Differentially-Expressed miRNAs in Ectopic Stromal Cells Contribute to Endometriosis Development: The Plausible Role of miR-139-5p and miR-375

 ,
,  ,
,
Abstract
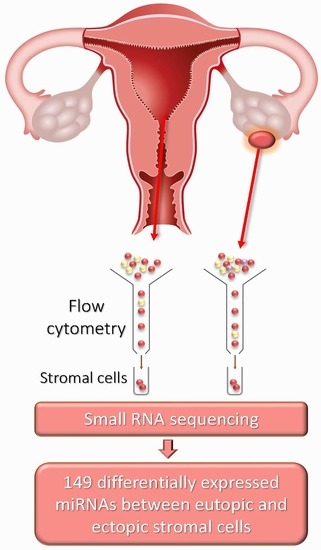
1. Introduction
2. Results
2.1. miRNA Profile of Eutopic and Ectopic Endometrium
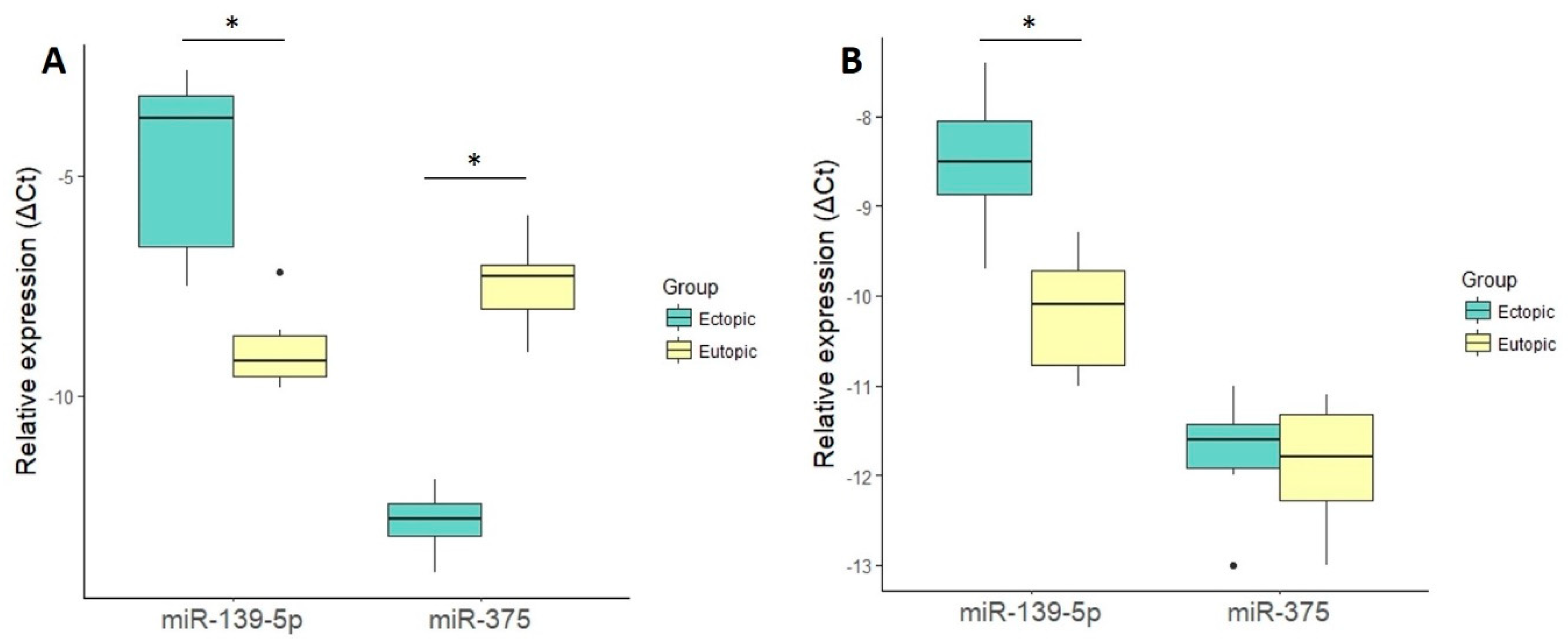
2.2. miRNA Validation by Quantitative Real-Time PCR (qRT-PCR)
2.3. Integrated miRNA–mRNA Analysis for Target Identification
2.4. miRNA Target Validation
3. Discussion
4. Materials and Methods
4.1. Patients and Sample Collection
4.2. Stromal Cell Isolation from Eutopic and Ectopic Endometria
4.3. Small RNA Sequencing
4.4. Sequencing Data Analysis
4.5. miRNA Target Prediction
4.6. miRNA Transfection
4.7. Primary Cultures of Human Endometrial Stromal Cells
4.8. qRT-PCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CPM | count per million |
| FACS | fluorescence-activated cell sorting |
| FC | fold change |
| FDR | false discovery rate |
| miRNA | microRNA |
| qRT-PCR | quantitative real-time PCR |
| TF | transcription factor |
References
- Vigano, P.; Candiani, M.; Monno, A.; Giacomini, E.; Vercellini, P.; Somigliana, E. Time to redefine endometriosis including its pro-fibrotic nature. Hum. Reprod. 2018, 33, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Barra, F.; Scala, C.; Mais, V.; Guerriero, S.; Ferrero, S. Investigational drugs for the treatment of endometriosis, an update on recent developments. Expert Opin. Investig. Drugs 2018, 27, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Klemmt, P.A.B.; Starzinski-Powitz, A. Molecular and Cellular Pathogenesis of Endometriosis. Curr. Womens Health Rev. 2018, 14, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Vigano, P. Endometriosis. Nat. Rev. Dis. Primers 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Arora, S.; Rana, R.; Chhabra, A.; Jaiswal, A.; Rani, V. miRNA-transcription factor interactions: A combinatorial regulation of gene expression. Mol. Genet. Genom. 2013, 288, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Braza-Boils, A.; Mari-Alexandre, J.; Gilabert, J.; Sanchez-Izquierdo, D.; Espana, F.; Estelles, A.; Gilabert-Estelles, J. MicroRNA expression profile in endometriosis: Its relation to angiogenesis and fibrinolytic factors. Hum. Reprod. 2014, 29, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Filigheddu, N.; Gregnanin, I.; Porporato, P.E.; Surico, D.; Perego, B.; Galli, L.; Patrignani, C.; Graziani, A.; Surico, N. Differential expression of microRNAs between eutopic and ectopic endometrium in ovarian endometriosis. J. Biomed. Biotechnol. 2010, 2010, 369549. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, S.M.; Creighton, C.J.; Han, D.Y.; Zariff, A.; Anderson, M.L.; Gunaratne, P.H.; Matzuk, M.M. Functional microRNA involved in endometriosis. Mol. Endocrinol. 2011, 25, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Saare, M.; Rekker, K.; Laisk-Podar, T.; Soritsa, D.; Roost, A.M.; Simm, J.; Velthut-Meikas, A.; Samuel, K.; Metsalu, T.; Karro, H.; et al. High-throughput sequencing approach uncovers the miRNome of peritoneal endometriotic lesions and adjacent healthy tissues. PLoS ONE 2014, 9, e112630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gu, C.; Ye, M.; Zhang, Z.; Li, L.; Fan, W.; Meng, Y. Integration analysis of microRNA and mRNA paired expression profiling identifies deregulated microRNA-transcription factor-gene regulatory networks in ovarian endometriosis. Reprod. Biol. Endocrinol. 2018, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Saare, M.; Rekker, K.; Laisk-Podar, T.; Rahmioglu, N.; Zondervan, K.; Salumets, A.; Gotte, M.; Peters, M. Challenges in endometriosis miRNA studies—From tissue heterogeneity to disease specific miRNAs. Biochim. Biophys. Acta 2017, 1863, 2282–2292. [Google Scholar] [CrossRef] [PubMed]
- Logan, P.C.; Yango, P.; Tran, N.D. Endometrial Stromal and Epithelial Cells Exhibit Unique Aberrant Molecular Defects in Patients With Endometriosis. Reprod. Sci. 2018, 25, 140–159. [Google Scholar] [CrossRef] [PubMed]
- Rekker, K.; Saare, M.; Eriste, E.; Tasa, T.; Kukuskina, V.; Roost, A.M.; Anderson, K.; Samuel, K.; Karro, H.; Salumets, A.; et al. High-throughput mRNA sequencing of stromal cells from endometriomas and endometrium. Reproduction 2017, 154, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, E.; Akahira, J.; Suzuki, F.; Nagase, S.; Ito, K.; Suzuki, T.; Sasano, H.; Yaegashi, N. Changes in microRNA expression levels correlate with clinicopathological features and prognoses in endometrial serous adenocarcinomas. Cancer Sci. 2010, 101, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, C.; Jiang, Y.; Wan, Y.; Zhou, S.; Cheng, W. Tumor-suppressor role of miR-139-5p in endometrial cancer. Cancer Cell Int. 2018, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.; Taylor, H. HOXA10 expression in ectopic endometrial tissue. Fertil. Steril. 2006, 85, 1386–1390. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Luo, X.; Bao, J.; Huang, X.; Jin, Y.; Chen, L.; Zheng, F. Decreased Expression of HOXA10 May Activate the Autophagic Process in Ovarian Endometriosis. Reprod. Sci. 2018, 25, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Geerts, D.; Bu, Z.; Ai, J.; Jin, L.; Li, Y.; Zhang, H.; Zhu, G. Regulation of endometrial receptivity by the highly expressed HOXA9, HOXA11 and HOXD10 HOX-class homeobox genes. Hum. Reprod. 2014, 29, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S.; Bagot, C.; Kardana, A.; Olive, D.; Arici, A. HOX gene expression is altered in the endometrium of women with endometriosis. Hum. Reprod. 1999, 14, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Celik, O.; Unlu, C.; Otlu, B.; Celik, N.; Caliskan, E. Laparoscopic endometrioma resection increases peri-implantation endometrial HOXA-10 and HOXA-11 mRNA expression. Fertil. Steril. 2015, 104, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Murakami, T.; Uehara, S.; Canis, M.; Sasano, H.; Okamura, K. Expression of estrogen receptor alpha and beta in peritoneal and ovarian endometriosis. Fertil. Steril. 2001, 75, 1198–1205. [Google Scholar] [CrossRef]
- Fujimoto, J.; Hirose, R.; Sakaguchi, H.; Tamaya, T. Expression of oestrogen receptor-alpha and -beta in ovarian endometriomata. Mol. Hum. Reprod. 1999, 5, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Smuc, T.; Pucelj, M.R.; Sinkovec, J.; Husen, B.; Thole, H.; Lanisnik Rizner, T. Expression analysis of the genes involved in estradiol and progesterone action in human ovarian endometriosis. Gynecol. Endocrinol. 2007, 23, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Akbas, G.E.; Song, J.; Taylor, H.S. A HOXA10 estrogen response element (ERE) is differentially regulated by 17 beta-estradiol and diethylstilbestrol (DES). J. Mol. Biol. 2004, 340, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Stow, L.R.; Jacobs, M.E.; Wingo, C.S.; Cain, B.D. Endothelin-1 gene regulation. FASEB J. 2011, 25, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, O.; Yamada-Nomoto, K.; Kobayashi, M.; Andoh, T.; Hongo, M.; Ono, Y.; Hasegawa-Idemitsu, A.; Sakai, A.; Osuga, Y.; Saito, S. Bradykinin system is involved in endometriosis-related pain through endothelin-1 production. Eur. J. Pain 2018, 22, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Pourjafar, M.; Saidijam, M.; Mansouri, K.; Malih, S.; Ranjbar Nejad, T.; Shabab, N.; Najafi, R. Cytoprotective effects of endothelin-1 on mesenchymal stem cells: An in vitro study. Clin. Exp. Pharmacol. Physiol. 2016, 43, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Cousins, F.L.; O, D.F.; Gargett, C.E. Endometrial stem/progenitor cells and their role in the pathogenesis of endometriosis. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 50, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Barragan, F.; Irwin, J.C.; Balayan, S.; Erikson, D.W.; Chen, J.C.; Houshdaran, S.; Piltonen, T.T.; Spitzer, T.L.; George, A.; Rabban, J.T.; et al. Human Endometrial Fibroblasts Derived from Mesenchymal Progenitors Inherit Progesterone Resistance and Acquire an Inflammatory Phenotype in the Endometrial Niche in Endometriosis. Biol. Reprod. 2016, 94, 118. [Google Scholar] [CrossRef] [PubMed]
- American Society for Reproductive. Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil. Steril. 1997, 67, 817–821. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, T.J. Generalized empirical Bayesian methods for discovery of differential data in high-throughput biology. Bioinformatics 2016, 32, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Bisognin, A.; Sales, G.; Coppe, A.; Bortoluzzi, S.; Romualdi, C. MAGIA(2): From miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update). Nucleic Acids Res. 2012, 40, W13–W21. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Maragkakis, M.; Alexiou, P.; Papadopoulos, G.L.; Reczko, M.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; Simossis, V.A.; et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinform. 2009, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Samalecos, A.; Reimann, K.; Wittmann, S.; Schulte, H.M.; Brosens, J.J.; Bamberger, A.M.; Gellersen, B. Characterization of a novel telomerase-immortalized human endometrial stromal cell line, St-T1b. Reprod. Biol. Endocrinol. 2009, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Kasvandik, S.; Samuel, K.; Peters, M.; Eimre, M.; Peet, N.; Roost, A.M.; Padrik, L.; Paju, K.; Peil, L.; Salumets, A. Deep Quantitative Proteomics Reveals Extensive Metabolic Reprogramming and Cancer-Like Changes of Ectopic Endometriotic Stromal Cells. J. Proteome Res. 2016, 15, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| miRNAs in Eutopic Stroma | Average Raw Read Count | miRNAs in Ectopic Stroma | Average Raw Read Count |
|---|---|---|---|
| let-7a-5p | 262,062 | let-7a-5p | 336,278 |
| miR-148a-3p | 251,377 | miR-10b-5p | 185,323 |
| let-7f-5p | 171,896 | miR-21-5p | 149,525 |
| miR-10b-5p | 119,737 | let-7f-5p | 97,019 |
| miR-21-5p | 116,936 | miR-148a-3p | 89,056 |
| miR-26a-5p | 102,057 | miR-99a-5p | 88,792 |
| miR-143-3p | 98,860 | miR-26a-5p | 88,348 |
| let-7i-5p | 89,804 | miR-143-3p | 67,660 |
| miR-99a-5p | 89,793 | let-7b-5p | 51,762 |
| miR-199a-3p | 76,412 | miR-126-3p | 49,937 |
| miRNA ID | log2FC | FDR (edgeR) | padj (DESeq2) | FDR.DE (BaySeq) | Average CPM Eutopic Stroma | Average CPM Ectopic Stroma |
|---|---|---|---|---|---|---|
| Upregulated miRNAs in ectopic stroma | ||||||
| hsa-miR-139-5p | 5.0 | 1.4 × 10−24 | 7.2 × 10−39 | 1.5 × 10−2 | 57 | 1292 |
| hsa-miR-139-3p | 6.1 | 4.9 × 10−24 | 8.5 × 10−29 | 1.6 × 10−2 | 6 | 242 |
| hsa-miR-202-5p | 9.3 | 2.8 × 10−19 | 5.8 × 10−11 | 7.2 × 10−3 | 0 | 51 |
| hsa-miR-506-3p | 5.8 | 1.4 × 10−17 | 9.9 × 10−17 | 1.8 × 10−2 | 4 | 204 |
| hsa-miR-150-5p | 4.3 | 2.5 × 10−14 | 7.7 × 10−17 | 9.5 × 10−3 | 14 | 203 |
| hsa-miR-202-3p | 9.1 | 3.1 × 10−14 | 3.5 × 10−9 | 3.9 × 10−2 | 0 | 41 |
| hsa-miR-150-3p | 7.3 | 6.5 × 10−12 | 5.2 × 10−6 | 4.7 × 10−3 | 0 | 15 |
| hsa-miR-513c-5p | 5.6 | 1.1 × 10−9 | 2.2 × 10−6 | 1.9 × 10−2 | 1 | 19 |
| hsa-miR-193a-5p | 2.7 | 1.2 × 10−9 | 3.5 × 10−14 | 3.8 × 10−2 | 44 | 194 |
| hsa-miR-584-5p | 3.1 | 9.1 × 10−7 | 6.5 × 10−5 | 3.4 × 10−2 | 3 | 23 |
| hsa-miR-371a-5p | 4.5 | 1.1 × 10−6 | 7.2 × 10−4 | 2.9 × 10−2 | 1 | 11 |
| hsa-miR-216b-5p | 4.3 | 7.5 × 10−5 | 1.8 × 10−3 | 4.5 × 10−2 | 1 | 9 |
| Downregulated miRNAs in ectopic stroma | ||||||
| hsa-miR-375 | −4.9 | 1.4 × 10−14 | 3.7 × 10−11 | 5.9 × 10−3 | 162 | 3 |
| hsa-miR-105-5p | −4.7 | 1.6 × 10−13 | 4.4 × 10−9 | 2.1 × 10−2 | 104 | 3 |
| hsa-miR-1298-5p | −5.8 | 2.5 × 10−9 | 5.6 × 10−5 | 1.3 × 10−2 | 18 | 0 |
| hsa-miR-6507-5p | −4.8 | 5.2 × 10−8 | 3.6 × 10−4 | 3.6 × 10−2 | 27 | 1 |
| hsa-miR-767-5p | −4.7 | 8.5 × 10−8 | 5.5 × 10−4 | 2.3 × 10−2 | 25 | 1 |
| hsa-miR-675-3p | −3.3 | 1.9 × 10−6 | 7.7 × 10−4 | 3.1 × 10−2 | 29 | 2 |
| hsa-miR-429 | −4.4 | 1.9 × 10−6 | 2.3 × 10−3 | 4.1 × 10−2 | 23 | 1 |
| hsa-miR-141-3p | −3.8 | 3.7 × 10−5 | 1.0 × 10−2 | 8.4 × 10−3 | 12 | 1 |
| hsa-miR-873-5p | −3.5 | 3.9 × 10−4 | 4.6 × 10−2 | 4.3 × 10−2 | 9 | 1 |
| Provisional ID | Average Read Count in Eutopic Stroma | Average Read Count in Ectopic Stroma | Similarities with Other Human miRNAs | Consensus Mature Sequence | Precursor Coordinate, Forward (+) or Reverse (−) Strand |
|---|---|---|---|---|---|
| 10_4598 | 1 | 2 | - | gucauagacuagugcuuccga | 10:106043903..106043987:− |
| 12_4331 | 630 | 67 | - | guucugggcuguagugagcuaugc | 12:24706803..24706886:+ |
| 11_4914 | 9 | 11 | - | aacugcucuucucuaauuuaa | 11:101346555..101346607:− |
| 19_4246 | 8 | 9 | hsa-miR-25-3p | gugugugcaccugugucugucugu | 19:18284682..18284741:+ |
| 3_22611 | 17 | 14 | - | cucugggcugcagugcgcuaugc | 3:49863521..49863597:− |
| 3_18752 | 4 | 0 | - | ugugguggcugcugcuggugc | 3:53763045..53763105:+ |
| 6_24262 | 4 | 17 | hsa-let-7b-5p | ugagguaguagguggugugc | 6:158493843..158493925:− |
| 8_30909 | 8 | 2 | hsa-miR-9903 | ccagccuacuggaggauaagagg | 8:98393666..98393724:− |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rekker, K.; Tasa, T.; Saare, M.; Samuel, K.; Kadastik, Ü.; Karro, H.; Götte, M.; Salumets, A.; Peters, M. Differentially-Expressed miRNAs in Ectopic Stromal Cells Contribute to Endometriosis Development: The Plausible Role of miR-139-5p and miR-375. Int. J. Mol. Sci. 2018, 19, 3789. https://doi.org/10.3390/ijms19123789
Rekker K, Tasa T, Saare M, Samuel K, Kadastik Ü, Karro H, Götte M, Salumets A, Peters M. Differentially-Expressed miRNAs in Ectopic Stromal Cells Contribute to Endometriosis Development: The Plausible Role of miR-139-5p and miR-375. International Journal of Molecular Sciences. 2018; 19(12):3789. https://doi.org/10.3390/ijms19123789
Chicago/Turabian StyleRekker, Kadri, Tõnis Tasa, Merli Saare, Külli Samuel, Ülle Kadastik, Helle Karro, Martin Götte, Andres Salumets, and Maire Peters. 2018. "Differentially-Expressed miRNAs in Ectopic Stromal Cells Contribute to Endometriosis Development: The Plausible Role of miR-139-5p and miR-375" International Journal of Molecular Sciences 19, no. 12: 3789. https://doi.org/10.3390/ijms19123789
APA StyleRekker, K., Tasa, T., Saare, M., Samuel, K., Kadastik, Ü., Karro, H., Götte, M., Salumets, A., & Peters, M. (2018). Differentially-Expressed miRNAs in Ectopic Stromal Cells Contribute to Endometriosis Development: The Plausible Role of miR-139-5p and miR-375. International Journal of Molecular Sciences, 19(12), 3789. https://doi.org/10.3390/ijms19123789