Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses


 ,
,
Abstract
1. Introduction
2. Results and Discussion
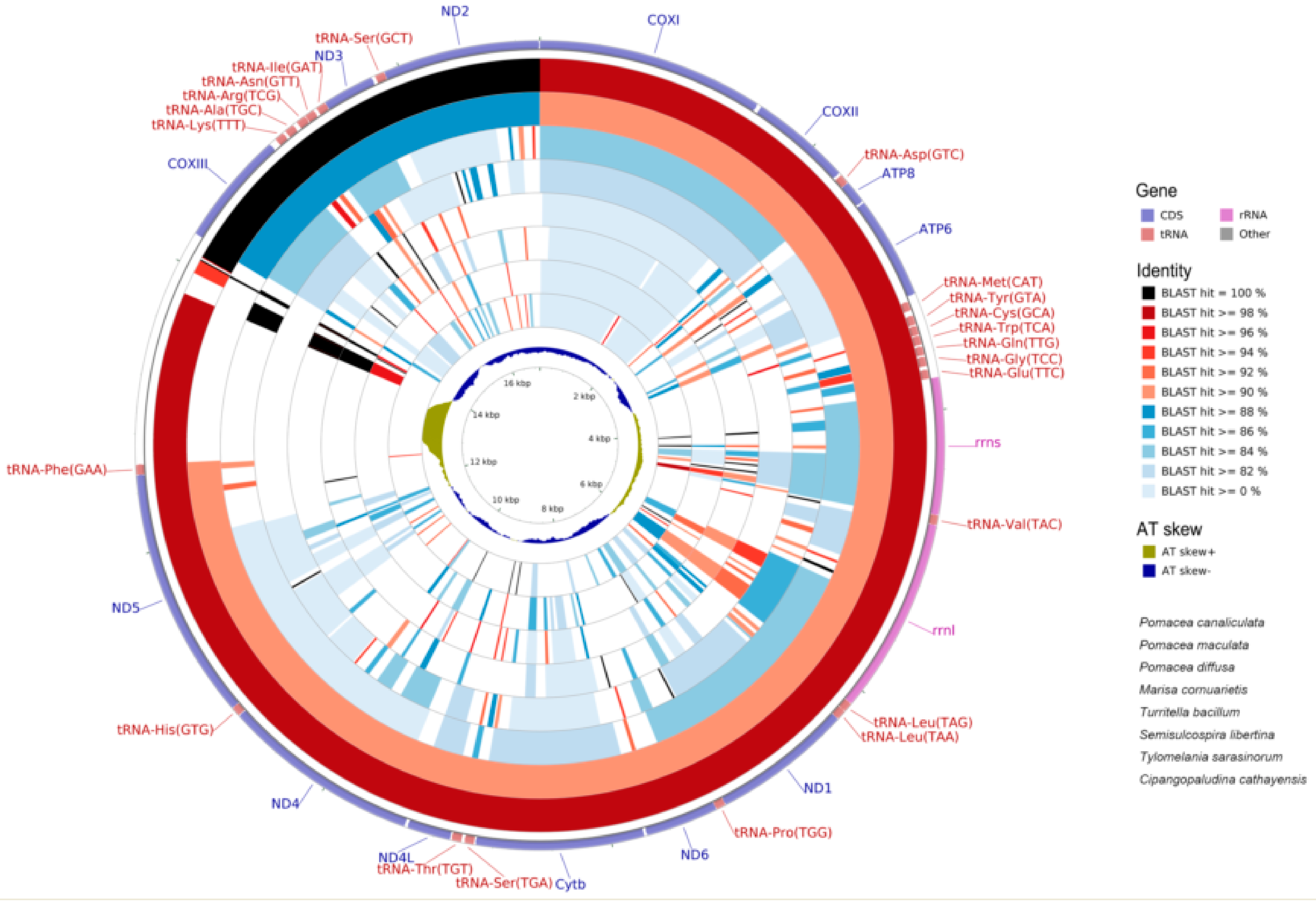
2.1. Genome Organization and Composition
2.2. Nucleotide Composition
2.3. Protein Coding Genes (PCG)
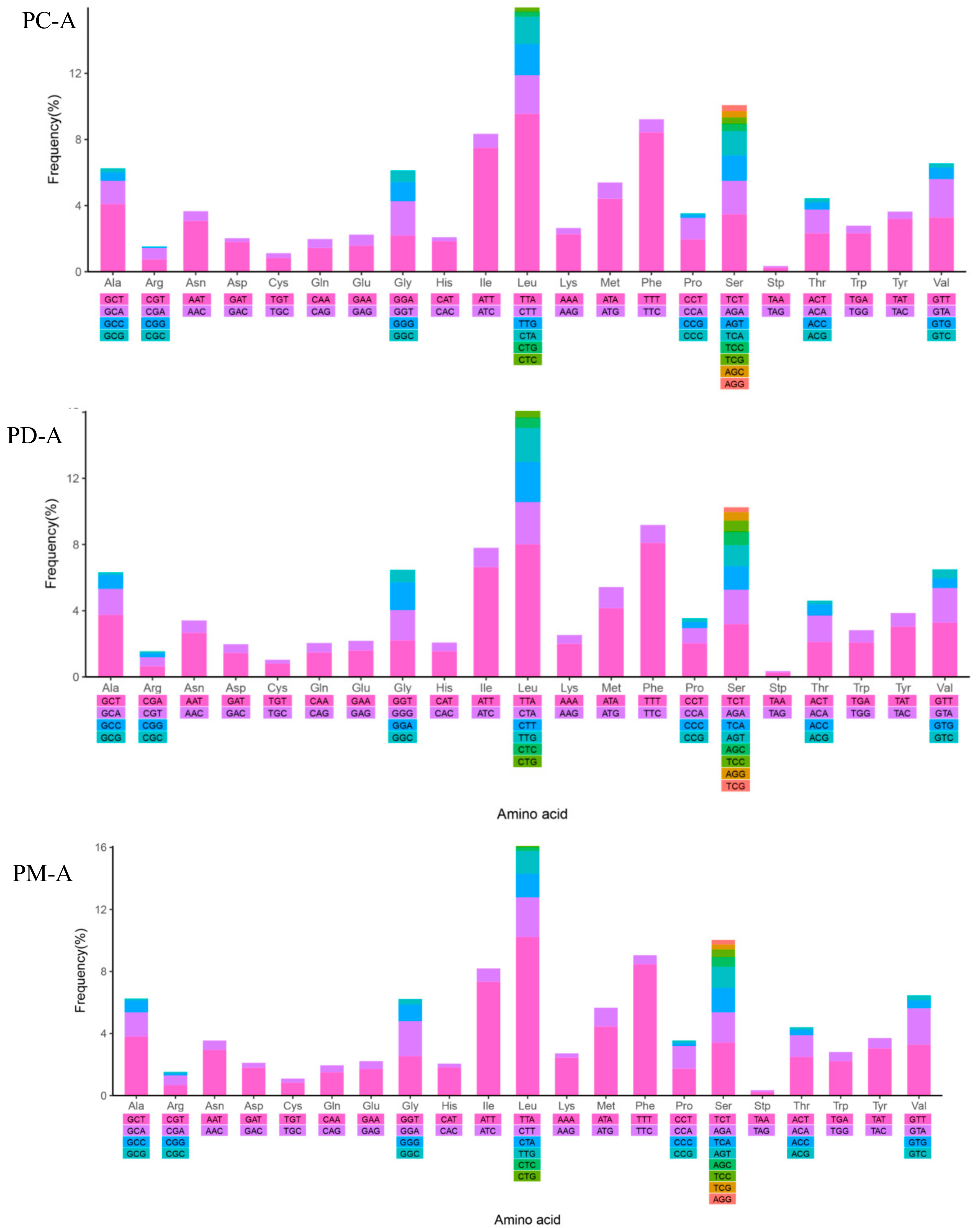
2.4. Mitochondrial Gene Codon Usage
2.5. Transfer and Ribosomal RNA Genes
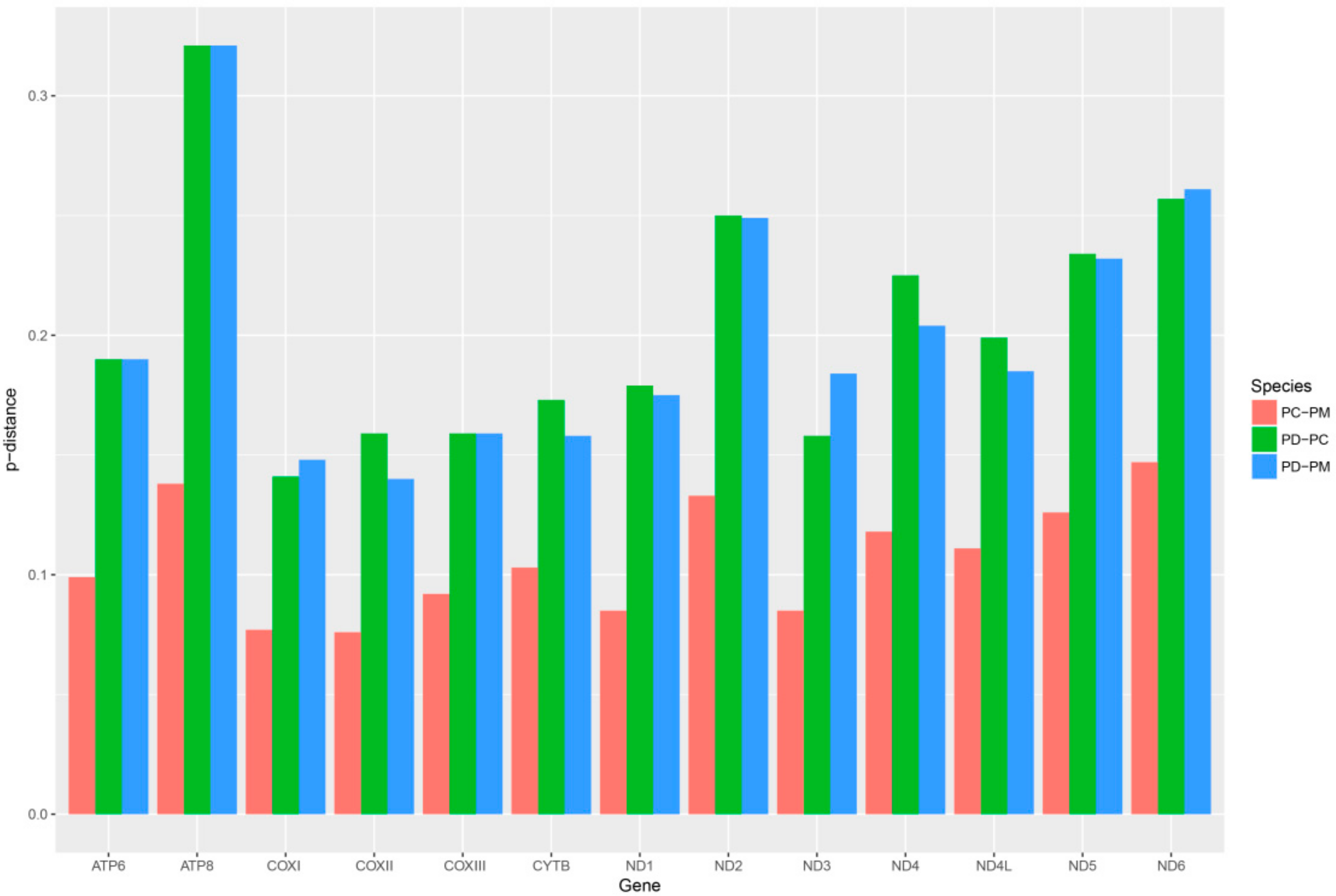
2.6. Mitogenome Comparisions
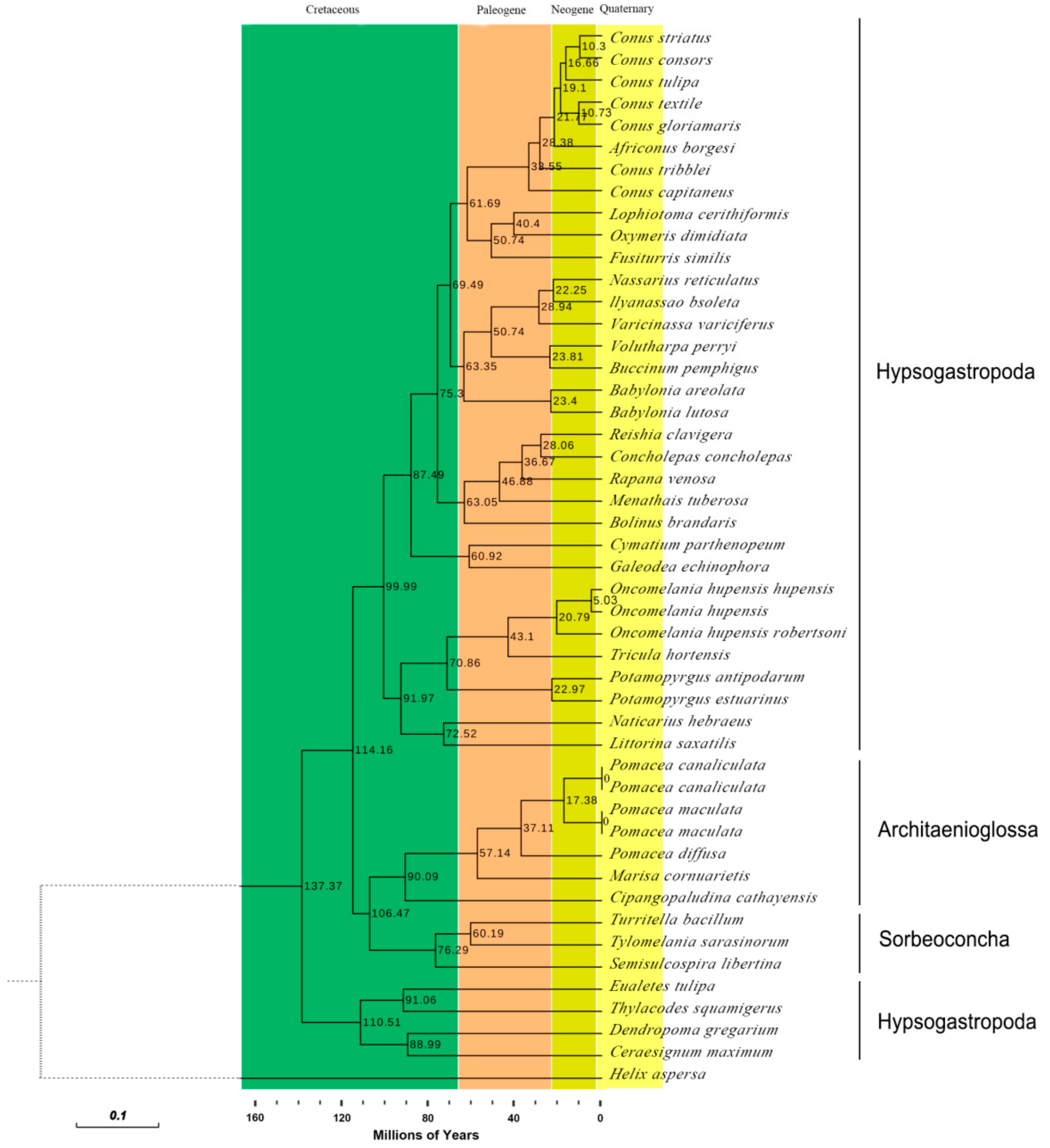
2.7. Phylogenetic Analyses
2.8. Divergence Times of the Caenogastropoda
3. Materials and Methods
3.1. Sampling and Materials
3.2. Library Preparation for Sequencing
3.3. Sequence Analysis and Annotation
3.4. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rawlings, T.A.; Hayes, K.A.; Cowie, R.H.; Collins, T.M. The identity, distribution, and impacts of non-native apple snails in the continental United States. BMC Evol. Biol. 2007, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Levin, P.C.R.; Taylor, J.M.; Hayes, K.A.; Burnett, K.M.; Ferguson, C.A. Apple snail invasions and the slow road to control: Ecological, economic, agricultural and cultural perspectives in hawaii. In Global Advances in Ecology and Management of Golden Apple Snails, 2nd ed.; Joshi, R.C., Sebastian, L.S., Eds.; Philippine Rice Research Institute: Nueva Ecija, Philippines, 2006; pp. 325–335. ISBN 9789719081319. [Google Scholar]
- Lowe, S.B.M.; Boudjelas, S.; De Poorter, M. 100 of the world’s worst invasive alien species: A selection from the global invasive species database. In The Invasive Species Specialists Group of the Species Survival Commission of the World Conservation Union; Hollands Printing: Auckland, New Zealand, 2000. [Google Scholar]
- Cazzaniga, N.J. Old species and new concepts in the taxonomy of pomacea (gastropoda: Ampullariidae). Europe PMC 2002, 26, 71–81. [Google Scholar]
- Cowie, R.H.; Hayes, K.A.; Thiengo, S.C. What are apple snails? Confused taxonomy and some preliminary resolution. In Global Advances in Ecology and Management of Golden Apple Snails, 2nd ed.; Joshi, R.C., Sebastian, L.S., Eds.; Philippine Rice Research Institute: Nueva Ecija, Philippines, 2006. [Google Scholar]
- RH, C. Apple snails (ampullariidae) as agricultural pests: Their biology, impacts and management. In Molluscs as Crop Pests; Barker, G.M., Ed.; CAB-International: Wallingford, UK, 2002. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- WM, B. The mitochondrial genome of animals. In Molecular Evolutionary Genetics; Mcintyre, R.J., Ed.; Plenum. Press: New York, NY, USA, 1985. [Google Scholar]
- Poulin, E.; Cardenas, L.; Hernandez, C.E.; Kornfield, I.; Ojeda, F.P. Resolution of the taxonomic status of chilean and californian jack mackerels using mitochondrial DNA sequence. J. Fish Biol. 2004, 65, 1160–1164. [Google Scholar] [CrossRef]
- Habib, M.; Lakra, W.S.; Mohindra, V.; Khare, P.; Barman, A.S.; Singh, A.; Lal, K.K.; Punia, P.; Khan, A.A. Evaluation of cytochrome b mtdna sequences in genetic diversity studies of channa marulius (channidae: Perciformes). Mol. Biol. Rep. 2011, 38, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Estebenet, A.L.; Martin, P.R. Pomacea canaliculata (gastropoda: Ampullariidae): Life-history traits and their plasticity. Biocell 2002, 26, 83–89. [Google Scholar] [PubMed]
- Liu, J.; He, Y.J.; Tan, J.C.; Xu, C.X.; Zhong, L.; Wang, Z.G.; Liao, Q.G. Characteristics of pomacea canaliculata reproduction under natural conditions. J. Appl. Ecol. 2012, 23, 559–565. [Google Scholar]
- Lv, S.; Zhang, Y.; Liu, H.X.; Zhang, C.W.; Steinmann, P.; Zhou, X.N.; Utzinger, J. Angiostrongylus cantonensis: Morphological and behavioral investigation within the freshwater snail pomacea canaliculata. Parasitol. Res. 2009, 104, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- San Martins, R.; Gelmi, C.; de Oliveira, J.V.; Galo, J.L.; Pranto, H. Use of a saponin based molluscicide to control pomacea canaliculata snails in southern brazil. Nat. Prod. Commun. 2009, 4, 1327–1330. [Google Scholar] [PubMed]
- Jarusiewicz, J.A.; Fried, B.; Sherma, J. Effects of diet on the carotenoid pigment and lipid content of pomacea bridgesii as determined by quantitative high performance thin layer chromatography. Comp. Biochem. Phys. Part B 2006, 143, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Olivier, H.M.; Jenkins, J.A.; Berhow, M.; Carter, J. A pilot study testing a natural and a synthetic molluscicide for controlling invasive apple snails (pomacea maculata). Bull. Environ. Contam. Toxicol. 2016, 96, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Pasquevich, M.Y.; Dreon, M.S.; Heras, H. The major egg reserve protein from the invasive apple snail pomacea maculata is a complex carotenoprotein related to those of pomacea canaliculata and pomacea scalaris. Comp. Biochem. Phys. Part B 2014, 169, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Pinto, H.A.; Cantanhede, S.P.; Thiengo, S.C.; de Melo, A.L.; Fernandez, M.A. The apple snail pomacea maculata (caenogastropoda: Ampullariidae) as the intermediate host of stomylotrema gratiosus (trematoda: Stomylotrematidae) in Brazil: The first report of a mollusc host of a stomylotrematid trematode. J. Parasitol. 2015, 101, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Souza Junior, E.; De Barros, J.C.; Paresque, K.; De Freitas, R.R. The effect of stocking density on the growth of apple snails native pomacea bridgesii and exotic pomacea lineata (mollusca, gastropoda). An. Acad. Bras. Cienc. 2013, 85, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, S.; Song, F.; Li, H.; Liu, J.; Liu, G.; Yu, X. The mitochondrial genome of pomacea maculata (gastropoda: Ampullariidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 2895–2896. [Google Scholar] [PubMed]
- Bian, Q.Q.; Li, X.Y.; Fang, Y.Q.; Jia, Y.Q.; Mu, X.D. Molecular identification of pomacea canaliculata and p. Insularum from rice paddy in different origins in china using mitochondrial adenosine triphosphate subunit 6 gene. Mitochondrial DNA 2015, 26, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Bian, Q.Q.; Zhao, G.H. Phylogenetic analysis of pomacea canaliculata isolates collected from rice fields in different origins of china by combined mitochondrial 12s and 16s genes. Mitochondrial DNA 2015, 26, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Y.; Zhu, S.; Xu, H.; Liu, Y.; Chen, L. The complete mitochondrial genome of pomacea canaliculata (gastropoda: Ampullariidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 884–885. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, P.; Rocroi, J.-P.; Frýda, J.; Hausdorf, B.; Ponder, W.; Valdés, Á.; Warén, A. Classification and Nomenclator of Gastropod Families; Malacologia ConchBooks: Hackenheim, Germany, 2005; Volume 47. [Google Scholar]
- Wang, M.; Qiu, J.W. Complete mitochondrial genome of the giant ramshorn snail marisa cornuarietis (gastropoda: Ampullariidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 1734–1735. [Google Scholar] [PubMed]
- Liu, G.H.; Wang, S.Y.; Huang, W.Y.; Zhao, G.H.; Wei, S.J.; Song, H.Q.; Xu, M.J.; Lin, R.Q.; Zhou, D.H.; Zhu, X.Q. The complete mitochondrial genome of galba pervia (gastropoda: Mollusca), an intermediate host snail of fasciola spp. PLoS ONE 2012, 7, e42172. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Yin, W.; Xia, R.; Fu, C.; Jin, B. Complete mitochondrial genome of a freshwater snail, semisulcospira libertina (cerithioidea: Semisulcospiridae). Mitochondrial DNA 2015, 26, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Peng, C.; Chen, Q.; Zhang, J.; Shi, Q. Mitochondrial genome sequencing of a vermivorous cone snail Conus quercinus supports the correlative analysis between phylogenetic relationships and dietary types of Conus species. PLoS ONE 2018, 13, e0193053. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.L.; Zardoya, R. Mitogenomic phylogeny of cone snails endemic to senegal. Mol. Phylogenet. Evol. 2017, 112, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Fontanilla, I.K.; Naggs, F.; Wade, C.M. Molecular phylogeny of the achatinoidea (mollusca: Gastropoda). Mol. Phylogenet. Evol. 2017, 114, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.R.; Bergthorsson, U.; Adema, C.M. Physella acuta: Atypical mitochondrial gene order among panpulmonates (gastropoda). J. Mollus. Stud. 2014, 80, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Osca, D.; Templado, J.; Zardoya, R. The mitochondrial genome of ifremeria nautilei and the phylogenetic position of the enigmatic deep-sea abyssochrysoidea (mollusca: Gastropoda). Gene 2014, 547, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Zhang, D.; Jakovlic, I.; Wang, W.M. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the viviparidae family (caenogastropoda). PLoS ONE 2017, 12, e0181699. [Google Scholar] [CrossRef] [PubMed]
- White, T.R.; Conrad, M.M.; Tseng, R.; Balayan, S.; Golding, R.; Martins, A.M.D.; Dayrat, B.A. Ten new complete mitochondrial genomes of pulmonates (mollusca: Gastropoda) and their impact on phylogenetic relationships. BMC Evol. Biol. 2011, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Sudbery, P. Human Molecular Genetics, 2nd ed.; Pearson Education: Iowa City, IA, USA, 2002. [Google Scholar]
- Arquez, M.; Colgan, D.; Castro, L.R. Sequence and comparison of mitochondrial genomes in the genus nerita (gastropoda: Neritimorpha: Neritidae) and phylogenetic considerations among gastropods. Mar. Genom. 2014, 15, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Gaitan-Espitia, J.D.; Nespolo, R.F.; Opazo, J.C. The complete mitochondrial genome of the land snail cornu aspersum (helicidae: Mollusca): Intra-specific divergence of protein-coding genes and phylogenetic considerations within euthyneura. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Osca, D.; Irisarri, I.; Todt, C.; Grande, C.; Zardoya, R. The complete mitochondrial genome of scutopus ventrolineatus (mollusca: Chaetodermomorpha) supports the aculifera hypothesis. BMC Evol. Biol. 2014, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.L.; Kirouac, L.E.; Thomas, W.K.; Ramsdell, J.S.; Lawlor, K.E.; Sharifi, O.; Grewal, S.; Baysdorfer, C.; Curr, K.; Naimie, A.A.; et al. The mitochondrial genomes of the nudibranch mollusks, melibe leonina and tritonia diomedea, and their impact on gastropod phylogeny. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xia, J.; Zhang, J.E.; Yang, J.; Zhao, H.; Wang, Q.; Sun, J.; Xue, H.; Wu, Y.; Chen, J.; et al. Characterization of the complete mitochondrial genome sequences of three croakers (perciformes, sciaenidae) and novel insights into the phylogenetics. Int. J. Mol. Sci. 2018, 19, 1741. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yang, H.R.; Zhang, C.X.; Xue, H.Y.; Xia, Y.; Zhang, J.E. Complete mitochondrial genome of the apple snail pomacea diffusa (gastropoda, ampullariidae) with phylogenetic consideration. Mitochondrial DNA B 2017, 2, 865–867. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.E.; Guo, J.; Deng, Z.; Luo, H.; Luo, M.; Zhao, B. The complete mitochondrial genome of the giant african snail achatina fulica (mollusca: Achatinidae). Mitochondrial. DNA A DNA Mapp. Seq. Anal. 2016, 27, 1622–1624. [Google Scholar] [PubMed]
- Yang, H.; Zhang, J.E.; Luo, H.; Luo, M.; Guo, J.; Deng, Z.; Zhao, B. The complete mitochondrial genome of the mudsnail cipangopaludina cathayensis (gastropoda: Viviparidae). Mitochondrial. DNA A DNA Mapp. Seq. Anal. 2016, 27, 1892–1894. [Google Scholar] [PubMed]
- Yang, H.R.; Zhang, J.E.; Deng, Z.X.; Luo, H.; Guo, J.; He, S.M.; Luo, M.Z.; Zhao, B.L. The complete mitochondrial genome of the golden apple snail pomacea canaliculata (gastropoda: Ampullariidae). Mitochondrial DNA B 2016, 1, 45–47. [Google Scholar] [CrossRef]
- Francino, M.P.; Ochman, H. Strand asymmetries in DNA evolution. Trends Genet. TIG 1997, 13, 240–245. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.T.; Foster, P.G.; Littlewood, D.T.J. The complete mitochondrial genome of a turbinid vetigastropod from miseq illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene 2014, 533, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Thaewnon-ngiw, B.; Klinbunga, S.; Phanwichien, K.; Sangduen, N.; Lauhachinda, N.; Menasveta, P. Genetic diversity and molecular markers in introduced and Thai native apple snails (Pomacea and Pila). J. Biochem. Mol. Biol. 2004, 37, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, T.A.; Maclnnis, M.J.; Bieler, R.; Boore, J.L.; Collins, T.M. Sessile snails, dynamic genomes: Gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genom. 2010, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Qu, M.; Zhang, X.; Ding, S. A comprehensive description and evolutionary analysis of 22 grouper (perciformes, epinephelidae) mitochondrial genomes with emphasis on two novel genome organizations. PLoS ONE 2013, 8, e73561. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Tian, P.; Lin, R.; Huang, D.; Wang, J. Characterization of the complete mitochondrial genome sequence of the globose head whiptail cetonurus globiceps (gadiformes: Macrouridae) and its phylogenetic analysis. PLoS ONE 2016, 11, e0153666. [Google Scholar] [CrossRef] [PubMed]
- Chao, Q.J.; Li, Y.D.; Geng, X.X.; Zhang, L.; Dai, X.; Zhang, X.; Li, J.; Zhang, H.J. Complete mitochondrial genome sequence of Marmota himalayana (rodentia: Sciuridae) and phylogenetic analysis within rodentia. Genet. Mol. Res. GMR 2014, 13, 2739–2751. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The trnascan-se, snoscan and snogps web servers for the detection of trnas and snornas. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of rna editing occurs in the mitochondrial trnas of the centipede lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, J.; Li, Y.; Cui, Y.; Xie, Q.; Bu, W.; Hillis, D.M. A mitochondrial genome of rhyparochromidae (hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, X.D.; Yang, R.H.; Hsiang, T.; Wang, K.; Liang, D.Q.; Liang, F.; Cao, D.M.; Zhou, F.; Wen, G.; et al. Complete mitochondrial genome of the medicinal fungus ophiocordyceps sinensis. Sci. Rep. 2015, 5, 13892. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Nadler, S.A.; Liu, S.S.; Podolska, M.; D’Amelio, S.; Shao, R.; Gasser, R.B.; Zhu, X.Q. Mitochondrial phylogenomics yields strongly supported hypotheses for ascaridomorph nematodes. Sci. Rep. 2016, 6, 39248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Estebenet, A.L.; Martin, P.R. Shell interpopulation variation and its origin in pomacea canaliculata (gastropoda: Ampullariidae) from Southern Pampas, Argentina. J. Mollus. Stud. 2003, 69, 301–310. [Google Scholar] [CrossRef]
- Matsukura, K.; Okuda, M.; Kubota, K.; Wada, T. Genetic divergence of the genus Pomacea (gastropoda: Ampullariidae) distributed in japan, and a simple molecular method to distinguish P. Canaliculata and P. Insularum. Appl. Entomol. Zool 2008, 43, 535–540. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012. [Google Scholar]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. Mitofish and mitoannotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. Arwen: A program to detect trna genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. Trnascan-se on-line: Integrating search and context for analysis of transfer rna genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, A.C.; Lessinger, A.C.; Torres, T.T.; da Silva, F.R.; Vettore, A.L.; Arruda, P.; Azeredo Espin, A.M. The mitochondrial genome of the blowfly Chrysomya chloropyga (diptera: Calliphoridae). Gene 2004, 339, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Issac, B.; Raghava, G.P.; Ramaswamy, R. Spectral repeat finder (srf): Identification of repetitive sequences using fourier transformation. Bioinformatics 2004, 20, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index--A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Prottest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res. 2012, 40, W569–W572. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. Timetree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Gene/Element | Strand | Size (bp) | GC-Content (%) | Amino Acids (aa) | Inferred Initiation Codon | Inferred Termination Codon | One Letter Code | Anti-Codon | Intergenic Nucleotide*(bp) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | COXI | H | 1536 | 33.27–36.07 | 511 | ATG | TAG/TAA | 27–32 | ||
| 2 | COXII | H | 687 | 31.3–34.79 | 228 | ATG | TAG/TAA | 17–21 | ||
| 3 | tRNA-Asp | H | 68 | 14.71–17.65 | D | GTC | 0 | |||
| 4 | ATP8 | H | 159 | 21.38–26.42 | 52 | ATG | TAA | 12–29 | ||
| 5 | ATP6 | H | 699–714 | 26.90–29.41 | 232–237 | ATG | TAG/TAA | 15–28 | ||
| 6 | tRNA-Met | L | 64–66 | 27.69–34.38 | M | CAT | 23–35 | |||
| 7 | tRNA-Tyr | L | 65–68 | 31.34–41.18 | Y | GTA | 8–25 | |||
| 8 | tRNA-Cys | L | 62–65 | 21.54–29.03 | C | GCA | 5–19 | |||
| 9 | tRNA-Trp | L | 64–68 | 27.94–35.38 | W | TCA | 11–119 | |||
| 10 | tRNA-Gln | L | 62–64 | 34.38–35.48 | Q | TTG | 9–10 | |||
| 11 | tRNA-Gly | L | 66–67 | 19.40–20.9 | G | TCC | 24–31 | |||
| 12 | tRNA-Glu | L | 65–67 | 20.90–24.62 | E | TTC | 0 | |||
| 13 | rrns | H | 929–950 | 28.09–29.68 | 0–2 | |||||
| 14 | tRNA-Val | H/L | 67–69 | 20.90–29.41 | V | TAC | 0–1 | |||
| 15 | rrnl | H | 1331–1345 | 25.47–27.14 | 0 | |||||
| 16 | tRNA-Leu | H | 64–66 | 23.44–27.27 | L | TAG | (−1)–0 | |||
| 17 | tRNA-Leu | H | 68–69 | 23.53–26.09 | L | TAA | 0 | |||
| 18 | ND1 | H | 945–960 | 28.02–31.96 | 314–319 | ATG | TAG/TAA | (−12)–9 | ||
| 19 | tRNA-Pro | H | 66–68 | 29.85–30.88 | P | TGG | 0 | |||
| 20 | ND6 | H | 495 | 23.64–26.87 | 164 | ATG | TAA | 10–19 | ||
| 21 | Cytb | H | 1140 | 28.51–32.02 | 379 | ATG | TAG/TAA | 9–17 | ||
| 22 | tRNA-Ser | H | 66 | 33.33–34.85 | S | TGA | 20–30 | |||
| 23 | tRNA-Thr | H/L | 68–71 | 30.99–35.29 | T | TGT | 9–14 | |||
| 24 | ND4L | H | 297–312 | 26.92–27.95 | 98–103 | ATA, ATG | TAG/TAA | (−7)–20 | ||
| 25 | ND4 | H | 1341–1362 | 25.73–29.30 | 446–453 | ATA, ATG | TAA | 3–9 | ||
| 26 | tRNA-His | H | 64–65 | 16.92–21.88 | H | GTG | 0 | |||
| 27 | ND5 | H | 1710–1728 | 28.13–31.42 | 569–575 | ATG | TAA | (−20)–0 | ||
| 28 | tRNA-Phe | H | 68–69 | 30.88–33.33 | F | GAA | 141–1617 | |||
| 29 | COXIII | H | 780 | 33.72–37.69 | 259 | ATA, ATG | TAA | 47–67 | ||
| 30 | tRNA-Lys | H | 67–68 | 29.85–32.35 | K | TTT | 10–18 | |||
| 31 | tRNA-Ala | H | 68–71 | 23.19–28.17 | A | TGC | 29–46 | |||
| 32 | tRNA-Arg | H | 67–69 | 29.85–33.33 | R | TCG | 2–5 | |||
| 33 | tRNA-Asn | H | 69–71 | 21.13–24.64 | N | GTT | 19–30 | |||
| 34 | tRNA-Ile | H | 70–72 | 30.56–34.29 | I | GAT | 0–6 | |||
| 35 | ND3 | H | 354 | 29.66–31.92 | 117 | ATG | TAA | 20–37 | ||
| 36 | tRNA-Ser | H | 68 | 30.88–32.35 | S | GCT | 0 | |||
| 37 | ND2 | H | 1062–1071 | 27.12–30.91 | 353–356 | ATG | TAG/TAA | 4–6 |
| Speics | Pomacea canaliculata | Pomacea diffusa | Pomacea maculata | Marisa cornuarietis | Cipangopaludina cathayensis | Turritella bacillum | Tylomelania sarasinorum | Semisulcospira libertina |
|---|---|---|---|---|---|---|---|---|
| Accession number | KU052865 | KY008698 | KY008699 | NC_025334.1 | NC_025577.1 | NC_029717.1 | NC_030263.1 | NC_023364.1 |
| Taxonomy | Caenogastropoda, Architaenioglossa | Caenogastropoda, Architaenioglossa | Caenogastropoda, Architaenioglossa | Caenogastropoda, Architaenioglossa | Caenogastropoda, Architaenioglossa | Caenogastropoda, Sorbeoconcha | Caenogastropoda, Sorbeoconcha | Caenogastropoda, Sorbeoconcha |
| Length (bp) | 16,965 | 16,640 | 15,516 | 15,923 | 17,157 | 15,868 | 16,632 | 15,432 |
| A (%) | 32.61 | 30.13 | 30.81 | 28.94 | 26.74 | 28.85 | 29.49 | 31.4 |
| T (%) | 39.85 | 39.62 | 41.13 | 41.04 | 44.51 | 35.88 | 35.65 | 34.76 |
| C (%) | 12.03 | 14.24 | 12.81 | 13.34 | 8.28 | 16.21 | 16.56 | 17.78 |
| G (%) | 15.51 | 16.02 | 15.25 | 16.68 | 20.48 | 19.06 | 18.3 | 16.06 |
| AT (%) | 72.46 | 69.75 | 71.94 | 69.98 | 71.25 | 64.73 | 65.14 | 66.16 |
| AT-skew | −0.0998 | −0.1361 | −0.1433 | −0.1729 | −0.2493 | −0.109 | −0.0946 | −0.0508 |
| GC-skew | 0.1263 | 0.059 | 0.0868 | 0.1113 | 0.4243 | 0.0809 | 0.0499 | −0.0507 |
| Length (aa) | 3724 | 3737 | 3733 | 3712 | 3727 | 3737 | 3755 | 3750 |
| AT (%) (all) | 70.71 | 67.98 | 71 | 68.98 | 70.29 | 63.89 | 64.29 | 65.66 |
| AT (%) (3rd) | 72.38 | 76.02 | 85.61 | 82.34 | 79.38 | 68.75 | 68.58 | 69.35 |
| AT-skew | −0.2099 | −0.2064 | −0.2022 | −0.2312 | −0.3243 | −0.119 | −0.1033 | −0.0642 |
| GC-skew | 0.0844 | 0.0466 | 0.0697 | 0.0764 | 0.3843 | 0.0862 | 0.0696 | −0.0397 |
| Length (bp) | 1334 | 1345 | 1331 | 1359 | 1387 | 1357 | 1382 | 1347 |
| AT (%) | 73.54 | 72.86 | 74.53 | 72.77 | 72.17 | 68.53 | 66.93 | 68.08 |
| AT-skew | 0.0051 | 0.0163 | 0.006 | −0.0253 | −0.1109 | −0.073 | −0.107 | −0.06 |
| GC-skew | 0.1501 | 0.1397 | 0.1622 | 0.2324 | 0.4456 | −0.002 | −0.1116 | −0.0791 |
| Length (bp) | 929 | 950 | 934 | 977 | 895 | 949 | 899 | 951 |
| AT (%) | 71.91 | 70.32 | 70.99 | 69.81 | 66.03 | 67.76 | 64.29 | 66.56 |
| AT-skew | 0.012 | 0.021 | 0.0377 | −0.0264 | −0.0592 | −0.154 | −0.1142 | −0.0142 |
| GC-skew | 0.249 | 0.1773 | 0.1956 | 0.2542 | 0.4474 | −0.033 | −0.1153 | −0.1509 |
| Length (bp) | 1473 | 1484 | 1479 | 1500 | 1471 | 1486 | 1482 | 1489 |
| AT (%) | 71.89 | 71.56 | 72.08 | 68.07 | 72.33 | 64.8 | 64.3 | 65.14 |
| AT-skew | −0.0293 | −0.0226 | −0.0263 | −0.0245 | −0.0602 | −0.09 | −0.0766 | −0.0124 |
| GC-skew | 0.0821 | 0.0616 | 0.0847 | 0.0814 | 0.2776 | 0.0554 | 0.0057 | −0.0405 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhang, J.-e.; Xia, J.; Yang, J.; Guo, J.; Deng, Z.; Luo, M. Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses. Int. J. Mol. Sci. 2018, 19, 3646. https://doi.org/10.3390/ijms19113646
Yang H, Zhang J-e, Xia J, Yang J, Guo J, Deng Z, Luo M. Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses. International Journal of Molecular Sciences. 2018; 19(11):3646. https://doi.org/10.3390/ijms19113646
Chicago/Turabian StyleYang, Huirong, Jia-en Zhang, Jun Xia, Jinzeng Yang, Jing Guo, Zhixin Deng, and Mingzhu Luo. 2018. "Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses" International Journal of Molecular Sciences 19, no. 11: 3646. https://doi.org/10.3390/ijms19113646
APA StyleYang, H., Zhang, J.-e., Xia, J., Yang, J., Guo, J., Deng, Z., & Luo, M. (2018). Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses. International Journal of Molecular Sciences, 19(11), 3646. https://doi.org/10.3390/ijms19113646