZ, tapinarof | Decreased disease severity | AHR−/− | Increased disease severity | Pro-inflammatory cytokine expression, keratinocyte interaction with adaptive immune system | [7,20,54,55] |
| Atherosclerosis | TCDD, BaP | Increased disease severity | AHR low affinity | Increased disease severity | Pro-inflammatory cytokine expression, reactive oxygen species, TCF21 interactions | [56,57,58,59,60,61,62] |
| Compound | Administration Method | Target Diseases | Comments |
|---|---|---|---|
| Tapinarof | Topical | Psoriasis | Currently in Phase III clinical trials; highly effective; safe |
| NOR | Depends | IBD, RA | Clinical safety studies will be required |
| I3C/DIM | Oral | IBD, MS, gut microbiome balance | Normally consumed in cruciferous vegetables |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. https://doi.org/10.3390/ijms19123851
Neavin DR, Liu D, Ray B, Weinshilboum RM. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. International Journal of Molecular Sciences. 2018; 19(12):3851. https://doi.org/10.3390/ijms19123851
Chicago/Turabian StyleNeavin, Drew R., Duan Liu, Balmiki Ray, and Richard M. Weinshilboum. 2018. "The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases" International Journal of Molecular Sciences 19, no. 12: 3851. https://doi.org/10.3390/ijms19123851
APA StyleNeavin, D. R., Liu, D., Ray, B., & Weinshilboum, R. M. (2018). The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. International Journal of Molecular Sciences, 19(12), 3851. https://doi.org/10.3390/ijms19123851