Hierarchical Clustering of DNA k-mer Counts in RNAseq Fastq Files Identifies Sample Heterogeneities


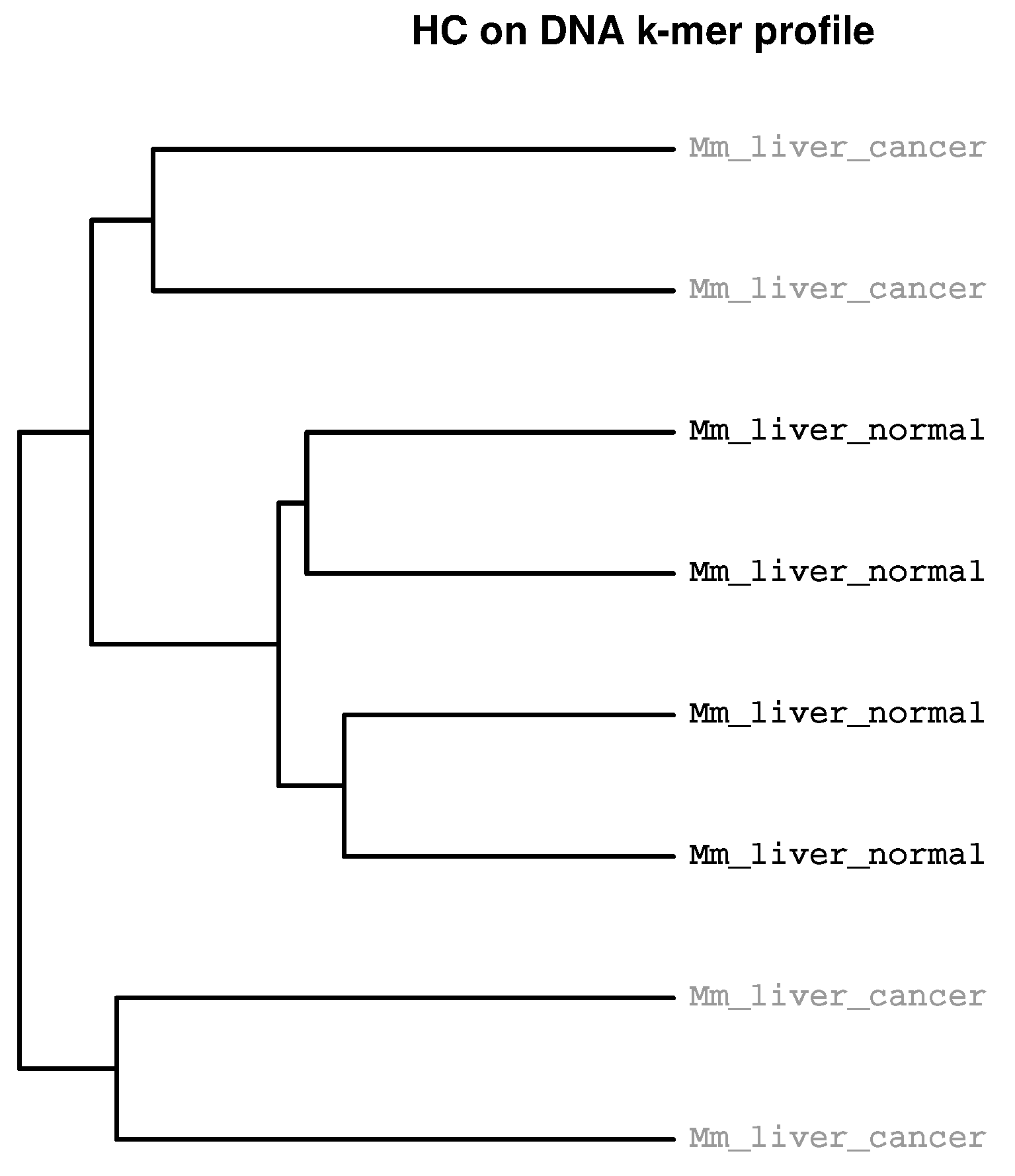{kind=link}
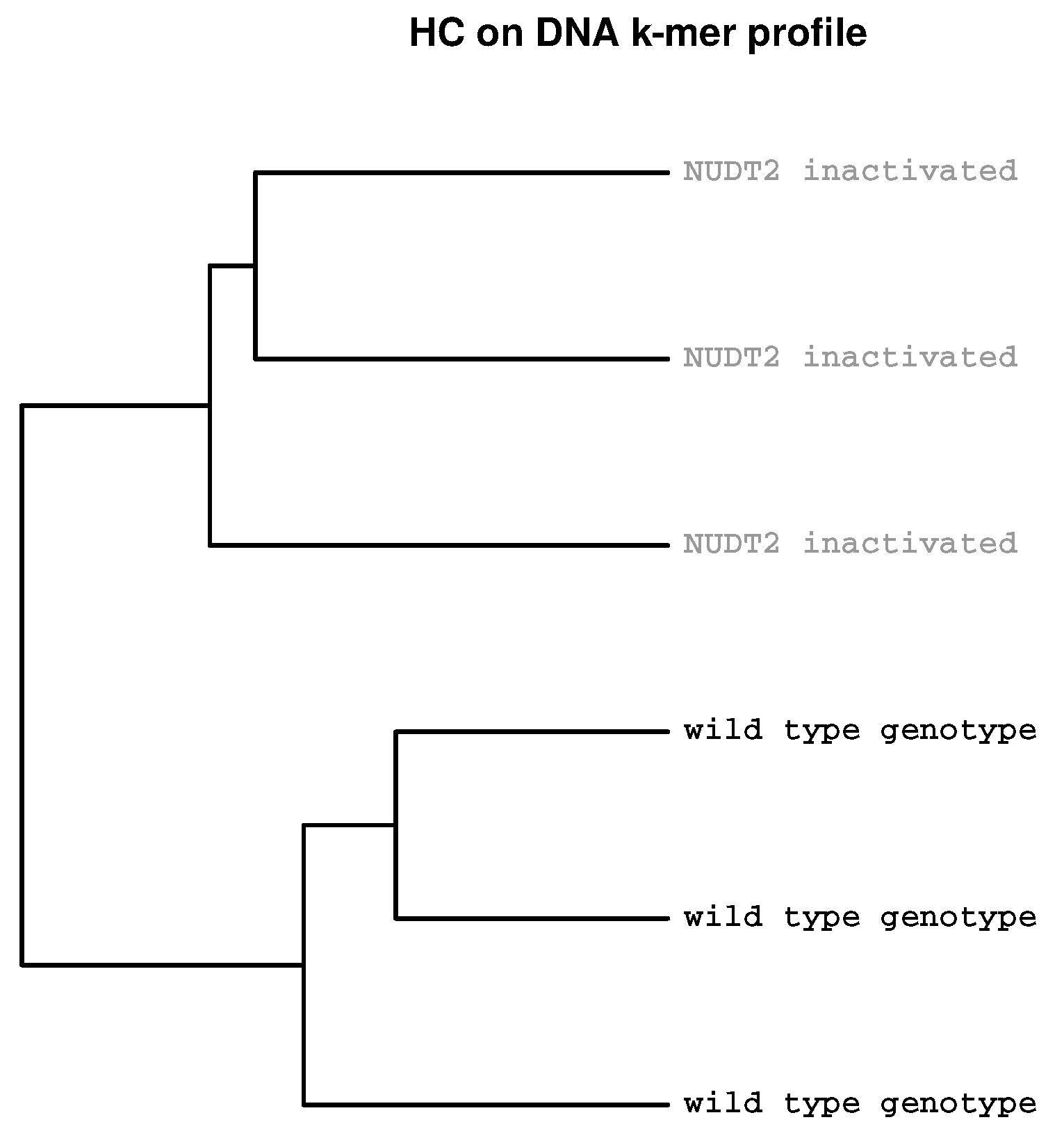{kind=link}
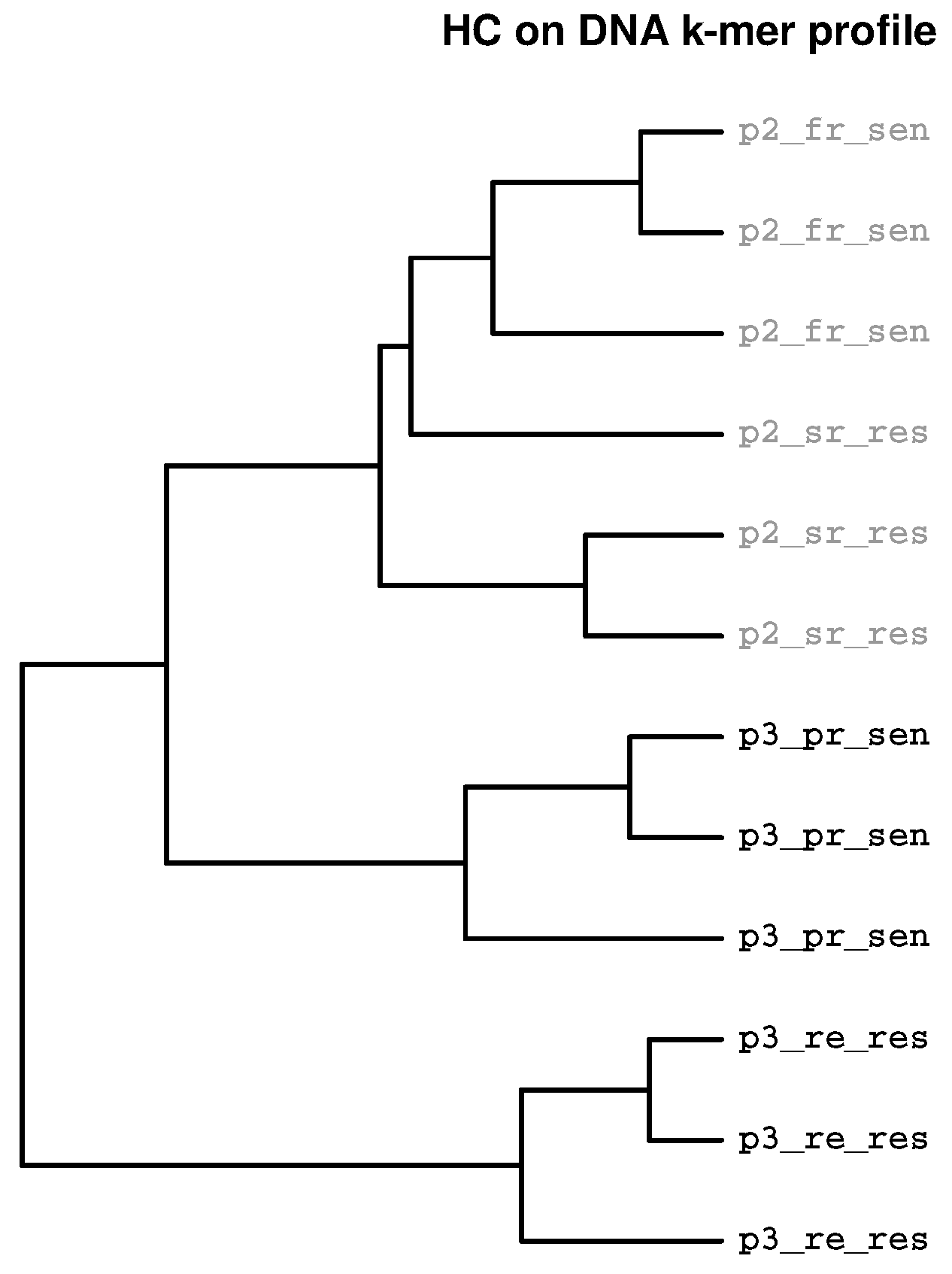{kind=link}
{kind=link}
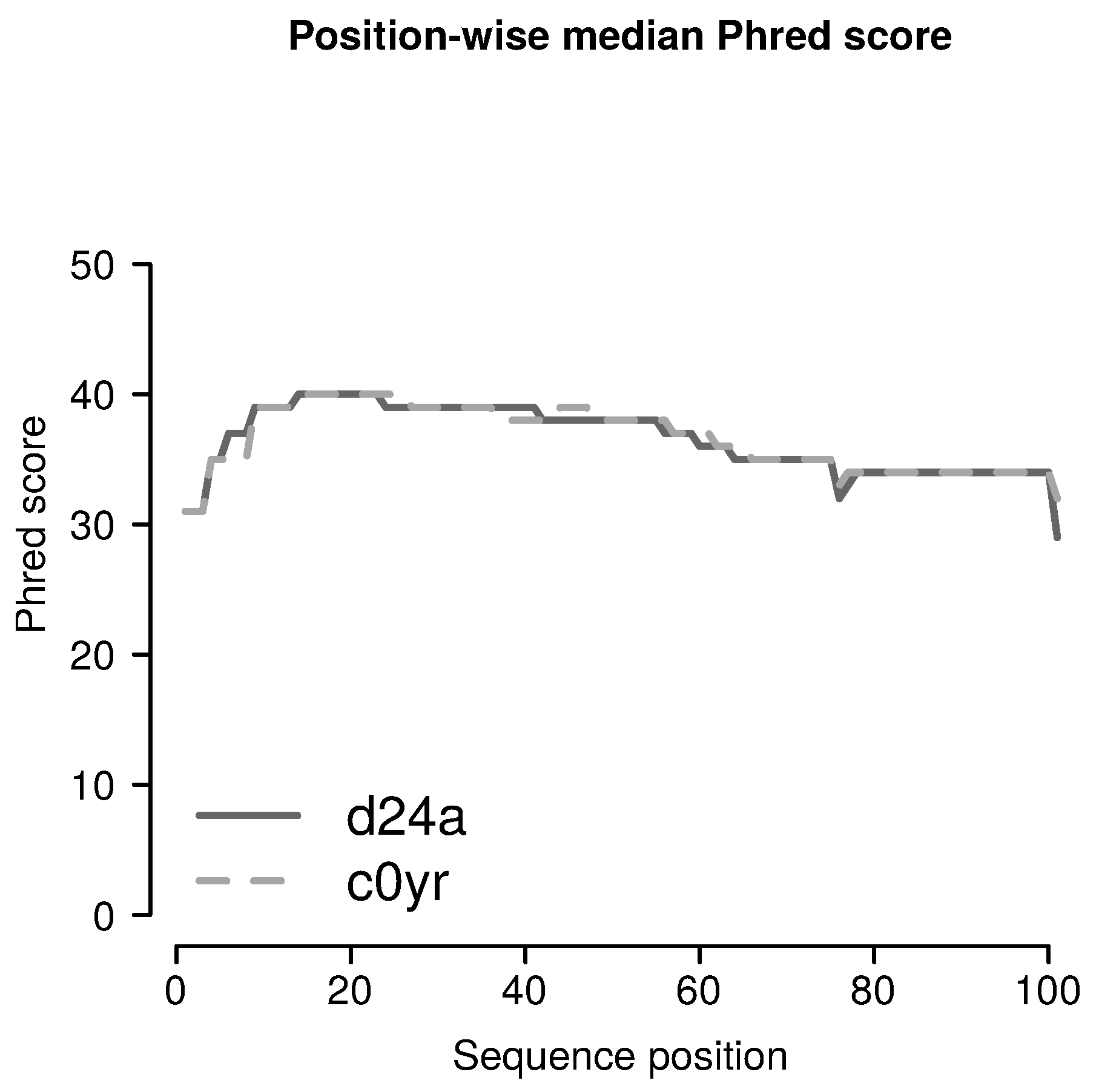{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Analysis of k-mer Counts
1.2. Quality Control of RNAseq Data
1.3. Word Sizes
1.4. Analysis of DNA k-mer Counts
1.5. HcKmer Analysis Algorithm
1.6. Analysed Samples
2. Results
2.1. Data Collection
2.2. Detection of Experimental Effects
2.3. Identification of Batch Effects
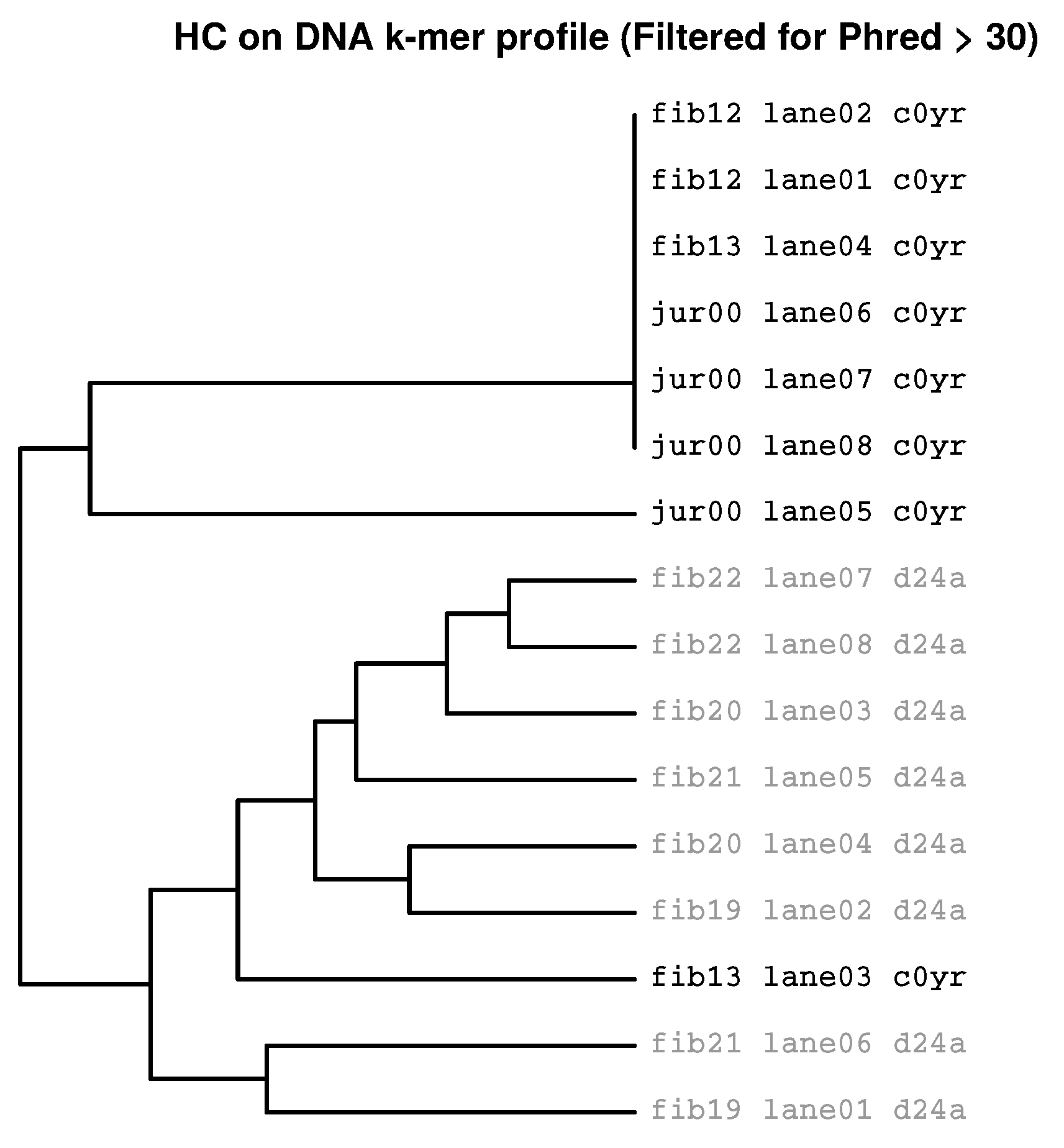
2.3.1. Filtering for Sufficient Phred Scores
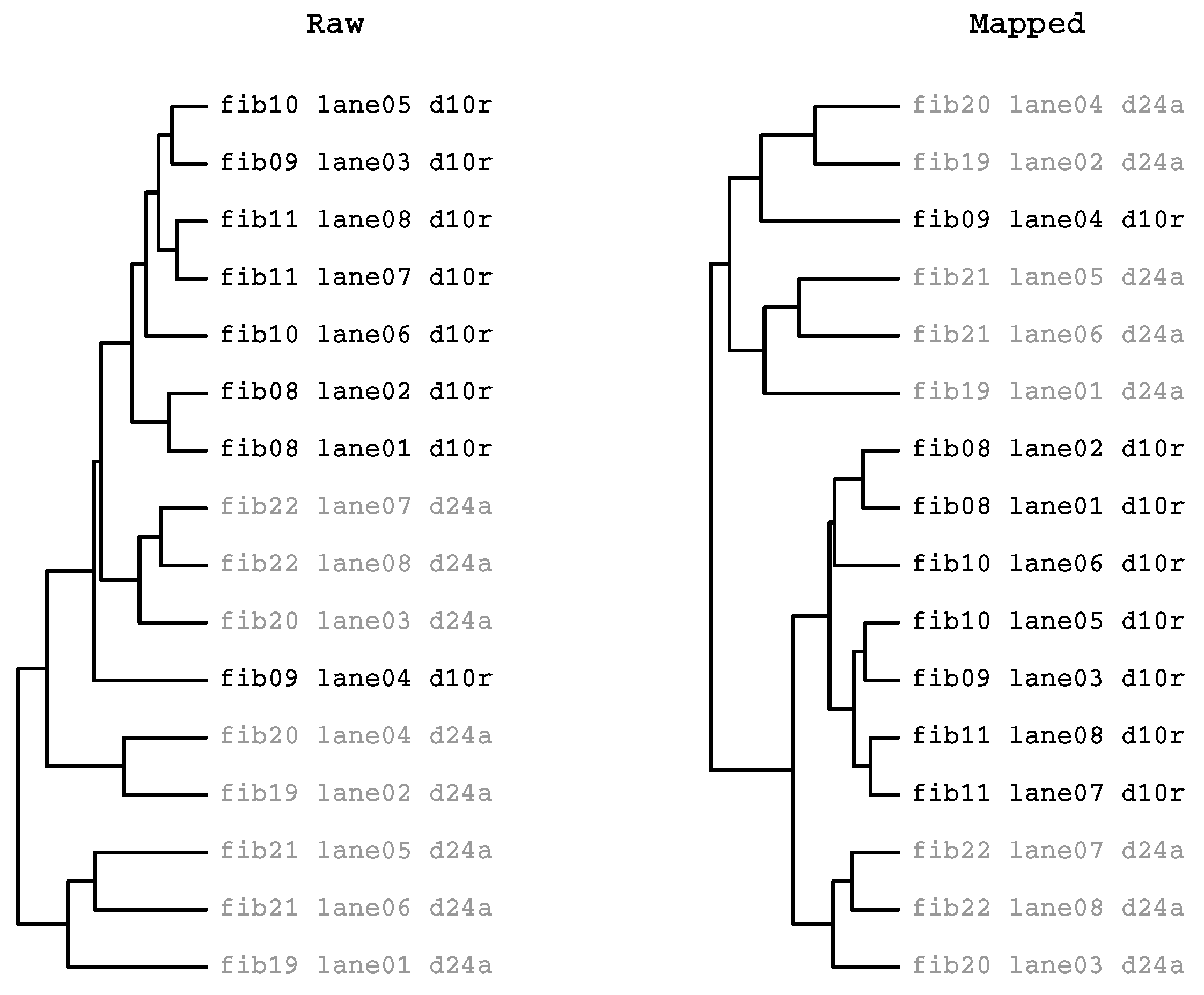
2.3.2. HcKmer on Sequences Aligned to the Human Genome
2.3.3. Prevalence of Batch Effects in RNAseq Data
2.3.4. Influence of Batch Effects on False Discovery Rate
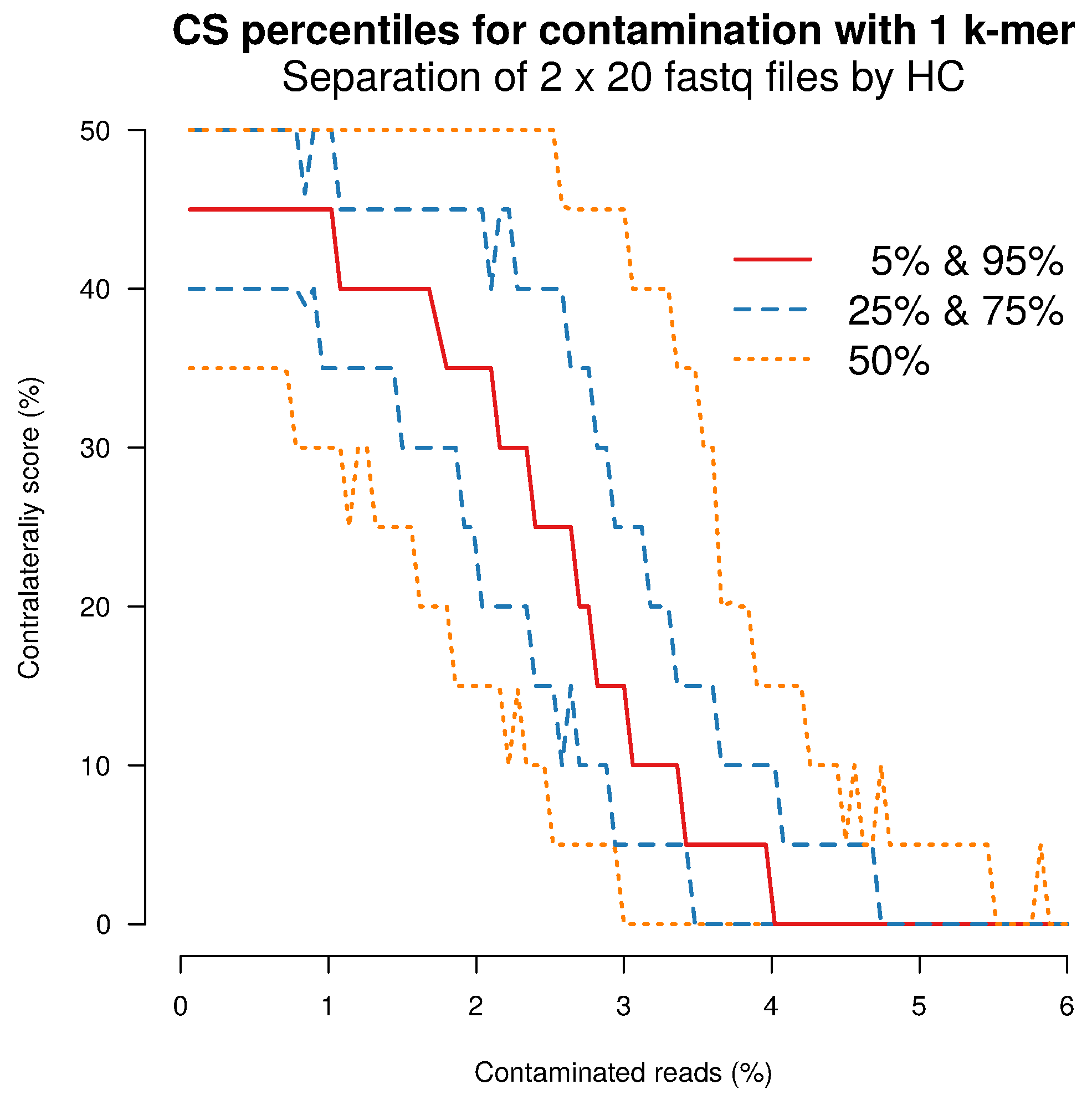
2.4. Separation Sensitivity on Simulated Data
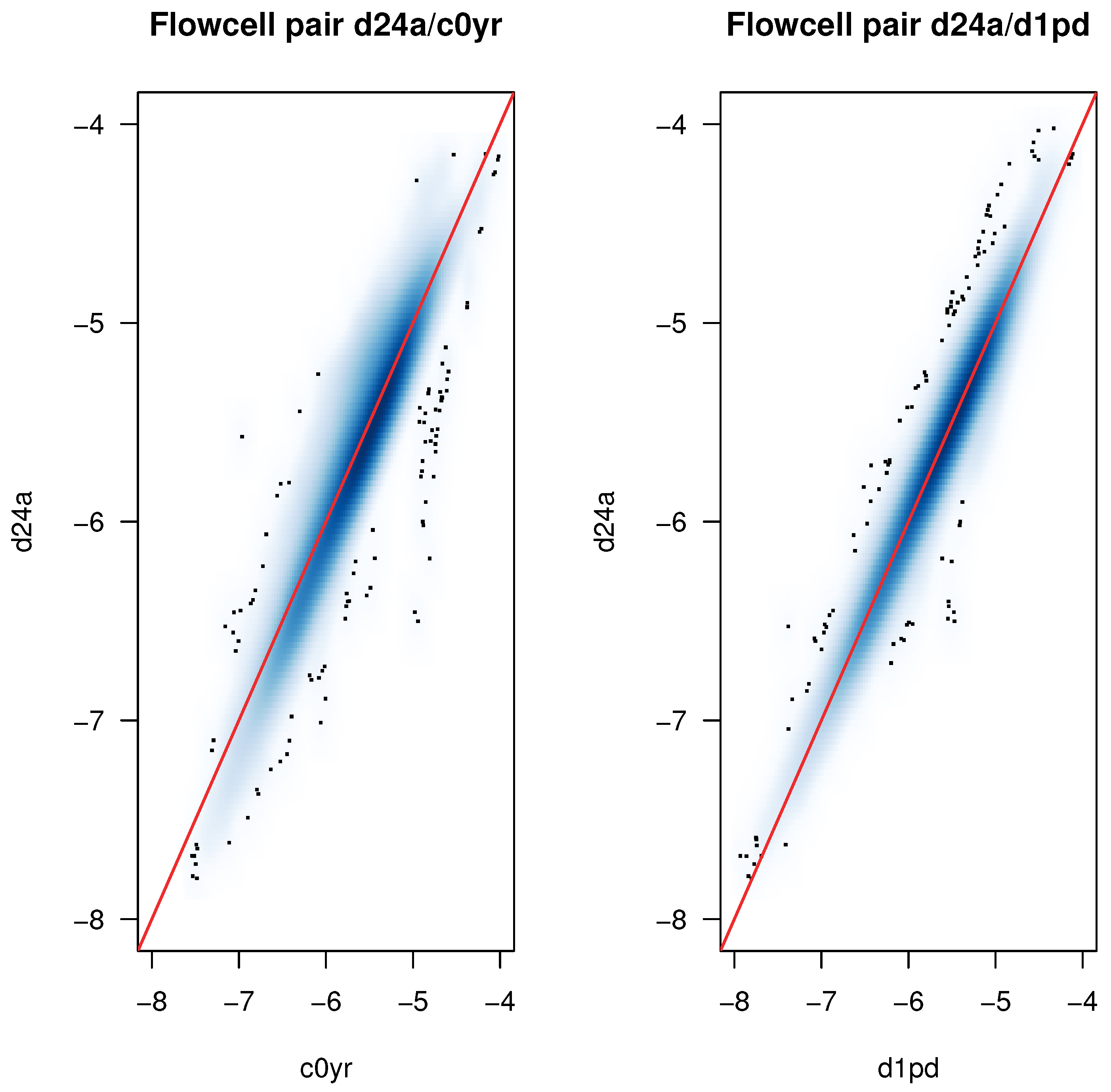
2.5. K-mer Spectrum Responsible for Tree Separation
3. Discussion
3.1. Indication of Batch Effects
3.2. Application of HcKmer
4. Materials and Methods
4.1. Algorithmic Framework
Sample Preparation, Sequencing and Alignment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BAM | Binary Alignment Map. File format for sequence alignment |
| CS | Contralaterality Score |
| CRAN | The Comprehensive R Archive Network |
| DEG | Differently expressed gene |
| Fastq | File format for sequencing data |
| FDR | False Discovery Rate |
| HC | Hierarchical clustering |
| HcKmer | Hierarchical clustering of DNA k-mer counts |
| Mm | Mus musculus |
| Phred score | Quality score for identification of nucleotides in sequencing data |
| QC | Quality control |
| RNAseq | Whole transcriptome sequencing |
| R, C | Computer programming languages |
| sd | standard deviation |
References
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bai, X.; Lu, Y.Y.; Tang, K.; Wang, Y.; Reinert, G.; Sun, F. Alignment-Free Sequence Analysis and Applications. Annu. Rev. Biomed. Data Sci. 2018, 1, 93–114. [Google Scholar] [CrossRef]
- Patro, R.; Mount, S.M.; Kingsford, C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014, 32, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A.; et al. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Denisov, G.; Walenz, B.; Halpern, A.L.; Miller, J.; Axelrod, N.; Levy, S.; Sutton, G. Consensus generation and variant detection by Celera Assembler. Bioinformatics 2008, 24, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, P.; Scott, E.; Kakaradov, B.; Pevzner, P. Error correction of high-throughput sequencing datasets with non-uniform coverage. Bioinformatics 2011, 27, i137–i141. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Schatz, M.C.; Salzberg, S.L. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol. 2010, 11, R116. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.B.; Dupim, E.G.; Goldstein, G. Improved assembly of noisy long reads by k-mer validation. Genome Res. 2016, 26, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2017, 33, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Melsted, P.; Pritchard, J.K. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinform. 2011, 12, 333. [Google Scholar] [CrossRef] [PubMed]
- Deorowicz, S.; Debudaj-Grabysz, A.; Grabowski, S. Disk-based k-mer counting on a PC. BMC Bioinform. 2013, 14, 160. [Google Scholar] [CrossRef] [PubMed]
- Deorowicz, S.; Kokot, M.; Grabowski, S.; Debudaj-Grabysz, A. KMC 2: Fast and resource-frugal k-mer counting. Bioinformatics 2015, 31, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Rizk, G.; Lavenier, D.; Chikhi, R. DSK: K-mer counting with very low memory usage. Bioinformatics 2013, 29, 652–653. [Google Scholar] [CrossRef] [PubMed]
- Manekar, S.C.; Sathe, S.R. A benchmark study of k-mer counting methods for high-throughput sequencing. Gigascience 2018. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- ’t Hoen, P.A.; Friedlander, M.R.; Almlof, J.; Sammeth, M.; Pulyakhina, I.; Anvar, S.Y.; Laros, J.F.; Buermans, H.P.; Karlberg, O.; Brannvall, M.; et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat. Biotechnol. 2013, 31, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Anvar, S.Y.; Khachatryan, L.; Vermaat, M.; van Galen, M.; Pulyakhina, I.; Ariyurek, Y.; Kraaijeveld, K.; den Dunnen, J.T.; de Knijff, P.; ’t Hoen, P.A.; et al. Determining the quality and complexity of next-generation sequencing data without a reference genome. Genome Biol. 2014, 15, 555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pell, J.; Canino-Koning, R.; Howe, A.C.; Brown, C.T. These are not the k-mers you are looking for: Efficient online k-mer counting using a probabilistic data structure. PLoS ONE 2014, 9, e101271. [Google Scholar] [CrossRef] [PubMed]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update–simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed]
- Bessman, M.J.; Frick, D.N.; O’Handley, S.F. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J. Biol. Chem. 1996, 271, 25059–25062. [Google Scholar] [CrossRef] [PubMed]
- Marriott, A.S.; Vasieva, O.; Fang, Y.; Copeland, N.A.; McLennan, A.G.; Jones, N.J. NUDT2 Disruption Elevates Diadenosine Tetraphosphate (Ap4A) and Down-Regulates Immune Response and Cancer Promotion Genes. PLoS ONE 2016, 11, e0154674. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Cooke, S.L.; Howe, K.; Newman, S.; Xian, J.; Temple, J.; Batty, E.M.; Pole, J.C.; Langdon, S.P.; Edwards, P.A.; et al. The role of tandem duplicator phenotype in tumour evolution in high-grade serous ovarian cancer. J. Pathol. 2012, 226, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Fare, T.L.; Coffey, E.M.; Dai, H.; He, Y.D.; Kessler, D.A.; Kilian, K.A.; Koch, J.E.; LeProust, E.; Marton, M.J.; Meyer, M.R.; et al. Effects of atmospheric ozone on microarray data quality. Anal. Chem. 2003, 75, 4672–4675. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Brenner, S.E.; Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38, e131. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-content normalization for RNA-Seq data. BMC Bioinform. 2011, 12, 480. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Fang, Z.; Zhang, K.; Zhi, D.; Cui, X. Length bias correction for RNA-seq data in gene set analyses. Bioinformatics 2011, 27, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.; Scharpf, R.; Bravo, H.; Simcha, D.; Langmead, B.; Johnson, W.; Geman, D.; Baggerly, K.; Irizarry, R. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Kaisers, W.; Boukamp, P.; Stark, H.J.; Schwender, H.; Tigges, J.; Krutmann, J.; Schaal, H. Age, gender and UV-exposition related effects on gene expression in in vivo aged short term cultivated human dermal fibroblasts. PLoS ONE 2017, 12, e0175657. [Google Scholar] [CrossRef] [PubMed]
- Kaisers, W. seqTools: Analysis of Nucleotide, Sequence and Quality Content on Fastq Files; R Package Version 1.0.0; Bioconductor: Redmond, WA, USA, 2014. [Google Scholar] [CrossRef]
- Lance, G.N.; Williams, W.T. Computer Programs for Hierarchical Polythetic Classification (“Similarity Analyses”). Comput. J. 1966, 9, 60–64. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Kaisers, W.; Schaal, H.; Schwender, H. rbamtools: An R interface to samtools enabling fast accumulative tabulation of splicing events over multiple RNA-seq samples. Bioinformatics 2015, 31, 1663–1664. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaisers , W.; Schwender, H.; Schaal , H. Hierarchical Clustering of DNA k-mer Counts in RNAseq Fastq Files Identifies Sample Heterogeneities. Int. J. Mol. Sci. 2018, 19, 3687. https://doi.org/10.3390/ijms19113687
Kaisers W, Schwender H, Schaal H. Hierarchical Clustering of DNA k-mer Counts in RNAseq Fastq Files Identifies Sample Heterogeneities. International Journal of Molecular Sciences. 2018; 19(11):3687. https://doi.org/10.3390/ijms19113687
Chicago/Turabian StyleKaisers , Wolfgang, Holger Schwender, and Heiner Schaal . 2018. "Hierarchical Clustering of DNA k-mer Counts in RNAseq Fastq Files Identifies Sample Heterogeneities" International Journal of Molecular Sciences 19, no. 11: 3687. https://doi.org/10.3390/ijms19113687
APA StyleKaisers , W., Schwender, H., & Schaal , H. (2018). Hierarchical Clustering of DNA k-mer Counts in RNAseq Fastq Files Identifies Sample Heterogeneities. International Journal of Molecular Sciences, 19(11), 3687. https://doi.org/10.3390/ijms19113687