Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease



{kind=link}
Abstract
1. Introduction
2. MR in Cardiac Adverse Remodeling
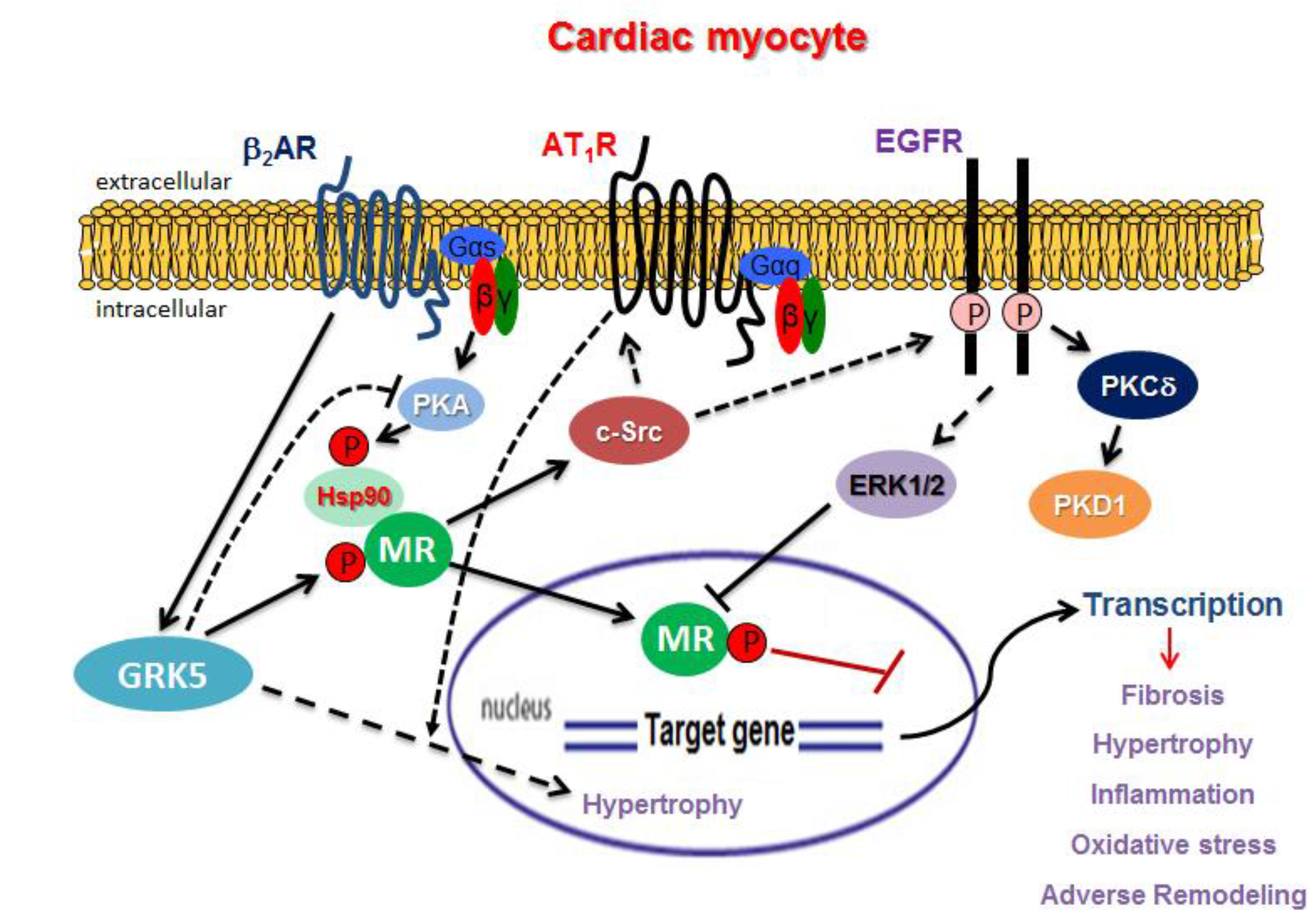
3. GPCR Signaling and MR Function
4. Therapeutic Implications of GPCR-MR Crosstalk for Heart Disease
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Young, M.J.; Rickard, A.J. Mineralocorticoid receptors in the heart: Lessons from cell-selective transgenic animals. J. Endocrinol. 2015, 224, R1–R13. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. Aldosterone in congestive heart failure. N. Engl. J. Med. 2001, 345, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Belden, Z.; Deiuliis, J.A.; Dobre, M.; Rajagopalan, S. The Role of the Mineralocorticoid Receptor in Inflammation: Focus on Kidney and Vasculature. Am. J. Nephrol. 2017, 46, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, J.; Jaisser, F.; Toto, R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension 2015, 65, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Hostetter, T.H.; Ibrahim, H.N. Aldosterone in chronic kidney and cardiac disease. J. Am. Soc. Nephrol. 2003, 14, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Andrew, R.; Cruden, N.L.; Kenyon, C.J.; Hughes, K.A.; Newby, D.E.; Hadoke, P.W.; Walker, B.R. Displacement of cortisol from human heart by acute administration of a mineralocorticoid receptor antagonist. J. Clin. Endocrinol. Metab. 2014, 99, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Funder, J. 30 years of the mineralocorticoid receptor: Mineralocorticoid receptor activation and specificity-conferring mechanisms: A brief history. J. Endocrinol. 2017, 234, T17–T21. [Google Scholar] [CrossRef] [PubMed]
- White, P.C. Aldosterone: Direct effects on and production by the heart. J. Clin. Endocrinol. Metab. 2003, 88, 2376–2383. [Google Scholar] [CrossRef] [PubMed]
- Davel, A.P.; Jaffe, I.Z.; Tostes, R.C.; Jaisser, F.; Belin de Chantemèle, E.J. New roles of aldosterone and mineralocorticoid receptors in cardiovascular disease: Translational and sex-specific effects. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H989–H999. [Google Scholar] [CrossRef] [PubMed]
- Ong, G.S.; Young, M.J. Mineralocorticoid regulation of cell function: The role of rapid signalling and gene transcription pathways. J. Mol. Endocrinol. 2017, 58, R33–R57. [Google Scholar] [CrossRef] [PubMed]
- Hermidorff, M.M.; de Assis, L.V.; Isoldi, M.C. Genomic and rapid effects of aldosterone: What we know and do not know thus far. Heart Fail. Rev. 2017, 22, 65–89. [Google Scholar] [CrossRef] [PubMed]
- Lother, A.; Moser, M.; Bode, C.; Feldman, R.D.; Hein, L. Mineralocorticoids in the heart and vasculature: New insights for old hormones. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.D. Aldosterone and blood pressure regulation: Recent milestones on the long and winding road from electrocortin to KCNJ5, GPER, and beyond. Hypertension 2014, 63, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Azibani, F.; Delcayre, C.; Jaisser, F. Aldosterone, mineralocorticoid receptor, and heart failure. Mol. Cell. Endocrinol. 2012, 350, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Queisser, N.; Amann, K.; Hey, V.; Habib, S.L.; Schupp, N. Blood pressure has only minor influence on aldosterone-induced oxidative stress and DNA damage in vivo. Free Radic. Biol. Med. 2013, 54, 17–25. [Google Scholar] [CrossRef] [PubMed]
- López-Andrés, N.; Martin-Fernandez, B.; Rossignol, P.; Zannad, F.; Lahera, V.; Fortuno, M.A.; Cachofeiro, V.; Díez, J. A role for cardiotrophin-1 in myocardial remodeling induced by aldosterone. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2372–H2382. [Google Scholar] [CrossRef]
- Martín-Fernández, B.; de las Heras, N.; Miana, M.; Ballesteros, S.; Delgado, C.; Song, S.; Hintze, T.; Cachofeiro, V.; Lahera, V. Structural, functional, and molecular alterations produced by aldosterone plus salt in rat heart: Association with enhanced serum and glucocorticoid-regulated kinase-1 expression. J. Cardiovasc. Pharmacol. 2011, 57, 114–121. [Google Scholar] [CrossRef]
- Neves, M.F.; Amiri, F.; Virdis, A.; Diep, Q.N.; Schiffrin, E.L. CIHR Multidisciplinary Research Group on Hypertension. Role of aldosterone in angiotensin II-induced cardiac and aortic inflammation, fibrosis, and hypertrophy. Can. J. Physiol. Pharmacol. 2005, 83, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Habibi, J.; DeMarco, V.G.; Ma, L.; Pulakat, L.; Rainey, W.E.; Whaley-Connell, A.T.; Sowers, J.R. Mineralocorticoid receptor blockade improves diastolic function independent of blood pressure reduction in a transgenic model of RAAS overexpression. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1484–H1491. [Google Scholar] [CrossRef] [PubMed]
- Kagiyama, S.; Matsumura, K.; Goto, K.; Otsubo, T.; Iida, M. Role of Rho kinase and oxidative stress in cardiac fibrosis induced by aldosterone and salt in angiotensin type 1a receptor knockout mice. Regul. Pept. 2010, 160, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.Y.; Pratt, J.H. Aldosterone increases plasminogen activator inhibitor-1 synthesis in rat cardiomyocytes. Mol. Cell. Biochem. 2005, 239, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Benard, L.; Milliez, P.; Ambroisine, M.L.; Messaoudi, S.; Samuel, J.L.; Delcayre, C. Effects of aldosterone on coronary function. Pharmacol. Rep. 2009, 61, 58–66. [Google Scholar] [CrossRef]
- Zhu, D.; Yu, H.; He, H.; Ding, J.; Tang, J.; Cao, D.; Hao, L. Spironolactone inhibits apoptosis in rat mesangial cells under hyperglycaemic conditions via the Wnt signalling pathway. Mol. Cell. Biochem. 2013, 380, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kitada, K.; Nakano, D.; Liu, Y.; Fujisawa, Y.; Hitomi, H.; Shibayama, Y.; Shibata, H.; Nagai, Y.; Mori, H.; Masaki, T.; et al. Oxidative stress induced glomerular mineralocorticoid receptor activation limits the benefit of salt reduction in Dahl salt-sensitive rats. PLoS ONE 2012, 7, e41896. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.; Campbell, R.C.; Warnock, D.G. Mineralocorticoid receptor blockers and chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Fraccarollo, D.; Berger, S.; Galuppo, P.; Kneitz, S.; Hein, L.; Schutz, G.; Frantz, S.; Ertl, G.; Bauersachs, J. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation 2011, 123, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Fraccarollo, D.; Galuppo, P.; Schraut, S.; Kneitz, S.; van Rooijen, N.; Ertl, G.; Bauersachs, J. Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension 2008, 51, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Lother, A.; Berger, S.; Gilsbach, R.; Rosner, S.; Ecke, A.; Barreto, F.; Bauersachs, J.; Schutz, G.; Hein, L. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension 2011, 57, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012, 60, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Gravez, B.; Tarjus, A.; Pelloux, V.; Ouvrard-Pascaud, A.; Delcayre, C.; Samuel, J.; Launay, J.M.; Sierra-Ramos, C.; Alvarez de la Rosa, D.; et al. Aldosterone-specific activation of cardiomyocyte mineralocorticoid receptor in vivo. Hypertension 2013, 61, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Buonafine, M.; Martínez-Martínez, E.; Amador, C.; Gravez, B.; Ibarrola, J.; Fernández-Celis, A.; El Moghrabi, S.; Rossignol, P.; López-Andrés, N.; Jaisser, F. Neutrophil Gelatinase-Associated Lipocalin from immune cells is mandatory for aldosterone-induced cardiac remodeling and inflammation. J. Mol. Cell. Cardiol. 2018, 115, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F.; Lenglet, S.; Vetterli, L.; Python, M.; Maturana, A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension 2008, 52, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F.; Python, M.; Maturana, A.D. Contribution of mineralocorticoid and glucocorticoid receptors to the chronotropic and hypertrophic actions of aldosterone in neonatal rat ventricular myocytes. Endocrinology 2010, 151, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Barbato, J.C.; Rashid, S.; Mulrow, P.J.; Shapiro, J.I.; Franco-Saenz, R. Mechanisms for aldosterone and spironolactone-induced positive inotropic actions in the rat heart. Hypertension 2004, 44, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Ouvrard-Pascaud, A.; Sainte-Marie, Y.; Benitah, J.P.; Perrier, R.; Soukaseum, C.; Nguyen Dinh Cat, A.; Royer, A.; Le Quang, K.; Charpentier, F.; Demolombe, S.; et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation 2005, 111, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Favre, J.; Gao, J.; Zhang, A.D.; Remy-Jouet, I.; Ouvrard-Pascaud, A.; Dautreaux, B.; Escoubet, B.; Thuillez, C.; Jaisser, F.; Richard, V. Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2035–H2043. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Tatsumi, T.; Matsunaga, S.; Hayashi, H.; Kimata, M.; Honsho, S.; Nishikawa, S.; Mano, A.; Shiraishi, J.; Yamada, H.; et al. Spironolactone modulates expressions of cardiac mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase 2 and prevents ventricular remodeling in post-infarct rat hearts. Hypertens. Res. 2007, 30, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Obata, K.; Xu, J.; Ichihara, S.; Noda, A.; Kimata, H.; Kato, T.; Izawa, H.; Murohara, T.; Yokota, M. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertension 2006, 47, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Selzer, C.; Schuster, P.; Lenski, M.; Adam, O.; Schäfers, H.J.; Böhm, M.; Laufs, U. The mineralocorticoid receptor promotes fibrotic remodeling in atrial fibrillation. J. Biol. Chem. 2014, 289, 6656–6668. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ma, J.; Tomita, T.; Morikawa, N.; Tanaka, N.; Masamura, K.; Kawai, Y.; Miyamori, I. Mineralocorticoid receptor is overexpressed in cardiomyocytes of patients with congestive heart failure. Congest. Heart Fail. 2005, 11, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Jin, D.; Muramatsu, M.; Kirimura, K.; Sakonjo, H.; Miyazaki, M. Eplerenone inhibits atherosclerosis in nonhuman primates. Hypertension 2005, 46, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Bodary, P.F.; Sambaziotis, C.; Wickenheiser, K.J.; Rajagopalan, S.; Pitt, B.; Eitzman, D.T. Aldosterone promotes thrombosis formation after arterial injury in mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 233. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.X.; Chen, X.Q.; Sun, X.N.; Sun, J.Y.; Zhang, W.C.; Zheng, X.J.; Zhang, Y.Y.; Shi, H.J.; Zhang, J.W.; Li, C.; et al. Mineralocorticoid receptor deficiency in macrophages inhibits atherosclerosis by affecting foam cell formation and efferocytosis. J. Biol. Chem. 2017, 292, 925–935. [Google Scholar] [CrossRef] [PubMed]
- De Rita, O.; Hackam, D.G.; Spence, J.D. Effects of aldosterone on human atherosclerosis: Plasma aldosterone and progression of carotid plaque. Can. J. Cardiol. 2012, 28, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Young, M.J. Corticosteroid receptors, macrophages and cardiovascular disease. J. Mol. Endocrinol. 2009, 42, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Muller, N.; Herold, M.J.; van den Brandt, J.; Reichardt, H.M. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 2007, 122, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, L.A.; Morgan, J.; Rickard, A.J.; Tesch, G.H.; Cranston, G.A.; Fletcher, E.K.; Delbridge, L.M.; Young, M.J. Macrophage mineralocorticoid receptor signaling plays a key role in aldosterone-independent cardiac fibrosis. Endocrinology 2012, 153, 3416–3425. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014, 63, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Usher, M.G.; Duan, S.Z.; Ivaschenko, C.Y.; Frieler, R.A.; Berger, S.; Schutz, G.; Lumeng, C.N.; Mortensen, R.M. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Investig. 2010, 120, 3350–3364. [Google Scholar] [CrossRef] [PubMed]
- Herrada, A.A.; Contreras, F.J.; Marini, N.P.; Amador, C.A.; Gonzalez, P.A.; Cortes, C.M.; Riedel, C.A.; Carvajal, C.A.; Figueroa, F.; Michea, L.F.; et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J. Immunol. 2010, 184, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.A.; Barrientos, V.; Pena, J.; Herrada, A.A.; Gonzalez, M.; Valdes, S.; Carrasco, L.; Alzamora, R.; Figueroa, F.; Kalergis, A.M.; et al. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T. lymphocytes. Hypertension 2014, 63, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Yang, J.; Young, M.J. 30 years of the mineralocorticoid receptor: Coregulators as mediators of mineralocorticoid receptor signaling diversity. J. Endocrinol. 2017, 234, T23–T34. [Google Scholar] [CrossRef] [PubMed]
- Faresse, N. Post-translational modifications of the mineralocorticoid receptor: How to dress the receptor according to the circumstances? J. Steroid Biochem. Mol. Biol. 2014, 143, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Ayuzawa, N.; Nagase, M.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Marumo, T.; Aiba, A.; Sakurai, T.; Shindo, T.; Fujita, T. Rac1-mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension 2016, 67, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Aukszi, B. Angiotensin receptor blocker drugs and inhibition of adrenal beta-arrestin-1-dependent aldosterone production: Implications for heart failure therapy. World J. Cardiol. 2017, 9, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic Nervous System in Heart Failure: Pathophysiology and Therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Garcia, D.; Walklett, K. Pharmacogenetics of cardiac inotropy. Pharmacogenomics 2014, 15, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Siryk-Bathgate, A.; Dabul, S.; Lymperopoulos, A. Current and future G protein-coupled receptor signaling targets for heart failure therapy. Drug Des. Dev. Ther. 2013, 7, 1209–1222. [Google Scholar] [CrossRef]
- Capote, L.A.; Mendez Perez, R.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Soltys, S.; Koch, W.J. An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5825–5830. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Koch, W.J. Adrenal beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J. Am. Coll. Cardiol. 2011, 57, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Sturchler, E.; Bathgate-Siryk, A.; Dabul, S.; Garcia, D.; Walklett, K.; Rengo, G.; McDonald, P.; Koch, W.J. Different potencies of angiotensin receptor blockers at suppressing adrenal β-Arrestin1-dependent post-myocardial infarction hyperaldosteronism. J. Am. Coll. Cardiol. 2014, 64, 2805–2806. [Google Scholar] [CrossRef] [PubMed]
- Dabul, S.; Bathgate-Siryk, A.; Valero, T.R.; Jafferjee, M.; Sturchler, E.; McDonald, P.; Koch, W.J.; Lymperopoulos, A. Suppression of adrenal βarrestin1-dependent aldosterone production by ARBs: Head-to-head comparison. Sci. Rep. 2015, 5, 8116. [Google Scholar] [CrossRef] [PubMed]
- Valero, T.R.; Sturchler, E.; Jafferjee, M.; Rengo, G.; Magafa, V.; Cordopatis, P.; McDonald, P.; Koch, W.J.; Lymperopoulos, A. Structure-activity relationship study of angiotensin II analogs in terms of β-arrestin-dependent signaling to aldosterone production. Pharmacol. Res. Perspect. 2016, 4, e00226. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Krug, A.W.; Freudinger, R.; Mildenberger, S.; Voelker, K.; Gekle, M. Aldosterone-induced EGFR expression: Interaction between the human mineralocorticoid receptor and the human EGFR promoter. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1790–E1800. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Husse, B.; Mildenberger, S.; Schreier, B.; Schuman, K.; Gekle, M. Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim. Biophys. Acta 2010, 1803, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Wuttke, M.; Ruhs, S.; Seiferth, A.; Mildenberger, S.; Rabe, S.; Schwerdt, G.; Gekle, M. Mineralocorticoid receptor inhibits CREB signaling by calcineurin activation. FASEB J. 2010, 24, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Dooley, R.; Harvey, B.J.; Thomas, W. Non-genomic actions of aldosterone: From receptors and signals to membrane targets. Mol. Cell. Endocrinol. 2012, 350, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.D.; Gros, R. Vascular effects of aldosterone: Sorting out the receptors and the ligands. Clin. Exp. Pharmacol. Physiol. 2013, 40, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Alzamora, R.; Brown, L.R.; Harvey, B.J. Direct binding and activation of protein kinase C isoforms by aldosterone and 17beta-estradiol. Mol. Endocrinol. 2007, 21, 2637–2650. [Google Scholar] [CrossRef] [PubMed]
- Manegold, J.C.; Falkenstein, E.; Wehling, M.; Christ, M. Rapid aldosterone effects on tyrosine phosphorylation in vascular smooth muscle cells. Cell. Mol. Biol. (Noisy-le-Grand, France) 1999, 45, 805–813. [Google Scholar]
- Ashton, A.W.; Le, T.Y.; Gomez-Sanchez, C.E.; Morel-Kopp, M.C.; McWhinney, B.; Hudson, A.; Mihailidou, A.S. Role of Nongenomic Signaling Pathways Activated by Aldosterone During Cardiac Reperfusion Injury. Mol. Endocrinol. 2015, 29, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Okoshi, M.P.; Yan, X.; Okoshi, K.; Nakayama, M.; Schuldt, A.J.; O’Connell, T.D.; Simpson, P.C.; Lorell, B.H. Aldosterone directly stimulates cardiac myocyte hypertrophy. J. Card. Fail. 2004, 10, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Eguchi, A.; Elliott, K.J.; Traynham, C.J.; Ibetti, J.; Eguchi, S.; Leosco, D.; Ferrara, N.; Rengo, G.; et al. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat. Commun. 2016, 7, 10877. [Google Scholar] [CrossRef] [PubMed]
- Callera, G.E.; Montezano, A.C.; Yogi, A.; Tostes, R.C.; He, Y.; Schiffrin, E.L.; Touyz, R.M. C-Src-dependent nongenomic signaling responses to aldosterone are increased in vascular myocytes from spontaneously hypertensive rats. Hypertension 2005, 46, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Willette, R.N.; Eybye, M.E.; Olzinski, A.R.; Behm, D.J.; Aiyar, N.; Maniscalco, K.; Bentley, R.G.; Coatney, R.W.; Zhao, S.; Westfall, T.D.; et al. Differential effects of p38 mitogen-activated protein kinase and cyclooxygenase 2 inhibitors in a model of cardiovascular disease. J. Pharmacol. Exp. Ther. 2009, 330, 964–970. [Google Scholar] [CrossRef] [PubMed]
- McEneaney, V.; Dooley, R.; Yusef, Y.R.; Keating, N.; Quinn, U.; Harvey, B.J.; Thomas, W. Protein kinase D1 modulates aldosterone-induced ENaC activity in a renal cortical collecting duct cell line. Mol. Cell. Endocrinol. 2010, 325, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Tsybouleva, N.; Zhang, L.; Chen, S.; Patel, R.; Lutucuta, S.; Nemoto, S.; DeFreitas, G.; Entman, M.; Carabello, B.A.; Roberts, R.; et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 2004, 109, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, A.; Isshiki, M.; Fujita, T. Aldosterone enhances ligand-stimulated nitric oxide production in endothelial cells. Hypertens. Res. 2008, 31, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.; Houard, N.; Lombès, M.; Barouki, R. Modulation of human mineralocorticoid receptor function by protein kinase A. Mol. Endocrinol. 1999, 13, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.; Fenne, I.S.; Cook, C.; Børud, B.; Bakke, M.; Lien, E.A.; Mellgren, G. cAMP-dependent protein kinase regulates ubiquitin–proteasome-mediated degradation and subcellular localization of the nuclear receptor coactivator GRIP1. J. Biol. Chem. 2004, 279, 49120–49130. [Google Scholar] [CrossRef] [PubMed]
- Faresse, N.; Vitagliano, J.J.; Staub, O. Differential ubiquitylation of the mineralocorticoid receptor is regulated by phosphorylation. FASEB J. 2012, 26, 4373–4382. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Rinehart, J.; Zhang, J.; Moeckel, G.; Castañeda-Bueno, M.; Stiegler, A.L.; Boggon, T.J.; Gamba, G.; Lifton, R.P. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013, 18, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.N.; Nguyen, K.; Lymperopoulos, A. GRK2 and Beta-Arrestins in Cardiovascular Disease: Established and Emerging Possibilities for Therapeutic Targeting. Curr. Mol. Pharmacol. 2012, 5, 317–326. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. GRK2 inhibition in heart failure: something old, something new. Curr. Pharm. Des. 2012, 18, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Rengo, G.; Lymperopoulos, A.; Koch, W.J. Future g protein-coupled receptor targets for treatment of heart failure. Curr. Treat. Opt. Cardiovasc. Med. 2009, 11, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Rengo, G.; Lymperopoulos, A.; Leosco, D.; Koch, W.J. GRK2 as a novel gene therapy target in heart failure. J. Mol. Cell. Cardiol. 2011, 50, 785–792. [Google Scholar] [CrossRef] [PubMed]
- McCrink, K.A.; Brill, A.; Lymperopoulos, A. Adrenal G protein-coupled receptor kinase-2 in regulation of sympathetic nervous system activity in heart failure. World J. Cardiol. 2015, 7, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Komolov, K.E.; Du, Y.; Duc, N.M.; Betz, R.M.; Rodrigues, J.P.G.L.M.; Leib, R.D.; Patra, D.; Skiniotis, G.; Adams, C.M.; Dror, R.O.; et al. Structural and Functional Analysis of a β2-Adrenergic Receptor Complex with GRK5. Cell 2017, 169, 407–421.e16. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.R.; Scott, M.G.; Pitcher, J.A. G protein-coupled receptor kinase 5 contains a DNA-binding nuclear localization sequence. Mol. Cell. Biol. 2004, 24, 10169–10179. [Google Scholar] [CrossRef] [PubMed]
- Martini, J.S.; Raake, P.; Vinge, L.E.; De George, B.R., Jr.; Chuprun, J.K.; Harris, D.M.; Gao, E.; Eckhart, A.D.; Pitcher, J.A.; Koch, W.J. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 12457–12462. [Google Scholar] [CrossRef] [PubMed]
- Hullmann, J.; Traynham, C.J.; Coleman, R.C.; Koch, W.J. The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development. Pharmacol. Res. 2016, 110, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Christ, M.; Wehling, M.; Kirsch, E.; Viengchareun, S.; Zennaro, M.C.; Lombès, M. Enhancement of beta-adrenergic cAMP-signaling by the mineralocorticoid receptor. Mol. Cell. Endocrinol. 2005, 231, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; McCrink, K.A.; Brill, A.; Maning, J.; Desimine, V.L.; Aukszi, B. Differential Roles of GRK2 and GRK5 in Cardiac Aldosterone Signaling Suggest GRK5-Mediated Cardio-protection Against Mineralocorticoids. Circulation 2017, 136, A12236. [Google Scholar]
- Lymperopoulos, A. Nova Southeastern University: Ft. Lauderdale, FL, USA, 2018; unpublished work.
- Liggett, S.B.; Cresci, S.; Kelly, R.J.; Syed, F.M.; Matkovich, S.J.; Hahn, H.S.; Diwan, A.; Martini, J.S.; Sparks, L.; Parekh, R.R.; et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat. Med. 2008, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.C.; Vallejos, X.; Siryk, A.; Rengo, G.; Cannavo, A.; Liccardo, D.; De Lucia, C.; Gao, E.; Leosco, D.; Koch, W.J.; et al. GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility. Cell Commun. Signal. 2013, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A. Autonomic Dysfunction in Critical Illness: ObNOX(2)ious (Baro)reflex Upregulation of G Protein-Coupled Receptor Kinase-2 Lets the Heart Down. Crit. Care Med. 2016, 44, 1621–1623. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Lymperopoulos, A.; Kennel, P.J.; Pollak, N.; Schulze, P.C.; Goldberg, I.J. Pathophysiology of sepsis-related cardiac dysfunction: Driven by inflammation, energy mismanagement, or both? Curr. Heart Fail. Rep. 2015, 12, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptional activity by inducing nuclear accumulation of IκB α. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.C.; Maurice, D.H.; Baillie, G.S. Targeting protein-protein interactions within the cyclic AMP signaling system as a therapeutic strategy for cardiovascular disease. Future Med. Chem. 2013, 5, 451–464. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, B.M.; Wertz, S.L.; Pollard, C.M.; Desimine, V.L.; Maning, J.; McCrink, K.A.; Lymperopoulos, A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int. J. Mol. Sci. 2018, 19, 3764. https://doi.org/10.3390/ijms19123764
Parker BM, Wertz SL, Pollard CM, Desimine VL, Maning J, McCrink KA, Lymperopoulos A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. International Journal of Molecular Sciences. 2018; 19(12):3764. https://doi.org/10.3390/ijms19123764
Chicago/Turabian StyleParker, Barbara M., Shelby L. Wertz, Celina M. Pollard, Victoria L. Desimine, Jennifer Maning, Katie A. McCrink, and Anastasios Lymperopoulos. 2018. "Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease" International Journal of Molecular Sciences 19, no. 12: 3764. https://doi.org/10.3390/ijms19123764
APA StyleParker, B. M., Wertz, S. L., Pollard, C. M., Desimine, V. L., Maning, J., McCrink, K. A., & Lymperopoulos, A. (2018). Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. International Journal of Molecular Sciences, 19(12), 3764. https://doi.org/10.3390/ijms19123764