NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
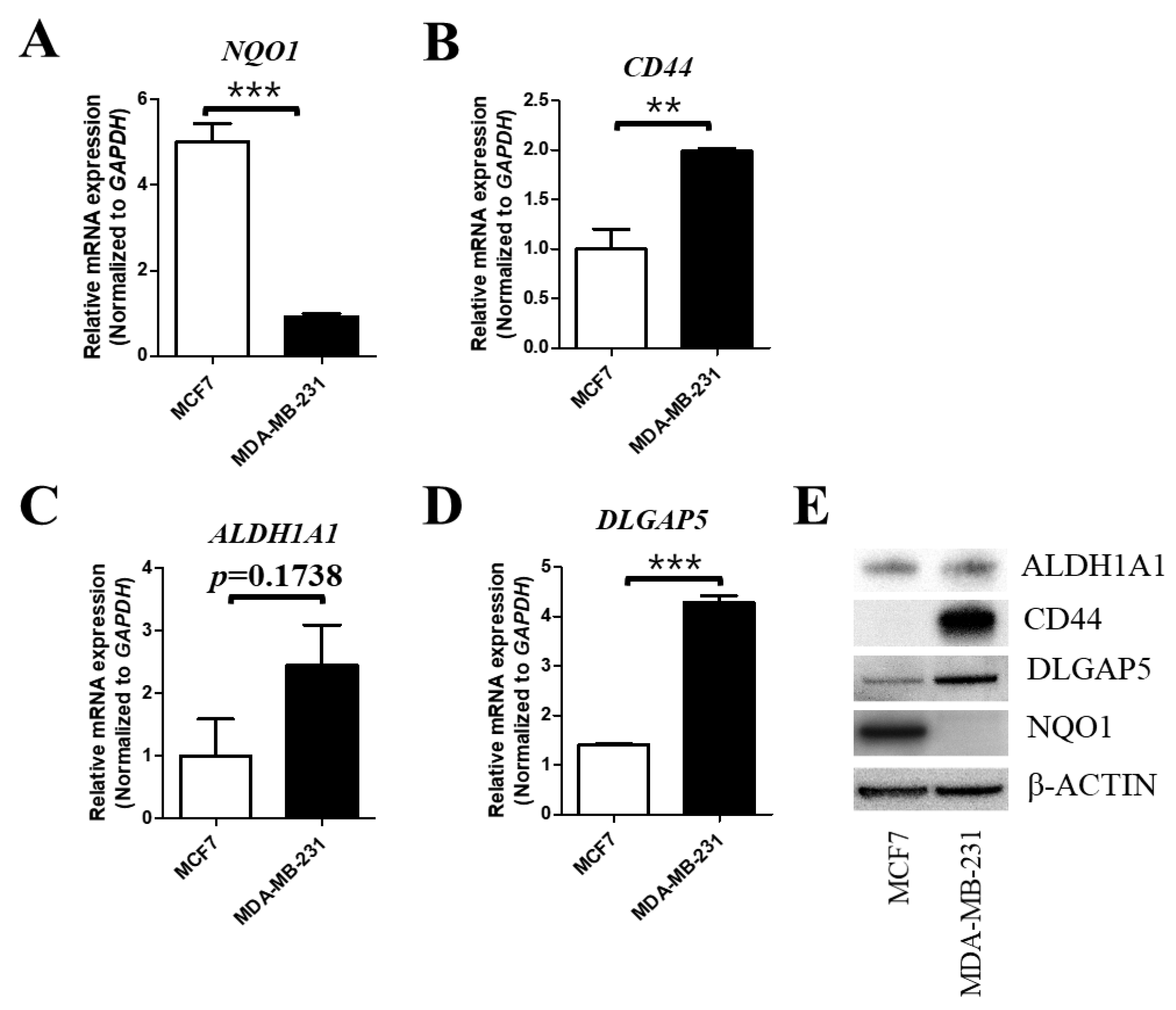
2.1. The Cellular Expression Level of NQO1 is Negatively Correlated with That of BCSC Markers
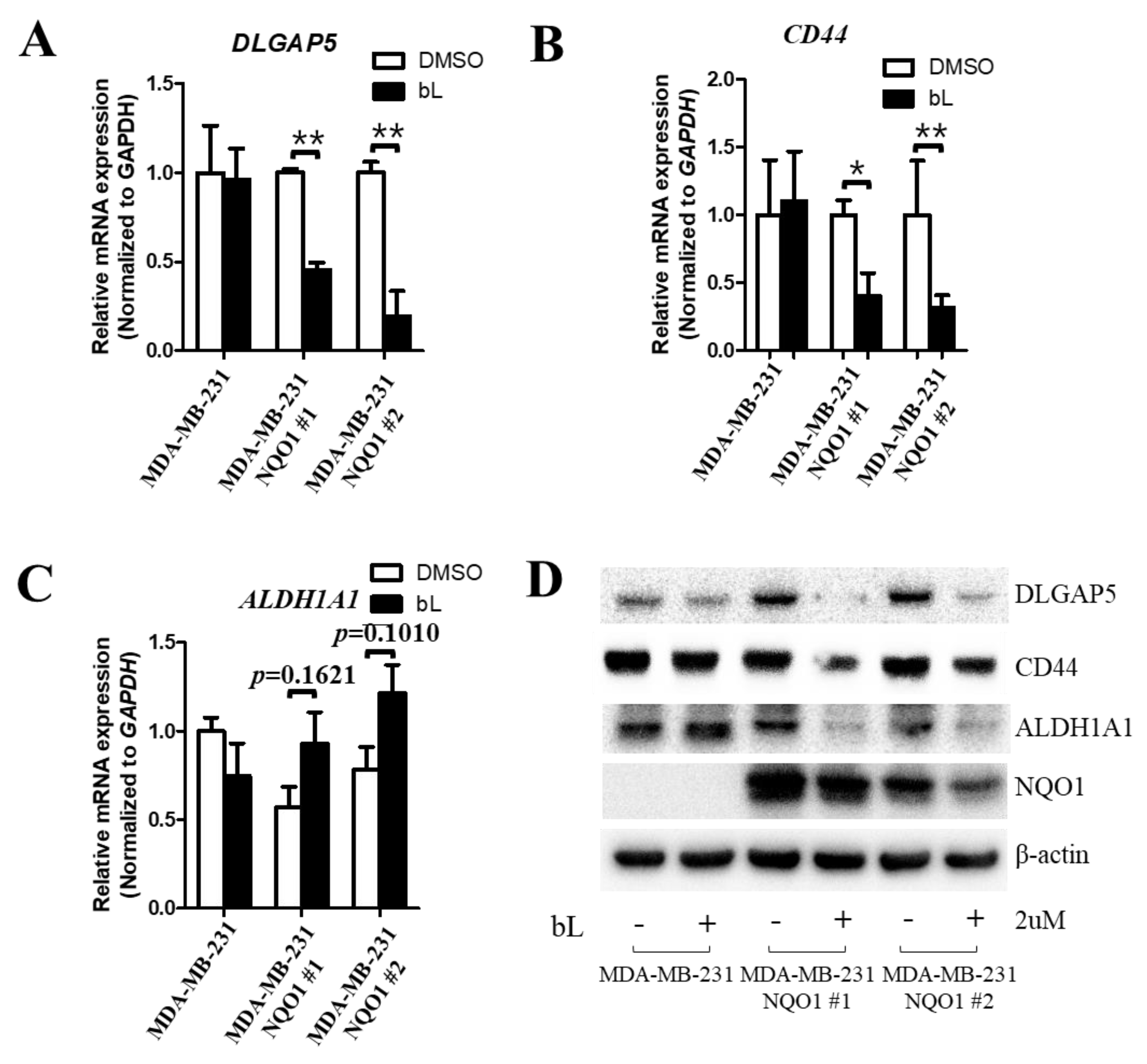
2.2. β-Lapachone-Mediated NQO1 Activation Regulates DLGAP5 and CD44 Expression Levels
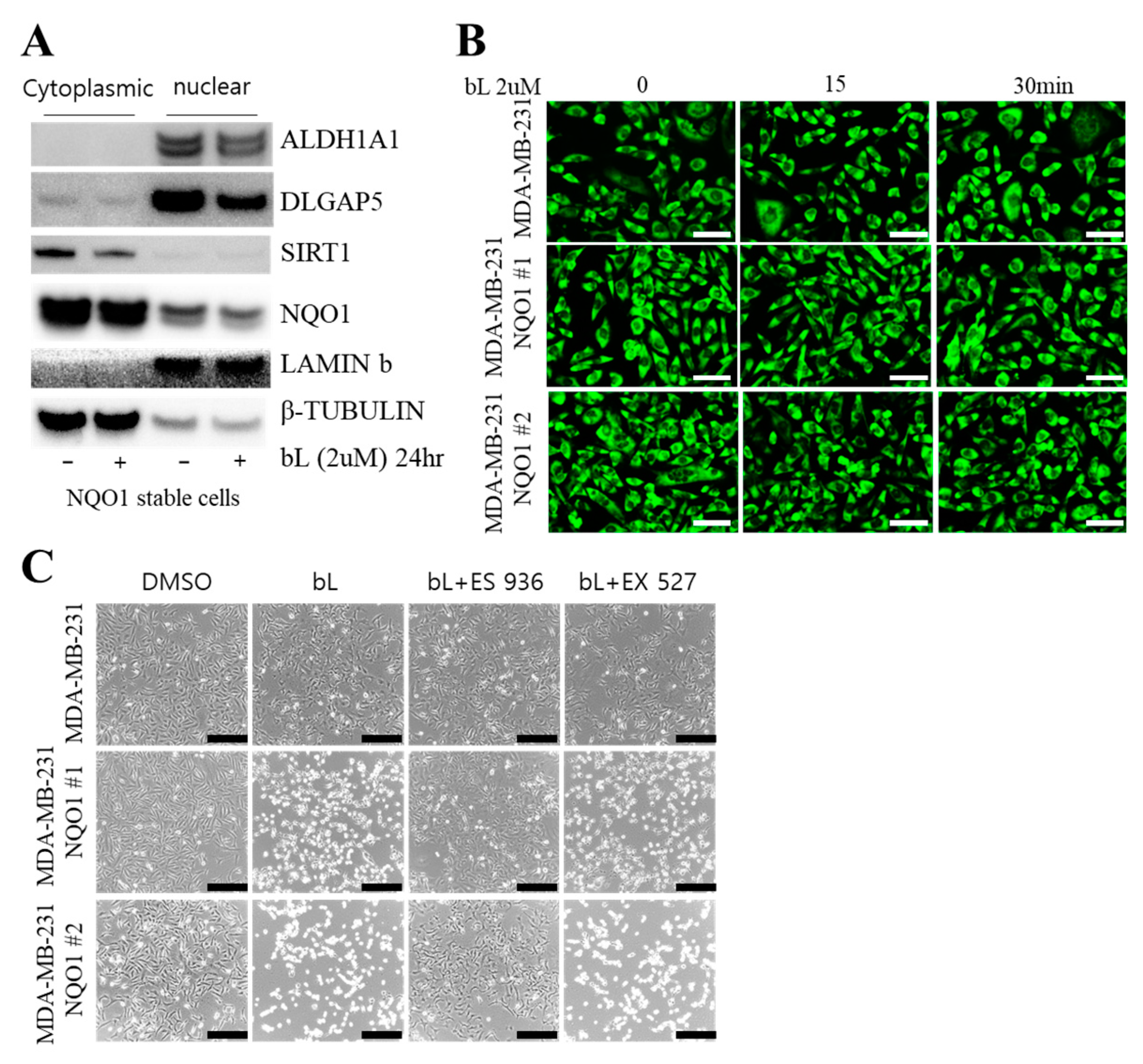
2.3. Sirtuin 1 (SIRT1) Is Not Involved in bL-NQO1-Mediated Gene Expression and Cell Death
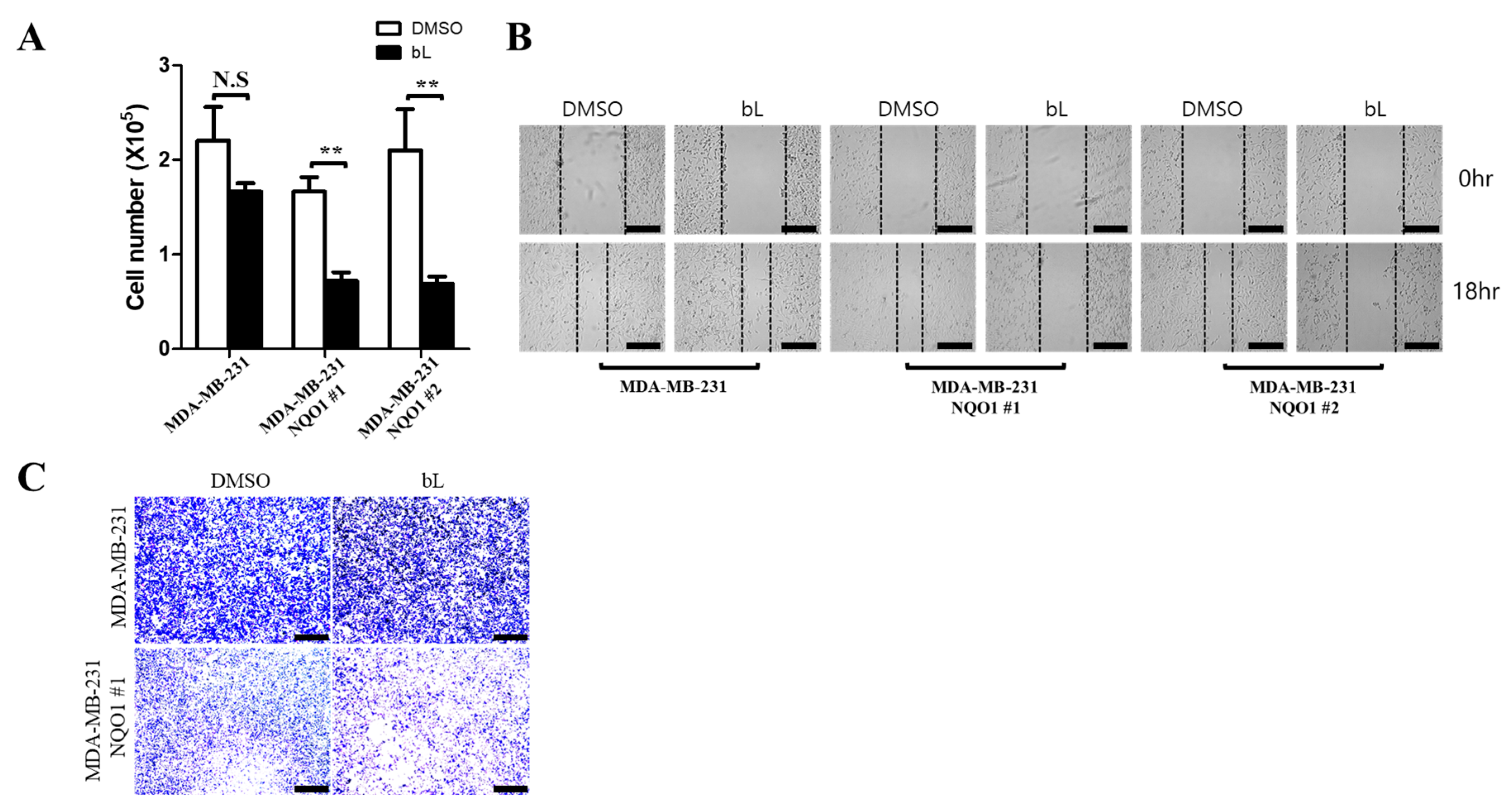
2.4. NQO1 Activated by bL Decreases Cell Proliferation and Migration
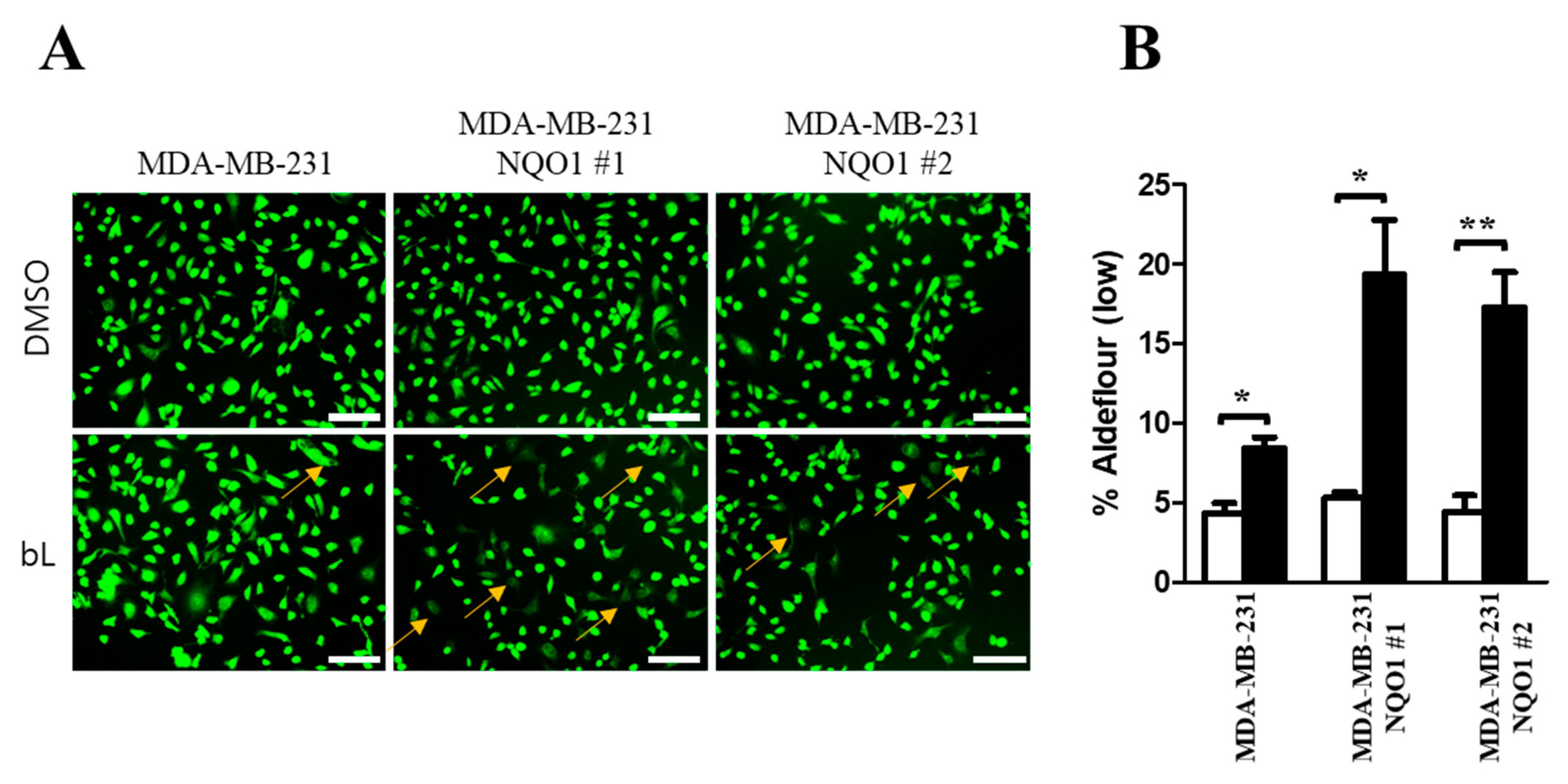
2.5. ALDH1 Activity Is Decreased by bL through NQO1
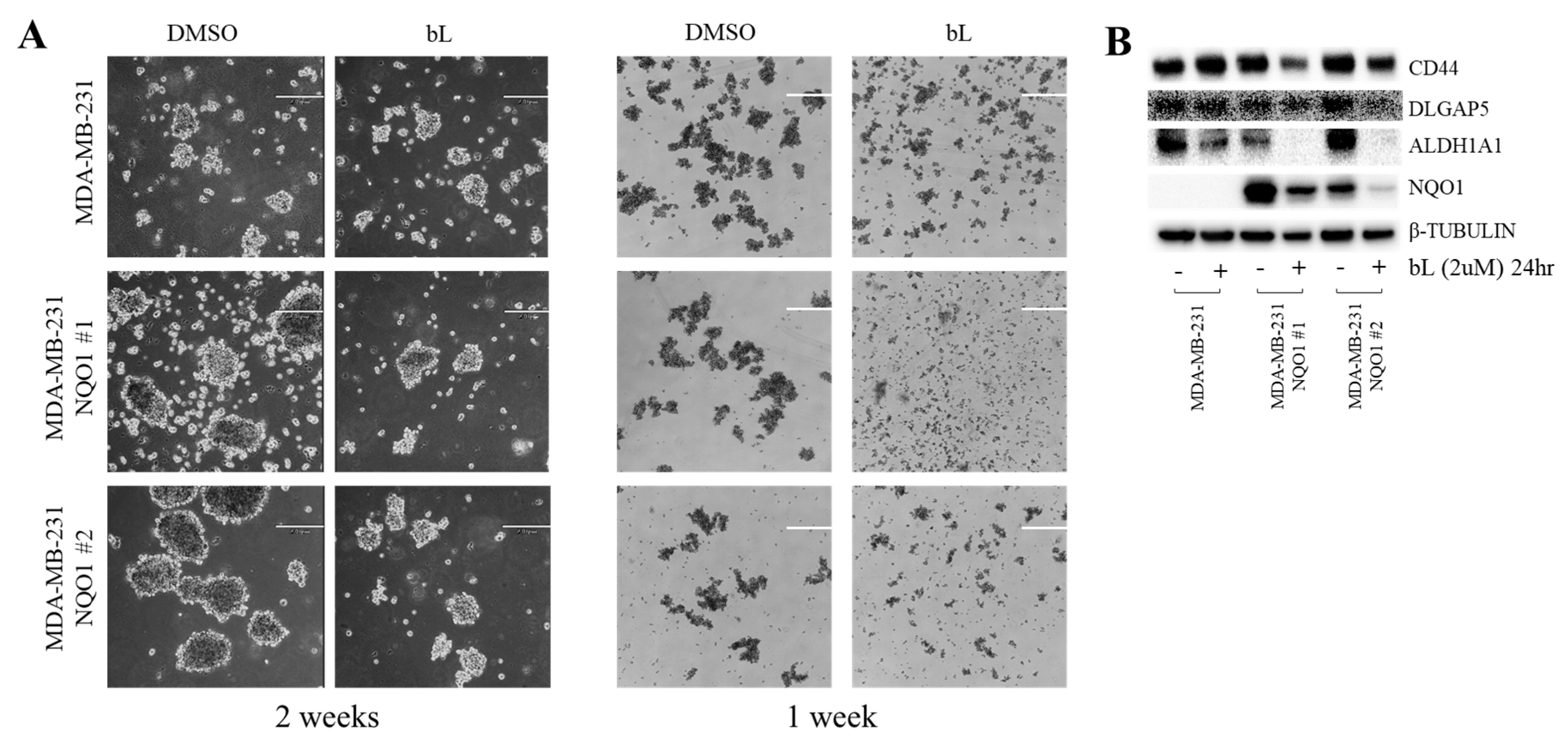
2.6. Mammosphere Formation Was Inhibited by bL-NQO1
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Stable Cell Line Establishment
4.2. Quantitative Real-Time PCR
4.3. Western Blot Analysis
4.4. Immunocytochemistry
4.5. Wound Healing Assay
4.6. ALDEFLUOR Assay
4.7. Migration Assay
4.8. Mammosphere Formation
4.9. Cytoplasmic and Nuclear Fractionation
4.10. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| bL | β-lapachone |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| CSCs | cancer stem cells |
| BCSCs | breast-cancer stem-cells |
| CD44 | CD44 molecules |
| DLGAP5 | DLG-associated protein 5 |
| ALDH1A1 | aldehyde dehydrogenase 1 family member A1 |
| SIRT1 | sirtuin 1 |
| NAD | nicotinamide adenine dinucleotide |
| ROS | reactive oxygen species |
References
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Lim, C.L. Breast cancer stem cells—From origins to targeted therapy. Stem Cell Investig. 2017, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, M.J.; Ahn, S.H.; Son, B.H.; Kim, S.B.; Ahn, J.H.; Noh, W.C.; Gong, G. Different prognostic significance of CD24 and CD44 expression in breast cancer according to hormone receptor status. Breast 2011, 20, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Ucar, D.; Han, S.; Amory, J.K.; Goldstein, A.S.; Ostmark, B.; Chang, L.J. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem.-Biol. Interact. 2012, 195, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.K.; Kanojia, D.; Liu, X.; Singh, U.P.; Berger, F.G.; Wang, Q.; Chen, H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFβ signaling. Oncogene 2012, 31, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Ross, D.D. Breast cancer resistance protein (BCRP/ABCG2): Its role in multidrug resistance and regulation of its gene expression. Chin. J. Cancer 2012, 31, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, E.; Alvarez, P.J.; Prados, J.; Melguizo, C.; Rama, A.R.; Aranega, A.; Rodriguez-Serrano, F. Cancer stem cells and their implication in breast cancer. Eur. J. Clin. Investig. 2014, 44, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013, 73, 7290–7300. [Google Scholar] [CrossRef] [PubMed]
- Van Oorschot, B.; Granata, G.; Di Franco, S.; Ten Cate, R.; Rodermond, H.M.; Todaro, M.; Medema, J.P.; Franken, N.A. Targeting DNA double strand break repair with hyperthermia and DNA-PKcs inhibition to enhance the effect of radiation treatment. Oncotarget 2016, 7, 65504–65513. [Google Scholar] [CrossRef] [PubMed]
- Czerwinska, P.; Kaminska, B. Regulation of breast cancer stem cell features. Contemp. Oncol. 2015, 19, A7–A15. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radio resistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.B.; Li, Y.Z.; Li, C.J. Cancer therapy with β-lapachone. Curr. Cancer Drug Targets 2002, 2, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Boothman, D.A.; Greer, S.; Pardee, A.B. Potentiation of halogenated pyrimidine radiosensitizers in human carcinoma cells by β-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran-5,6-dione), a novel DNA repair inhibitor. Cancer Res. 1987, 47, 5361–5366. [Google Scholar] [PubMed]
- Costa, M.P.; Feitosa, A.C.; Oliveira, F.C.; Cavalcanti, B.C.; da Silva, E.N.; Dias, G.G.; Sales, F.A.; Sousa, B.L.; Barroso-Neto, I.L.; Pessoa, C.; et al. Controlled Release of Nor-β-lapachone by PLGA Microparticles: A Strategy for Improving Cytotoxicity against Prostate Cancer Cells. Molecules 2016, 21, 873. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Bey, E.A.; Li, L.S.; Kabbani, W.; Yan, J.; Xie, X.J.; Hsieh, J.T.; Gao, J.; Boothman, D.A. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010, 70, 8088–8096. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Wang, C.; Pardee, A.B. Induction of apoptosis by β-lapachone in human prostate cancer cells. Cancer Res. 1995, 55, 3712–3715. [Google Scholar] [PubMed]
- Li, C.J.; Li, Y.Z.; Pinto, A.V.; Pardee, A.B. Potent inhibition of tumor survival in vivo by β-lapachone plus taxol: Combining drugs imposes different artificial checkpoints. Proc. Natl. Acad. Sci. USA 1999, 96, 13369–13374. [Google Scholar] [CrossRef] [PubMed]
- Kee, J.Y.; Han, Y.H.; Park, J.; Kim, D.S.; Mun, J.G.; Ahn, K.S.; Kim, H.J.; Um, J.Y.; Hong, S.H. β-Lapachone Inhibits Lung Metastasis of Colorectal Cancer by Inducing Apoptosis of CT26 Cells. Integr. Cancer Ther. 2017, 16, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Q.; Cheng, X.; Wang, H.; Wang, G.; Hao, H. UDP-glucuronosyltransferase 1A determinates intracellular accumulation and anti-cancer effect of β-lapachone in human colon cancer cells. PLoS ONE 2015, 10, e0117051. [Google Scholar] [CrossRef] [PubMed]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by β-lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, X.; Xu, M.; Piao, J.; Zhang, Y.; Lin, Z.; Chen, L. β-lapachone suppresses tumour progression by inhibiting epithelial-to-mesenchymal transition in NQO1-positive breast cancers. Sci. Rep. 2017, 7, 2681. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Averboukh, L.; Pardee, A.B. β-Lapachone, a novel DNA topoisomerase I inhibitor with a mode of action different from camptothecin. J. Biol. Chem. 1993, 268, 22463–22468. [Google Scholar] [PubMed]
- Li, Y.Z.; Li, C.J.; Pinto, A.V.; Pardee, A.B. Release of mitochondrial cytochrome C in both apoptosis and necrosis induced by β-lapachone in human carcinoma cells. Mol. Med. 1999, 5, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.S.; Aubry, D.; Ginet, V.; Puyal, J.; Heulot, M.; Widmann, C.; Duchosal, M.A.; Nahimana, A. Combinative effects of β-Lapachone and APO866 on pancreatic cancer cell death through reactive oxygen species production and PARP-1 activation. Biochimie 2015, 116, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Min, K.J.; Lee, T.J.; Yoo, Y.H.; Kim, Y.S.; Kwon, T.K. β-Lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis. 2014, 5, e1230. [Google Scholar] [CrossRef] [PubMed]
- Boothman, D.A.; Pardee, A.B. Inhibition of radiation-induced neoplastic transformation by β-lapachone. Proc. Natl. Acad. Sci. USA 1989, 86, 4963–4967. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Bang, W.; Shin, J.C.; Park, S.M.; Cho, J.J.; Choi, Y.H.; Seo, K.S.; Choi, N.J.; Shim, J.H.; Chae, J.I. Downregulation of Sp1 is involved in β-lapachone-induced cell cycle arrest and apoptosis in oral squamous cell carcinoma. Int. J. Oncol. 2015, 46, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Shiah, S.G.; Chuang, S.E.; Chau, Y.P.; Shen, S.C.; Kuo, M.L. Activation of c-Jun NH2-terminal kinase and subsequent CPP32/Yama during topoisomerase inhibitor β-lapachone-induced apoptosis through an oxidation-dependent pathway. Cancer Res. 1999, 59, 391–398. [Google Scholar] [PubMed]
- Yu, H.Y.; Kim, S.O.; Jin, C.Y.; Kim, G.Y.; Kim, W.J.; Yoo, Y.H.; Choi, Y.H. β-lapachone-Induced Apoptosis of Human Gastric Carcinoma AGS Cells Is Caspase-Dependent and Regulated by the PI3K/Akt Pathway. Biomol. Ther. 2014, 22, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Silvers, M.A.; Deja, S.; Singh, N.; Egnatchik, R.A.; Sudderth, J.; Luo, X.; Beg, M.S.; Burgess, S.C.; DeBerardinis, R.J.; Boothman, D.A.; et al. The NQO1 bioactivatable drug, β-lapachone, alters the redox state of NQO1+ pancreatic cancer cells, causing perturbation in central carbon metabolism. J. Biol. Chem. 2017, 292, 18203–18216. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Chin, S.F.; Blanco, E.; Bey, E.A.; Kabbani, W.; Xie, X.J.; Bornmann, W.G.; Boothman, D.A.; Gao, J. Intratumoral delivery of β-lapachone via polymer implants for prostate cancer therapy. Clin. Cancer Res. 2009, 15, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of β-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.P.; Shiah, S.G.; Don, M.J.; Kuo, M.L. Involvement of hydrogen peroxide in topoisomerase inhibitor β-lapachone-induced apoptosis and differentiation in human leukemia cells. Free Radic. Biol. Med. 1998, 24, 660–670. [Google Scholar] [CrossRef]
- Cruz, F.S.; Docampo, R.; Boveris, A. Generation of superoxide anions and hydrogen peroxide from β-lapachone in bacteria. Antimicrob. Agents Chemother. 1978, 14, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Rauth, A.M.; Goldberg, Z.; Misra, V. DT-diaphorase: Possible roles in cancer chemotherapy and carcinogenesis. Oncol. Res. 1997, 9, 339–349. [Google Scholar] [PubMed]
- Planchon, S.M.; Pink, J.J.; Tagliarino, C.; Bornmann, W.G.; Varnes, M.E.; Boothman, D.A. β-Lapachone-induced apoptosis in human prostate cancer cells: Involvement of NQO1/xip3. Exp. Cell Res. 2001, 267, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sun, J.; Tan, Y.; Li, Z.; Kong, F.; Shen, Y.; Liu, C.; Chen, L. Prognostic implication of NQO1 overexpression in hepatocellular carcinoma. Hum. Pathol. 2017, 69, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Sandoval, J.M.; Dejeans, N.; Ameye, G.; Poirel, H.A.; Verrax, J.; Calderon, P.B. Overexpression of NAD(P)H:quinone oxidoreductase 1 (NQO1) and genomic gain of the NQO1 locus modulates breast cancer cell sensitivity to quinones. Life Sci. 2016, 145, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Wu, Q.; Cui, X.; Lin, Z.; Liu, S.; Chen, L. Clinical implications of high NQO1 expression in breast cancers. J. Exp. Clin. Cancer Res. 2014, 33, 14. [Google Scholar] [CrossRef] [PubMed]
- Dayem, A.A.; Choi, H.Y.; Kim, J.H.; Cho, S.G. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers 2010, 2, 859–884. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Lee, S.H.; Kwak, M.K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid. Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, K.O.; Thorsteinsson, L.; Sigurjonsson, O.E.; Keller, J.R.; Olafsson, K.; Egeland, T.; Gudmundsson, S.; Rafnar, T. Gene expression analysis of hematopoietic progenitor cells identifies Dlg7 as a potential stem cell gene. Stem Cells 2007, 25, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Loo, L.W.; Cheng, I.; Tiirikainen, M.; Lum-Jones, A.; Seifried, A.; Dunklee, L.M.; Church, J.M.; Gryfe, R.; Weisenberger, D.J.; Haile, R.W.; et al. cis-Expression QTL analysis of established colorectal cancer risk variants in colon tumors and adjacent normal tissue. PLoS ONE 2012, 7, e30477. [Google Scholar] [CrossRef] [PubMed]
- Tsou, A.P.; Yang, C.W.; Huang, C.Y.; Yu, R.C.; Lee, Y.C.; Chang, C.W.; Chen, B.R.; Chung, Y.F.; Fann, M.J.; Chi, C.W.; et al. Identification of a novel cell cycle regulated gene, HURP, overexpressed in human hepatocellular carcinoma. Oncogene 2003, 22, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Eissa, S.; Matboli, M.; Mansour, A.; Mohamed, S.; Awad, N.; Kotb, Y.M. Evaluation of urinary HURP mRNA as a marker for detection of bladder cancer: Relation to bilharziasis. Med. Oncol. 2014, 31, 804. [Google Scholar] [CrossRef] [PubMed]
- Choupani, J.; Mansoori Derakhshan, S.; Bayat, S.; Alivand, M.R.; Shekari Khaniani, M. Narrower insight to SIRT1 role in cancer: A potential therapeutic target to control epithelial-mesenchymal transition in cancer cells. J. Cell. Physiol. 2018, 233, 4443–4457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Sauve, A.A. Regulatory Effects of NAD(+) Metabolic Pathways on Sirtuin Activity. Prog. Mol. Biol. Transl. sci. 2018, 154, 71–104. [Google Scholar] [PubMed]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Arnal, L.; Katada, S.; Orozco-Solis, R.; Sassone-Corsi, P. NAD(+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat. Struct. Mol. Biol. 2015, 22, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Thiaville, M.M.; Chen, L.; Stoeck, A.; Xuan, J.; Gao, M.; Shih Ie, M.; Wang, T.L. Defining NOTCH3 target genes in ovarian cancer. Cancer Res. 2012, 72, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, H.P.; Ho, F.M.; Chao, K.F.; Kuo, M.L.; Lin-Shiau, S.Y.; Liu, S.H. β-Lapachone reduces endotoxin-induced macrophage activation and lung edema and mortality. Am. J. Respir. Crit. Care Med. 2003, 168, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Ko, H.M.; Jeong, Y.H.; Park, E.M.; Kim, H.S. β-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J. Neuroinflamm. 2015, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Podar, K.; Tai, Y.T.; Lin, B.; Hideshima, T.; Akiyama, M.; LeBlanc, R.; Catley, L.; Mitsiades, N.; Mitsiades, C.; et al. β-lapachone, a novel plant product, overcomes drug resistance in human multiple myeloma cells. Exp. Hematol. 2002, 30, 711–720. [Google Scholar] [CrossRef]
- Lee, B.N.R.; Son, Y.S.; Lee, D.; Choi, Y.J.; Kwon, S.M.; Chang, H.K.; Kim, P.H.; Cho, J.Y. Hedgehog-Interacting Protein (HIP) Regulates Apoptosis Evasion and Angiogenic Function of Late Endothelial Progenitor Cells. Sci. Rep. 2017, 7, 12449. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Jeong, S.; Yoon, K.A.; Sung, H.J.; Cho, H.S.; Kim, D.W.; Cho, J.Y. Deficiency of DGCR8 increases bone formation through downregulation of miR-22 expression. Bone 2017, 103, 287–294. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.W.; Cho, J.-Y. NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity. Int. J. Mol. Sci. 2018, 19, 3813. https://doi.org/10.3390/ijms19123813
Kim DW, Cho J-Y. NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity. International Journal of Molecular Sciences. 2018; 19(12):3813. https://doi.org/10.3390/ijms19123813
Chicago/Turabian StyleKim, Dong Wook, and Je-Yoel Cho. 2018. "NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity" International Journal of Molecular Sciences 19, no. 12: 3813. https://doi.org/10.3390/ijms19123813
APA StyleKim, D. W., & Cho, J.-Y. (2018). NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity. International Journal of Molecular Sciences, 19(12), 3813. https://doi.org/10.3390/ijms19123813