YAP Activation Drives Liver Regeneration after Cholestatic Damage Induced by Rbpj Deletion

 , , ,
, , ,
Abstract
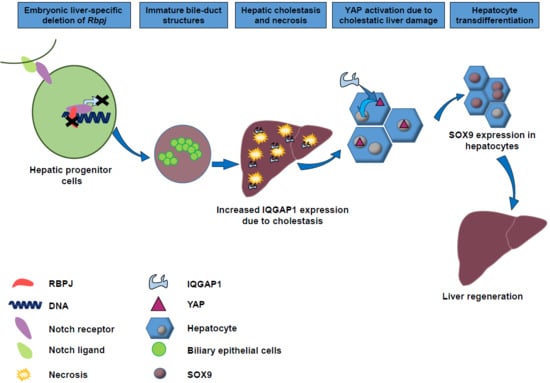
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
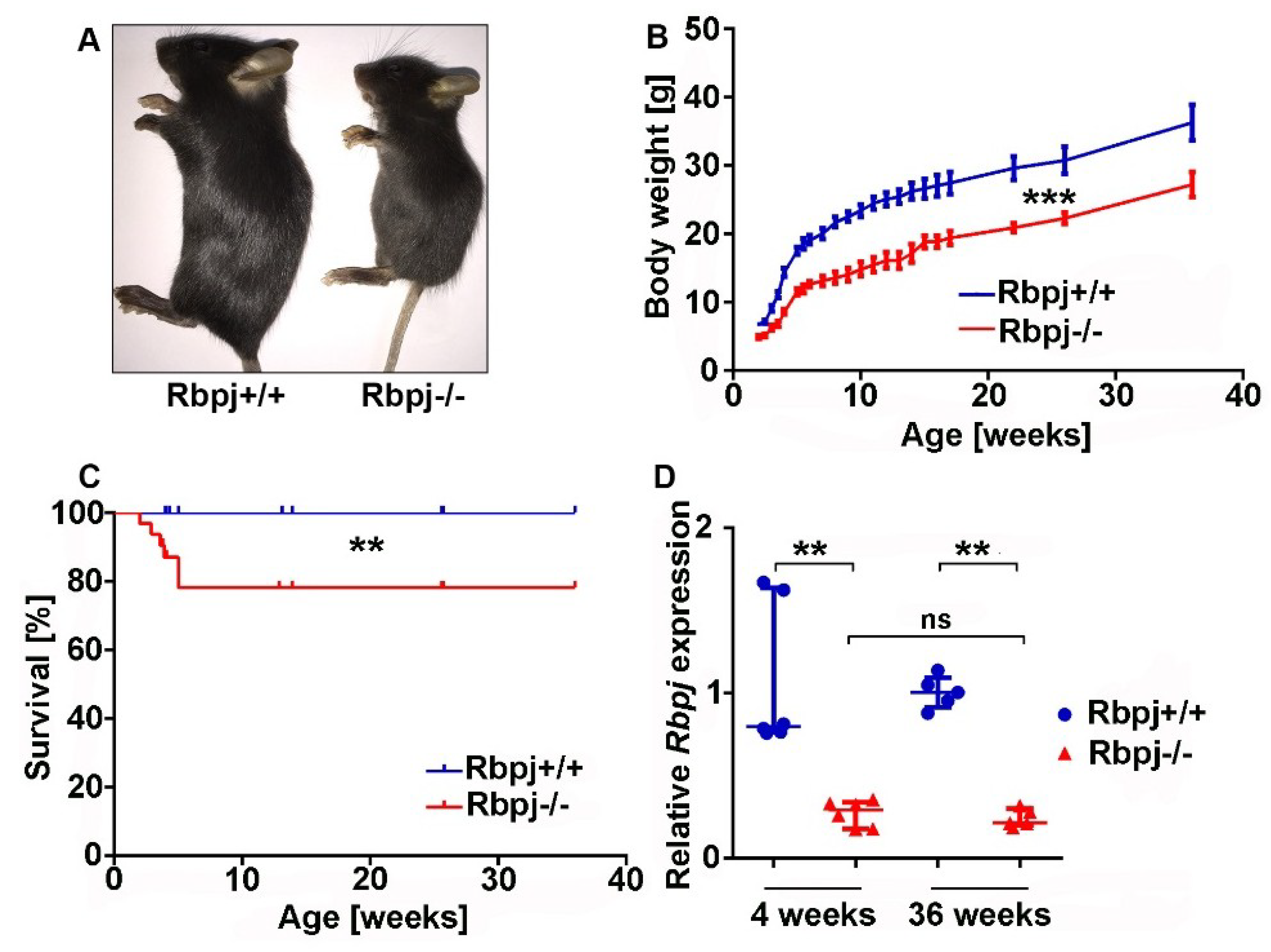
2.1. Liver-Specific Embryonic Deletion of Rbpj Results in Reduced Body Size and Impaired Survival
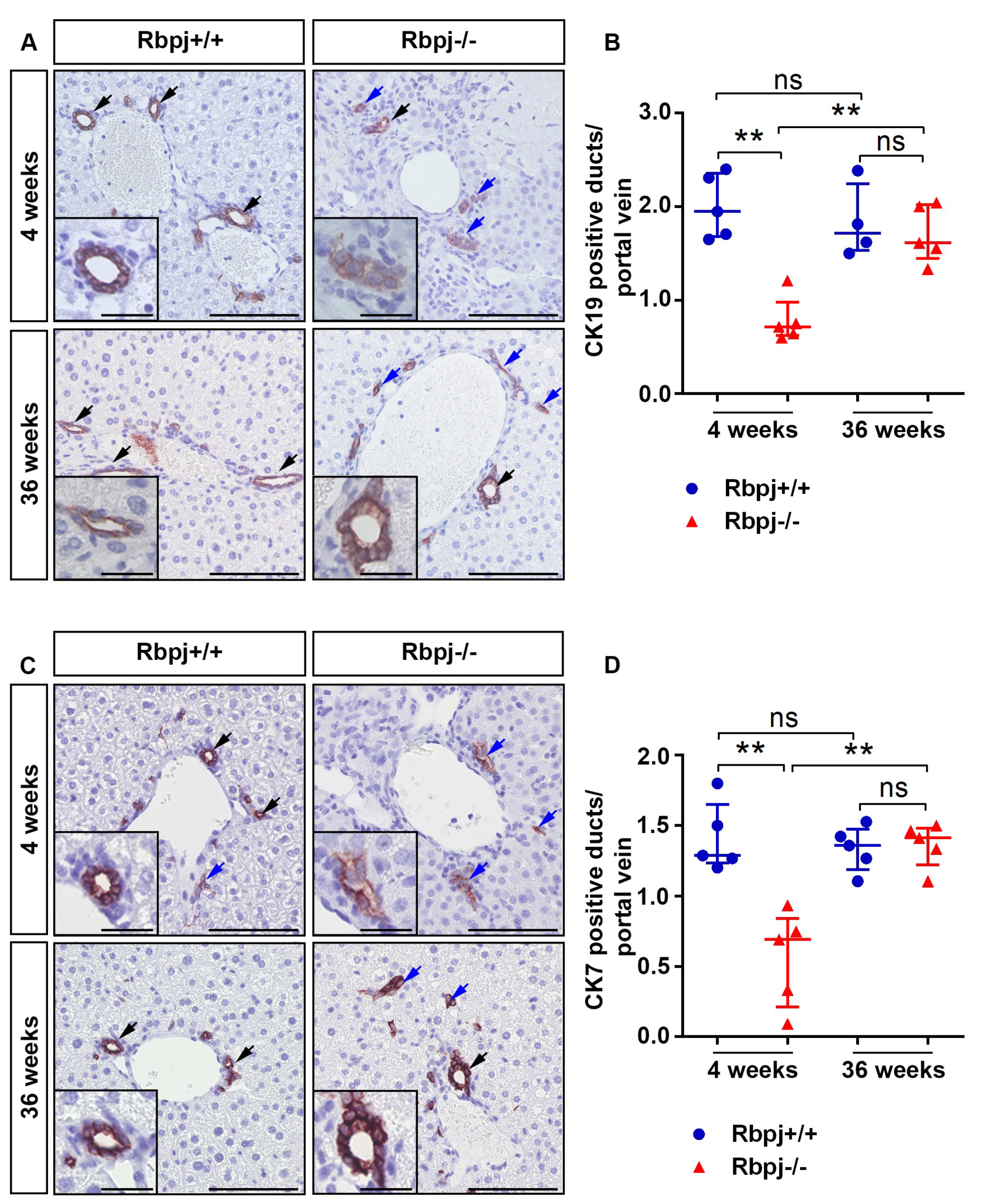
2.2. Loss of RBPJ Results in Impaired Maturation of IHBD
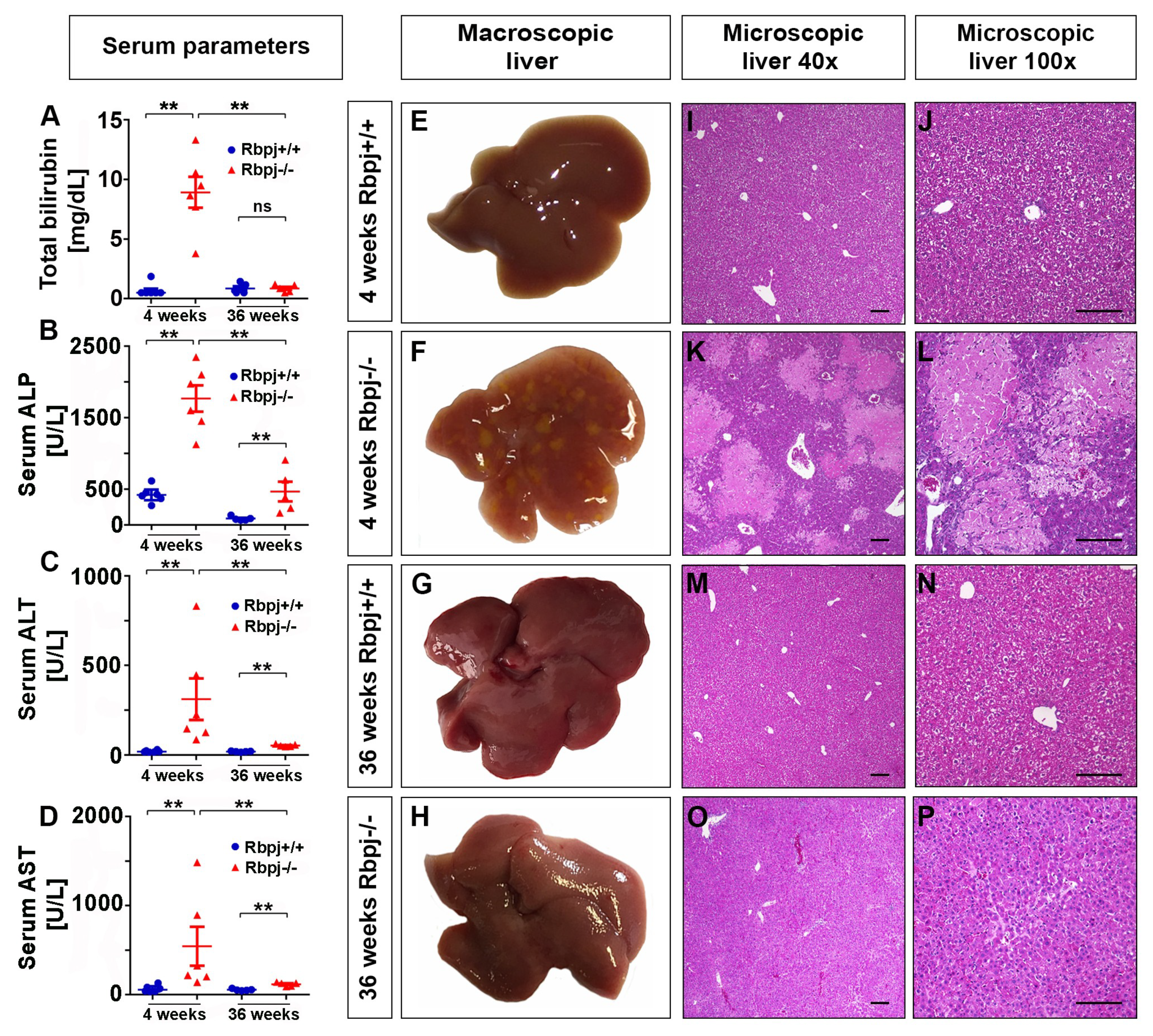
2.3. Loss of RBPJ Results in Hepatic Cholestasis and Necrosis
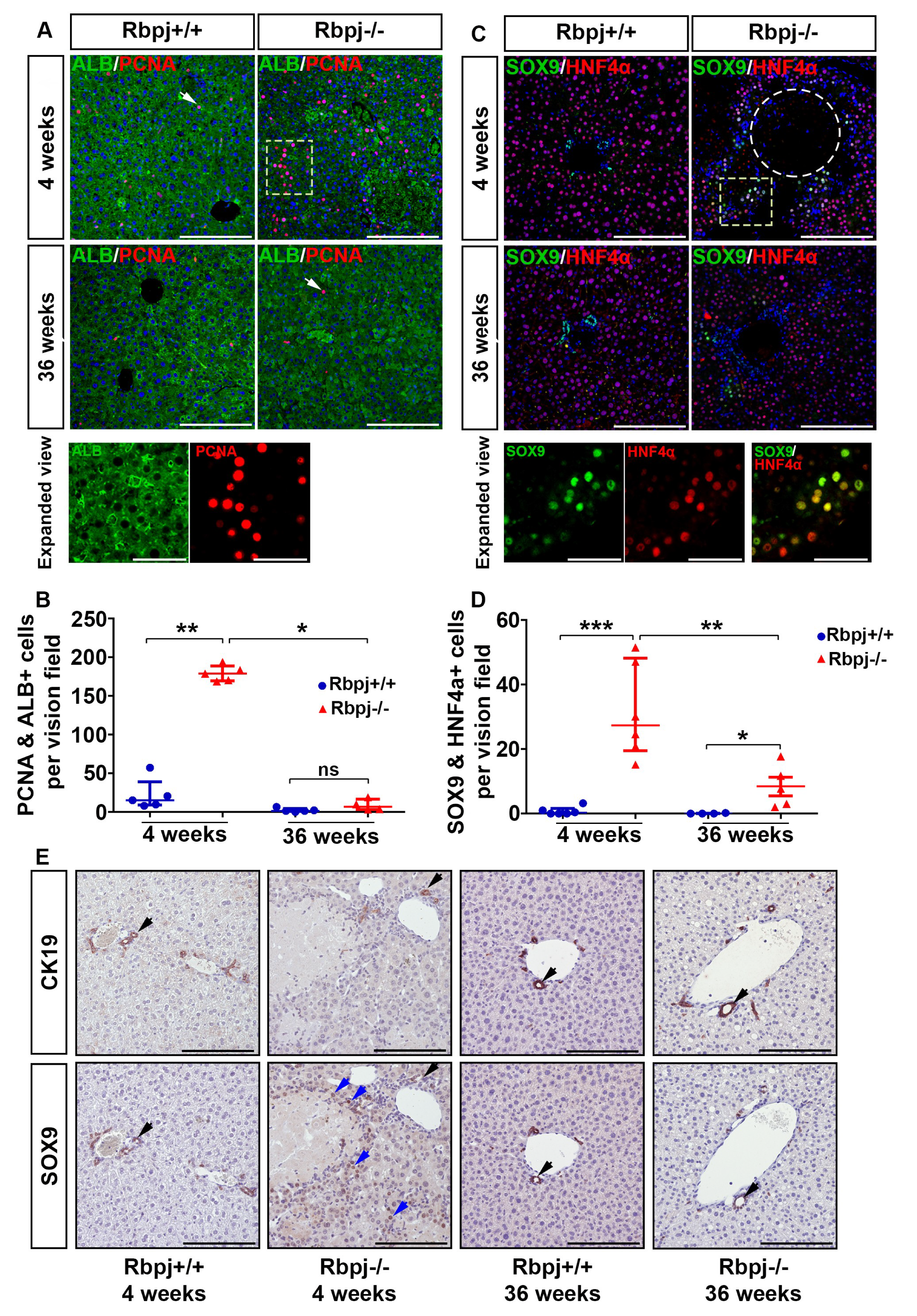
2.4. Increased Hepatocyte Proliferation and Induction of Hepatocyte Plasticity Due to RBPJ Deficiency-Induced Cholestasis
2.5. YAP Activation to Bypass Notch Pathway Deficiency
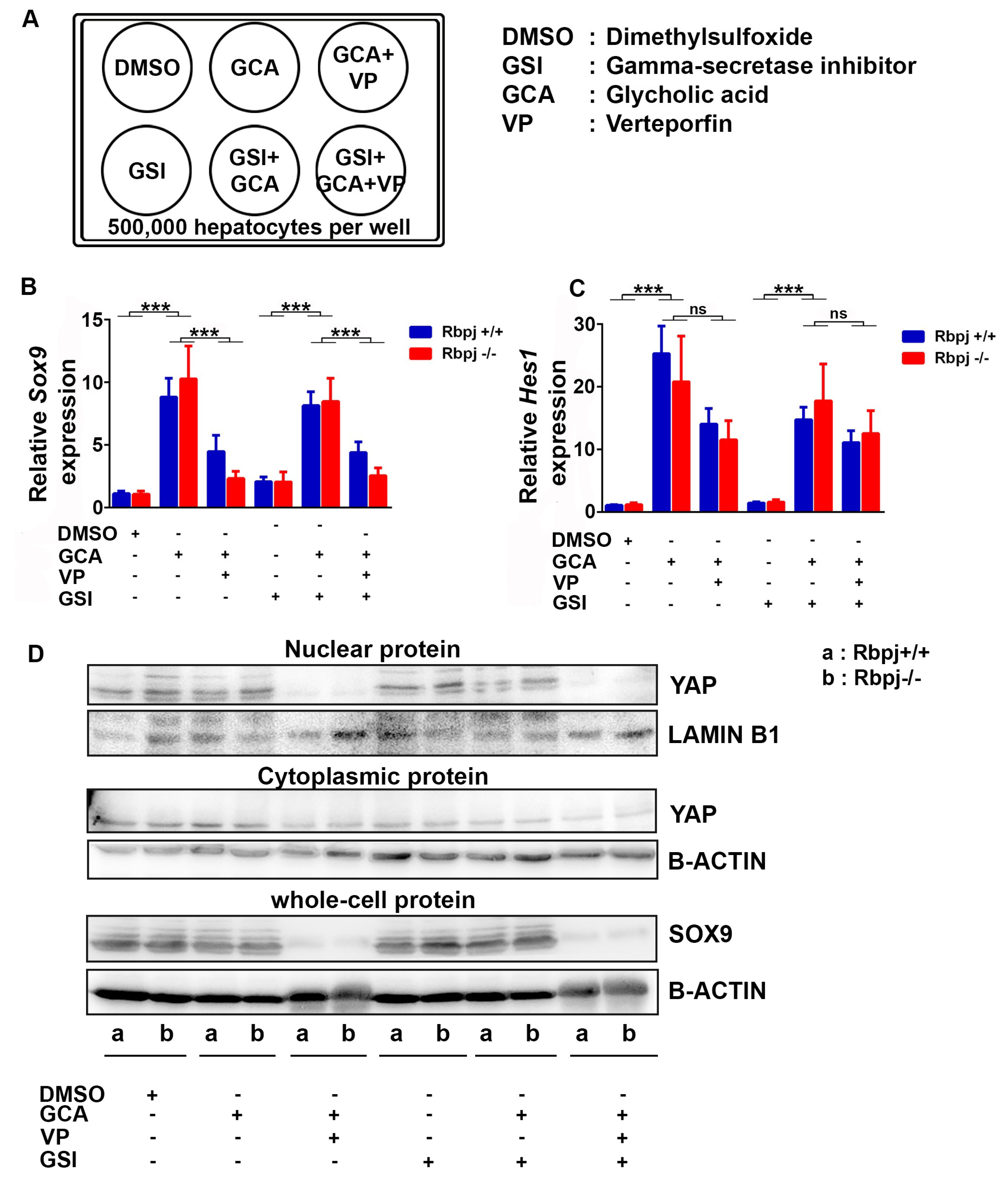
2.6. Induction of Cholestatic Liver Injury In Vitro Induces Sox9 and Hes1 Expression Independent of Notch Signalling
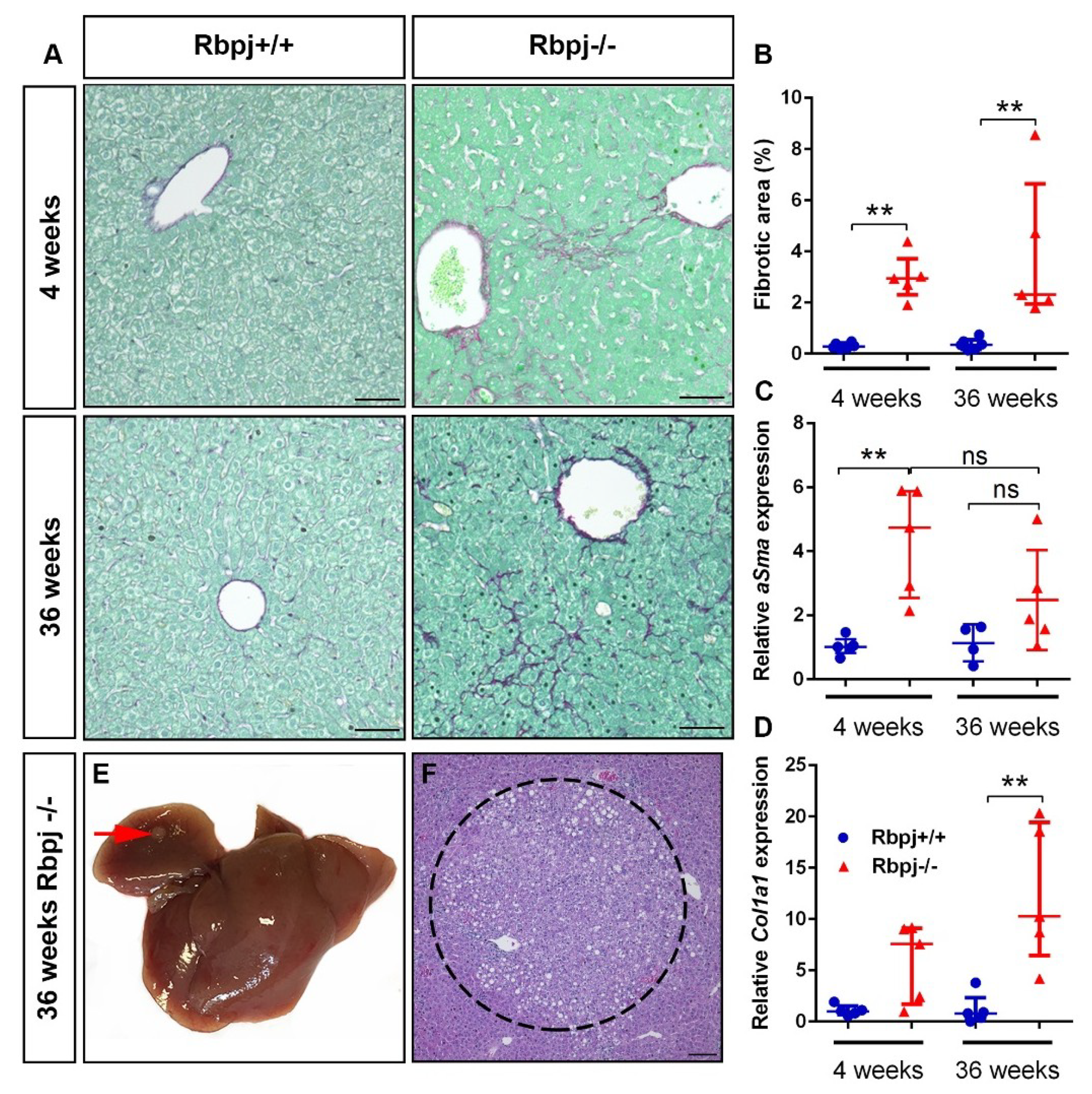
2.7. Chronic Liver Damage Subsequently Induced by the Loss of RBPJ Results in Liver Fibrosis
3. Discussion
4. Materials and Methods
4.1. Mouse Model
4.2. Liver Histology
4.3. Immunostaining
4.4. Protein Isolation and Western Blotting
4.5. RNA Isolation
4.6. Quantitative Real-Time PCR (qRT-PCR)
4.7. Serum Parameters
4.8. Gene Expression Analysis
4.9. Gene Set Enrichment Analysis (GSEA)
4.10. Hepatocyte Isolation and Cultivation
4.11. Cholestatic Liver Injury In Vitro
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Burden of liver disease in Europe. Available online: http://www.easl.eu/medias/EASLimg/Discover/EU/54ae845caec619f_file.pdf (accessed on 27 November 2018).
- Udompap, P.; Kim, D.; Kim, W.R. Current and Future Burden of Chronic Nonmalignant Liver Disease. Clin. Gastroenterol. Hepatol. 2015, 13, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar] [PubMed]
- Kodama, Y.; Hijikata, M.; Kageyama, R.; Shimotohno, K.; Chiba, T. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology 2004, 127, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Geisler, F.; Nagl, F.; Mazur, P.K.; Lee, M.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Schmid, R.M.; Siveke, J.T. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 2008, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Lozier, J.; McCright, B.; Gridley, T. Notch signaling regulates bile duct morphogenesis in mice. PLoS ONE 2008, 3, e1851. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Panikkar, A.; Xu, J.; Antoniou, A.; Raynaud, P.; Lemaigre, F.; Stanger, B.Z. Notch signaling controls liver development by regulating biliary differentiation. Development 2009, 136, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Sparks, E.E.; Huppert, K.A.; Brown, M.A.; Washington, M.K.; Huppert, S.S. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 2010, 51, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Cadamuro, M.; Guido, M.; Spirli, C.; Fiorotto, R.; Colledan, M.; Torre, G.; Alberti, D.; Sonzogni, A.; Okolicsanyi, L.; et al. Analysis of liver repair mechanisms in Alagille syndrome and biliary atresia reveals a role for notch signaling. Am. J. Pathol. 2007, 171, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Van Rooijen, N.; et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 2012, 18, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, R.; Raizner, A.; Morell, C.M.; Torsello, B.; Scirpo, R.; Fabris, L.; Spirli, C.; Strazzabosco, M. Notch signaling regulates tubular morphogenesis during repair from biliary damage in mice. J. Hepatol. 2013, 59, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Vanderpool, C.; Sparks, E.E.; Huppert, K.A.; Gannon, M.; Means, A.L.; Huppert, S.S. Genetic interactions between hepatocyte nuclear factor-6 and Notch signaling regulate mouse intrahepatic bile duct development in vivo. Hepatology 2012, 55, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Walter, T.J.; Vanderpool, C.; Cast, A.E.; Huppert, S.S. Intrahepatic bile duct regeneration in mice does not require Hnf6 or Notch signaling through Rbpj. Am. J. Pathol. 2014, 184, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Jeliazkova, P.; Jörs, S.; Lee, M.; Zimber-Strobl, U.; Ferrer, J.; Schmid, R.M.; Siveke, J.T.; Geisler, F. Canonical Notch2 signaling determines biliary cell fates of embryonic hepatoblasts and adult hepatocytes independent of Hes1. Hepatology 2013, 57, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Tanigaki, K.; Han, H.; Yamamoto, N.; Tashiro, K.; Ikegawa, M.; Kuroda, K.; Suzuki, A.; Nakano, T.; Honjo, T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 2002, 3, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kellendonk, C.; Opherk, C.; Anlag, K.; Schütz, G.; Tronche, F. Hepatocyte-specific expression of Cre recombinase. Genesis 2000, 26, 151–153. [Google Scholar] [CrossRef]
- Falix, F.A.; Weeda, V.B.; Labruyere, W.T.; Poncy, A.; de Waart, D.R.; Hakvoort, T.B.; Lemaigre, F.; Gaemers, I.C.; Aronson, D.C.; Lamers, W.H. Hepatic Notch2 deficiency leads to bile duct agenesis perinatally and secondary bile duct formation after weaning. Dev. Biol. 2014, 396, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Schmid, M.; Müssel, C.; Sträng, J.E.; Buske, C.; Bullinger, L.; Kraus, J.M.; Kestler, H.A. GiANT: Gene set uncertainty in enrichment analysis. Bioinformatics 2016, 32, 1891–1894. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Bessho, K.; Mourya, R.; Shivakumar, P.; Walters, S.; Magee, J.C.; Rao, M.; Jegga, A.G.; Bezerra, J.A. Gene expression signature for biliary atresia and a role for interleukin-8 in pathogenesis of experimental disease. Hepatology 2014, 60, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, N.; Xu, Y.; Chen, Q.; Khan, M.; Potter, J.J.; Nayar, S.K.; Cornish, T.; Alpini, G.; Bronk, S.; et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 2012, 56, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe-Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Anakk, S.; Bhosale, M.; Schmidt, V.A.; Johnson, R.L.; Finegold, M.J.; Moore, D.D. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013, 5, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Critical Factors in the Assessment of Cholestatic Liver Injury In Vitro. Methods Mol. Biol. 2015, 1250, 363–376. [Google Scholar] [PubMed]
- Wang, C.; Zhu, X.; Feng, W.; Yu, Y.; Jeong, K.; Guo, W.; Lu, Y.; Mills, G.B. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 2015, 6, 27–37. [Google Scholar] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Poncy, A.; Antoniou, A.; Cordi, S.; Pierreux, C.E.; Jacquemin, P.; Lemaigre, F.P. Transcription factors SOX4 and SOX9 cooperatively control development of bile ducts. Dev. Biol. 2015, 404, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Solé, M.; Thung, S.; Stanger, B.Z.; et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012, 143, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Kulic, I.; Robertson, G.; Chang, L.; Baker, J.H.; Lockwood, W.W.; Mok, W.; Fuller, M.; Fournier, M.; Wong, N.; Chou, V.; et al. Loss of the Notch effector RBPJ promotes tumorigenesis. J. Exp. Med. 2015, 212, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.J.; Zovein, A.C.; Koh, H.; Radtke, F.; Weinmaster, G.; Iruela-Arispe, M.L. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: Insights into Alagille syndrome. Development 2010, 137, 4061–4072. [Google Scholar] [CrossRef] [PubMed]
- Just, P.A.; Poncy, A.; Charawi, S.; Dahmani, R.; Traore, M.; Dumontet, T.; Drouet, V.; Dumont, F.; Gilgenkrantz, H.; Colnot, S.; et al. LKB1 and Notch Pathways Interact and Control Biliary Morphogenesis. PLoS ONE 2015, 10, e0145400. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Chivukula, I.V.; Hankeova, S.; Sjöqvist, M.; Tsoi, Y.L.; Ramsköld, D.; Masek, J.; Elmansuri, A.; Hoogendoorn, A.; Vazquez, E.; et al. Mouse Model of Alagille Syndrome and Mechanisms of Jagged1 Missense Mutations. Gastroenterology 2018, 154, 1080–1095. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Elkahloun, A.G.; Pike, B.L.; Okajima, K.; Krantz, I.D.; Genin, A.; Piccoli, D.A.; Meltzer, P.S.; Spinner, N.B.; Collins, F.S.; et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 1997, 16, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Long, X.D.; Xia, Q. Recent Advances in Etiology of Biliary Atresia. Clin. Pediatr. 2015, 54, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Zagory, J.A.; Dietz, W.; Park, A.; Fenlon, M.; Xu, J.; Utley, S.; Mavila, N.; Wang, K.S. Notch signaling promotes ductular reactions in biliary atresia. J. Surg. Res. 2017, 215, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Mašek, J.; Andersson, E.R. The developmental biology of genetic Notch disorders. Development 2017, 144, 1743–1763. [Google Scholar] [CrossRef] [PubMed]
- Hindley, C.J.; Mastrogiovanni, G.; Huch, M. The plastic liver: Differentiated cells, stem cells, every cell? J. Clin. Investig. 2014, 124, 5099–5102. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; Grompe, M.; Sander, M. Stem cells versus plasticity in liver and pancreas regeneration. Nat. Cell Biol. 2016, 18, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, M.; Nyabi, O.; Sid, B.; Evraerts, J.; El Malmi, I.; Heremans, Y.; Dollé, L.; Benton, C.; Calderon, P.B.; van Grunsven, L.; et al. Downregulation of Sox9 expression associates with hepatogenic differentiation of human liver mesenchymal stem/progenitor cells. Stem. Cells Dev. 2014, 23, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Berthiaume, F.; Tilles, A.W.; Yarmush, M.L. A new technique for primary hepatocyte expansion in vitro. Biotechnol. Bioeng. 2008, 101, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, A.; Pace, A.; Rockwell, C.E.; Roth, K.J.; Chow, A.; O’Brien, K.M.; Albee, R.; Kelly, K.; Towery, K.; Luyendyk, J.P.; et al. Hepatic stellate cells orchestrate clearance of necrotic cells in a hypoxia-inducible factor-1α-dependent manner by modulating macrophage phenotype in mice. J. Immunol. 2014, 192, 3847–3857. [Google Scholar] [CrossRef] [PubMed]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Dill, M.T.; Tornillo, L.; Fritzius, T.; Terracciano, L.; Semela, D.; Bettler, B.; Heim, M.H.; Tchorz, J.S. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 2013, 57, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, M.; Bozko, P.; Palagani, V.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Activation of Notch signaling is required for cholangiocarcinoma progression and is enhanced by inactivation of p53 in vivo. PLoS ONE 2013, 8, e77433. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Wiemann, S.U.; Buer, J.; Lauber, J.; Dittmar, K.E.; Wüstefeld, T.; Blasco, M.A.; Manns, M.P.; Rudolph, K.L. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003, 22, 4003–4013. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tharehalli, U.; Svinarenko, M.; Kraus, J.M.; Kühlwein, S.D.; Szekely, R.; Kiesle, U.; Scheffold, A.; Barth, T.F.E.; Kleger, A.; Schirmbeck, R.; et al. YAP Activation Drives Liver Regeneration after Cholestatic Damage Induced by Rbpj Deletion. Int. J. Mol. Sci. 2018, 19, 3801. https://doi.org/10.3390/ijms19123801
Tharehalli U, Svinarenko M, Kraus JM, Kühlwein SD, Szekely R, Kiesle U, Scheffold A, Barth TFE, Kleger A, Schirmbeck R, et al. YAP Activation Drives Liver Regeneration after Cholestatic Damage Induced by Rbpj Deletion. International Journal of Molecular Sciences. 2018; 19(12):3801. https://doi.org/10.3390/ijms19123801
Chicago/Turabian StyleTharehalli, Umesh, Michael Svinarenko, Johann M. Kraus, Silke D. Kühlwein, Robin Szekely, Ute Kiesle, Annika Scheffold, Thomas F.E. Barth, Alexander Kleger, Reinhold Schirmbeck, and et al. 2018. "YAP Activation Drives Liver Regeneration after Cholestatic Damage Induced by Rbpj Deletion" International Journal of Molecular Sciences 19, no. 12: 3801. https://doi.org/10.3390/ijms19123801
APA StyleTharehalli, U., Svinarenko, M., Kraus, J. M., Kühlwein, S. D., Szekely, R., Kiesle, U., Scheffold, A., Barth, T. F. E., Kleger, A., Schirmbeck, R., Kestler, H. A., Seufferlein, T., Oswald, F., Katz, S.-F., & Lechel, A. (2018). YAP Activation Drives Liver Regeneration after Cholestatic Damage Induced by Rbpj Deletion. International Journal of Molecular Sciences, 19(12), 3801. https://doi.org/10.3390/ijms19123801