Revisiting Bacterial Ubiquitin Ligase Effectors: Weapons for Host Exploitation


 ,
,  ,
, 

Abstract
1. Introduction
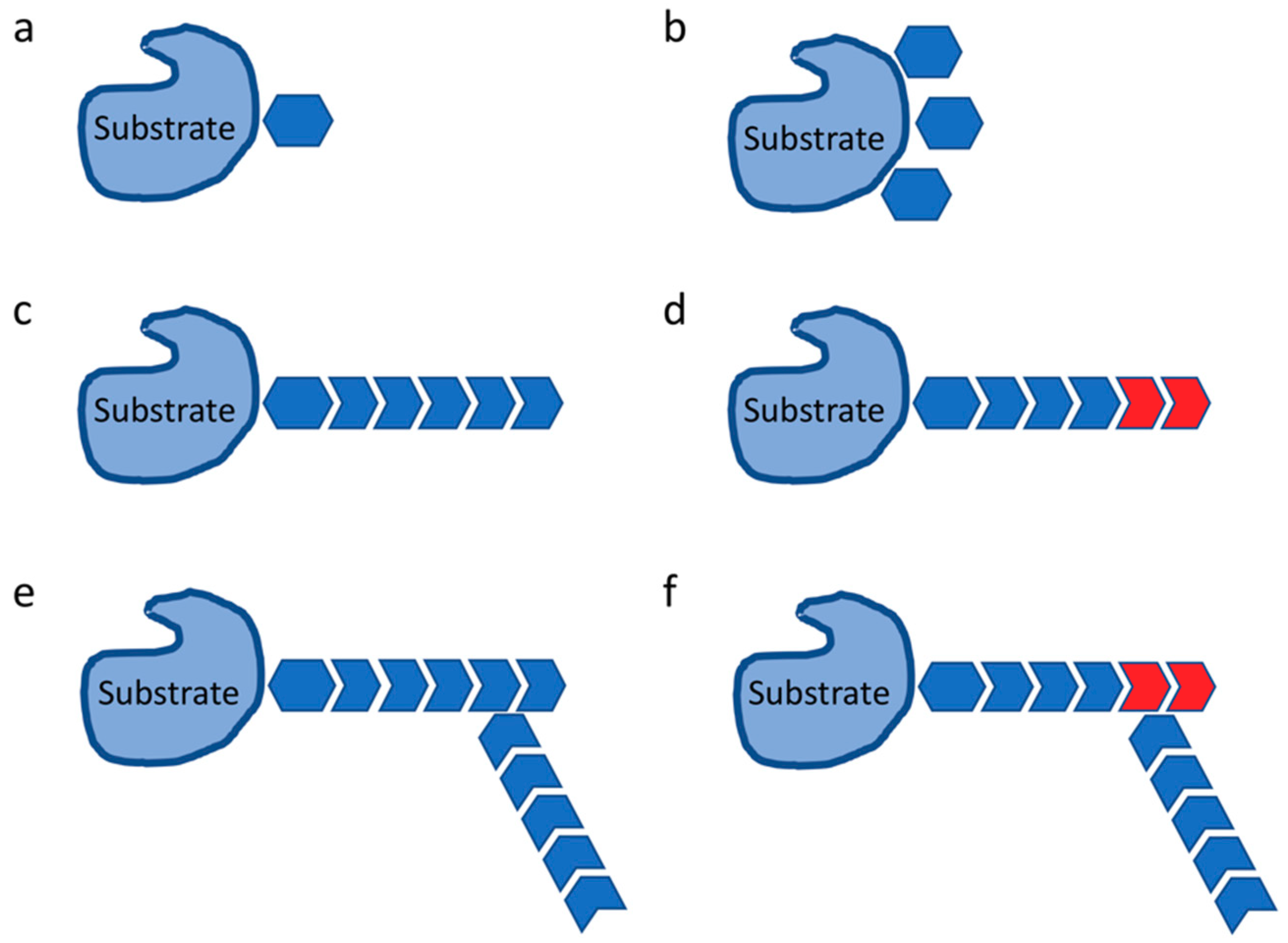
2. Ubiquitylation in Immune Response
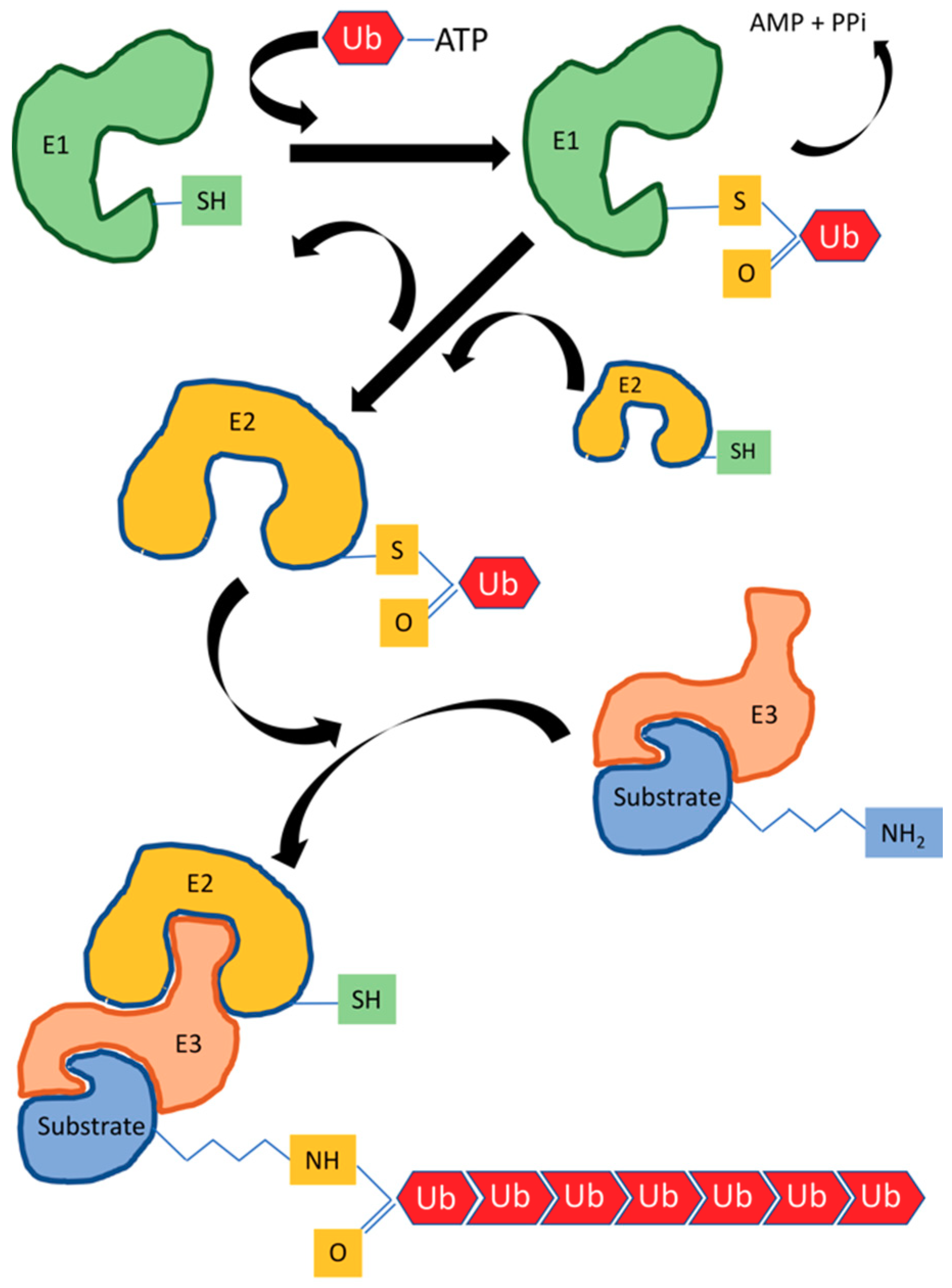
3. HECT E3 Ubiquitin Ligases
4. RING/U-Box Ubiquitin Ligases
5. F-Box Proteins
6. Novel E3 Ligases
7. New Effector Bacterial Ubiquitin Ligases
8. Host Cell Ubiquitin Ligase Modulated by Bacterial Virulence Factors
9. Conclusions
Funding
Conflicts of Interest
References
- Laney, J.D.; Hochstrasser, M. Substrate targeting in the ubiquitin system. Cell 1999, 97, 427–430. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Sarcevic, B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the e2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Ceglia, S.; Palmieri, C.; Vecchio, E.; Fiume, G.; de Laurentiis, A.; Mimmi, S.; Falcone, C.; Iaccino, E.; Scialdone, A.; et al. Crl3ibtk regulates the tumor suppressor pdcd4 through ubiquitylation coupled to proteasomal degradation. J. Biol. Chem. 2015, 290, 13958–13971. [Google Scholar] [CrossRef] [PubMed]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Pinto, M.P.; Grou, C.P.; Alencastre, I.S.; Fransen, M.; Sa-Miranda, C.; Azevedo, J.E. Ubiquitination of mammalian pex5p, the peroxisomal import receptor. J. Biol. Chem. 2007, 282, 31267–31272. [Google Scholar] [CrossRef] [PubMed]
- Ishikura, S.; Weissman, A.M.; Bonifacino, J.S. Serine residues in the cytosolic tail of the t-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J. Biol. Chem. 2010, 285, 23916–23924. [Google Scholar] [CrossRef] [PubMed]
- Tuccillo, F.M.; Palmieri, C.; Fiume, G.; de Laurentiis, A.; Schiavone, M.; Falcone, C.; Iaccino, E.; Galandrini, R.; Capuano, C.; Santoni, A.; et al. Cancer-associated cd43 glycoforms as target of immunotherapy. Mol. Cancer Ther. 2014, 13, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an e1-e2-e3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Schindelin, H. Structural insights into e1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 2008, 134, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. Ring-type e3 ligases: Master manipulators of e2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C. A new gun in town: The u box is a ubiquitin ligase domain. Sci. Signal. 2002, 2002, pe4. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Lauwers, E.; Jacob, C.; Andre, B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J. Cell Biol. 2009, 185, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-ring ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bosu, D.R.; Kipreos, E.T. Cullin-ring ubiquitin ligases: Global regulation and activation cycles. Cell Div. 2008, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Gophna, U.; Ron, E.Z.; Graur, D. Bacterial type III secretion systems are ancient and evolved by multiple horizontal-transfer events. Gene 2003, 312, 151–163. [Google Scholar] [CrossRef]
- Fronzes, R.; Christie, P.J.; Waksman, G. The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 2009, 7, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Sasakawa, C. Bacterial e3 ligase effectors exploit host ubiquitin systems. Curr. Opin. Microbiol. 2017, 35, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kang, J.; Zhang, L.; Liang, Z.; Tang, X.; Yan, Y.; Qian, H.; Zhang, X.; Xu, W.; Mao, F. Ubiquitination regulation of inflammatory responses through nf-κb pathway. Am. J. Transl. Res. 2018, 10, 881–891. [Google Scholar] [PubMed]
- Budhidarmo, R.; Zhu, J.; Middleton, A.J.; Day, C.L. The ring domain of ring finger 11 (rnf11) protein binds ubc13 and inhibits formation of polyubiquitin chains. FEBS Lett. 2018, 592, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chai, Q.Y.; Liu, C.H. The ubiquitin system: A critical regulator of innate immunity and pathogen-host interactions. Cell. Mol. Immunol. 2016, 13, 560–576. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Kumar, H. Viral deubiquitinases: Role in evasion of anti-viral innate immunity. Crit. Rev. Microbiol. 2018, 44, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, S.; Fichtner, M.; Watters, O.; Konig, H.G.; Prehn, J.H.M. Increased a20-e3 ubiquitin ligase interactions in bid-deficient glia attenuate tlr3- and tlr4-induced inflammation. J. Neuroinflamm. 2018, 15, 130. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. Nf-κb signaling in inflammation. Signal Transduct. Targeted Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Sun, S.C. Tumor necrosis factor receptor-associated factor regulation of nuclear factor κb and mitogen-activated protein kinase pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Xie, P. Traf molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Fiume, G.; Scialdone, A.; Albano, F.; Rossi, A.; Tuccillo, F.M.; Rea, D.; Palmieri, C.; Caiazzo, E.; Cicala, C.; Bellevicine, C.; et al. Impairment of t cell development and acute inflammatory response in hiv-1 tat transgenic mice. Sci. Rep. 2015, 5, 13864. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Sun, S.C. Ubiquitin signaling in immune responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef] [PubMed]
- Vander Ark, A.; Cao, J.; Li, X. Tgf-beta receptors: In and beyond tgf-beta signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.H.; Landstrom, M. Traf6 stimulates the tumor-promoting effects of tgfbeta type I receptor through polyubiquitination and activation of presenilin 1. Sci. Signal. 2014, 7, ra2. [Google Scholar] [CrossRef] [PubMed]
- Sundar, R.; Gudey, S.K.; Heldin, C.H.; Landstrom, M. Traf6 promotes tgfbeta-induced invasion and cell-cycle regulation via lys63-linked polyubiquitination of lys178 in tgfbeta type I receptor. Cell Cycle 2015, 14, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; Garcia de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. Traf4 promotes tgf-beta receptor signaling and drives breast cancer metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Schmukle, A.C.; Walczak, H. The emerging role of linear ubiquitination in cell signaling. Sci. Signal. 2011, 4, re5. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F. Linear ubiquitination signals in adaptive immune responses. Immunol. Rev. 2015, 266, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Izrael-Tomasevic, A.; Yu, K.; Bustos, D.; Goncharov, T.; Belmont, L.D.; Masselot, A.; Bakalarski, C.E.; Kirkpatrick, D.S.; Vucic, D. Ubiquitination profiling identifies sensitivity factors for iap antagonist treatment. Biochem. J. 2015, 466, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Fiume, G.; Rossi, A.; de Laurentiis, A.; Falcone, C.; Pisano, A.; Vecchio, E.; Pontoriero, M.; Scala, I.; Scialdone, A.; Masci, F.F.; et al. Eukaryotic initiation factor 4h is under transcriptional control of p65/nf-κb. PLoS ONE 2013, 8, e66087. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the tnf-r1 signaling complex and is required for tnf-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Escoll, P.; Mondino, S.; Rolando, M.; Buchrieser, C. Targeting of host organelles by pathogenic bacteria: A sophisticated subversion strategy. Nat. Rev. Microbiol. 2016, 14, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R.F. Usp19 modulates autophagy and antiviral immune responses by deubiquitinating beclin-1. EMBO J. 2016, 35, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, L.; Hong, Z.; Lv, Z.; Mao, Z.; Tang, Y.; Kong, X.; Li, S.; Cui, Y.; Liu, H.; et al. The e3 ubiquitin ligase rnf185 facilitates the cgas-mediated innate immune response. PLoS Pathog. 2017, 13, e1006264. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Dikic, I. Regulation of salmonella-host cell interactions via the ubiquitin system. Int. J. Med. Microbiol. 2017, 308, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Noad, J.; von der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. Lubac-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and nf-κb. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, S.J.L.; Fricke, F.; Herhaus, L.; Gupta, J.; Hotte, K.; Pampaloni, F.; Grumati, P.; Kaulich, M.; Sou, Y.S.; Komatsu, M.; et al. Linear ubiquitination of cytosolic salmonella typhimurium activates nf-κb and restricts bacterial proliferation. Nat. Microbiol. 2017, 2, 17066. [Google Scholar] [CrossRef] [PubMed]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. Otulin antagonizes lubac signaling by specifically hydrolyzing met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian autophagy: How does it work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Suzuki, T.; Mimuro, H.; Mizushima, T.; Sasakawa, C. Shigella hijacks the glomulin-ciaps-inflammasome axis to promote inflammation. EMBO Rep. 2018, 19, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Higashide, W.M.; McCormick, B.A.; Chen, J.; Zhou, D. The inflammation-associated salmonella sopa is a hect-like e3 ubiquitin ligase. Mol. Microbiol. 2006, 62, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, 2177–2189. [Google Scholar] [CrossRef] [PubMed]
- Kamanova, J.; Sun, H.; Lara-Tejero, M.; Galan, J.E. The salmonella effector protein sopa modulates innate immune responses by targeting trim e3 ligase family members. PLoS Pathog. 2016, 12, e1005552. [Google Scholar] [CrossRef] [PubMed]
- Fiskin, E.; Bhogaraju, S.; Herhaus, L.; Kalayil, S.; Hahn, M.; Dikic, I. Structural basis for the recognition and degradation of host trim proteins by salmonella effector sopa. Nat. Commun. 2017, 8, 14004. [Google Scholar] [CrossRef] [PubMed]
- Battle, S.E.; Brady, M.J.; Vanaja, S.K.; Leong, J.M.; Hecht, G.A. Actin pedestal formation by enterohemorrhagic escherichia coli enhances bacterial host cell attachment and concomitant type III translocation. Infect. Immun. 2014, 82, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Piscatelli, H.; Kotkar, S.A.; McBee, M.E.; Muthupalani, S.; Schauer, D.B.; Mandrell, R.E.; Leong, J.M.; Zhou, D. The ehec type III effector nlel is an e3 ubiquitin ligase that modulates pedestal formation. PLoS ONE 2011, 6, e19331. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; You, Q.; Zhu, H.; Chang, Z.; Li, Q.; Wang, H.; Wang, C.; Hui, L.; Du, C.; Xie, X.; et al. Bacterial effector nlel promotes enterohemorrhagic e. Coli-induced attaching and effacing lesions by ubiquitylating and inactivating jnk. PLoS Pathog. 2017, 13, e1006534. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wasilko, D.J.; Liu, Y.; Sun, J.; Wu, X.; Luo, Z.Q.; Mao, Y. Structure of the legionella virulence factor, sidc reveals a unique pi(4)p-specific binding domain essential for its targeting to the bacterial phagosome. PLoS Pathog. 2015, 11, e1004965. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Skarina, T.; Yee, A.; Jobin, M.C.; Dileo, R.; Semesi, A.; Fares, C.; Lemak, A.; Coombes, B.K.; Arrowsmith, C.H.; et al. Nleg type 3 effectors from enterohaemorrhagic escherichia coli are u-box e3 ubiquitin ligases. PLoS Pathog. 2010, 6, e1000960. [Google Scholar] [CrossRef] [PubMed]
- Quaile, A.T.; Urbanus, M.L.; Stogios, P.J.; Nocek, B.; Skarina, T.; Ensminger, A.W.; Savchenko, A. Molecular characterization of lubx: Functional divergence of the u-box fold by legionella pneumophila. Structure 2015, 23, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Kubori, T.; Hyakutake, A.; Nagai, H. Legionella translocates an e3 ubiquitin ligase that has multiple u-boxes with distinct functions. Mol. Microbiol. 2008, 67, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Kubori, T.; Shinzawa, N.; Kanuka, H.; Nagai, H. Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 2010, 6, e1001216. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Doms, A.G.; Cheng, E.; Kim, B.; Evans, T.R.; Machner, M.P. Host cell-catalyzed s-palmitoylation mediates golgi targeting of the legionella ubiquitin ligase gobx. J. Biol. Chem. 2015, 290, 25766–25781. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, M.; Li, X.; Stefanovic, S.; Sharma, A.; Mateja, A.; Keenan, R.J.; Hegde, R.S. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 2010, 466, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Ensminger, A.W.; Isberg, R.R. E3 ubiquitin ligase activity and targeting of bat3 by multiple legionella pneumophila translocated substrates. Infect. Immun. 2010, 78, 3905–3919. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Machner, M.P. Exploitation of the host cell ubiquitin machinery by microbial effector proteins. J. Cell Sci. 2017, 130, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Rohde, J.R.; Breitkreutz, A.; Chenal, A.; Sansonetti, P.J.; Parsot, C. Type iii secretion effectors of the ipah family are e3 ubiquitin ligases. Cell Host Microbe 2007, 1, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Keszei, A.F.; Sicheri, F. Mechanism of catalysis, e2 recognition, and autoinhibition for the ipah family of bacterial e3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2017, 114, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Quezada, C.M.; Hicks, S.W.; Galan, J.E.; Stebbins, C.E. A family of salmonella virulence factors functions as a distinct class of autoregulated e3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2009, 106, 4864–4869. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.C.; Keszei, A.F.; Rohde, J.R.; Tyers, M.; Sicheri, F. Conserved structural mechanisms for autoinhibition in ipah ubiquitin ligases. J. Biol. Chem. 2012, 287, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, M.M.; Goldberg, M.B.; Rose, D.J.; Grotbeck, E.J.; Burland, V.; Blattner, F.R. Complete DNA sequence and analysis of the large virulence plasmid of shigella flexneri. Infect. Immun. 2001, 69, 3271–3285. [Google Scholar] [CrossRef] [PubMed]
- Toyotome, T.; Suzuki, T.; Kuwae, A.; Nonaka, T.; Fukuda, H.; Imajoh-Ohmi, S.; Toyofuku, T.; Hori, M.; Sasakawa, C. Shigella protein ipah(9.8) is secreted from bacteria within mammalian cells and transported to the nucleus. J. Biol. Chem. 2001, 276, 32071–32079. [Google Scholar] [CrossRef] [PubMed]
- Okuda, J.; Toyotome, T.; Kataoka, N.; Ohno, M.; Abe, H.; Shimura, Y.; Seyedarabi, A.; Pickersgill, R.; Sasakawa, C. Shigella effector ipah9.8 binds to a splicing factor u2af(35) to modulate host immune responses. Biochem. Biophys. Res. Commun. 2005, 333, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Kim, M.; Schmidt-Supprian, M.; Ma, A.; Ogawa, M.; Sasakawa, C. A bacterial e3 ubiquitin ligase ipah9.8 targets nemo/ikkgamma to dampen the host nf-κb-mediated inflammatory response. Nat. Cell Biol. 2010, 12, 66–73, Suppl. pp. 61–69. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jiang, W.; Yu, Q.; Liu, W.; Zhou, P.; Li, J.; Xu, J.; Xu, B.; Wang, F.; Shao, F. Ubiquitination and degradation of gbps by a shigella effector to suppress host defence. Nature 2017, 551, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Nakano, H.; Sasakawa, C. Shigella ipah0722 e3 ubiquitin ligase effector targets traf2 to inhibit pkc-nf-κb activity in invaded epithelial cells. PLoS Pathog. 2013, 9, e1003409. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, A.P.; Brown, N.F.; Stoepel, J.; Wiermer, M.; Martin, D.D.; Hsu, K.J.; Imami, K.; Ross, C.J.; Hayden, M.R.; Foster, L.J.; et al. The salmonella type III effector ssph2 specifically exploits the nlr co-chaperone activity of sgt1 to subvert immunity. PLoS Pathog. 2013, 9, e1003518. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Bayard, J.; Ramos-Morales, F. Salmonella type III secretion effector slrp is an e3 ubiquitin ligase for mammalian thioredoxin. J. Biol. Chem. 2009, 284, 27587–27595. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Bayard, J.; Cardenal-Munoz, E.; Ramos-Morales, F. The salmonella type III secretion effector, salmonella leucine-rich repeat protein (slrp), targets the human chaperone erdj3. J. Biol. Chem. 2010, 285, 16360–16368. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Vacratsis, P.O.; Bliska, J.B.; Dixon, J.E. The yersinia virulence factor yopm forms a novel protein complex with two cellular kinases. J. Biol. Chem. 2003, 278, 18514–18523. [Google Scholar] [CrossRef] [PubMed]
- LaRock, C.N.; Cookson, B.T. The yersinia virulence effector yopm binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 2012, 12, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.Q. Ubiquitination independent of e1 and e2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.; Kwon, D.H.; Kim, B.H.; Kim, J.; Park, M.R.; Park, Z.Y.; Song, H.K. Structural and biochemical study of the mono-adp-ribosyltransferase domain of sdea, a ubiquitylating/deubiquitylating enzyme from legionella pneumophila. J. Mol. Biol. 2018, 430, 2843–2856. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 2016, 167, 1636–1649 e1613. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, S.; Dikic, I. Cell biology: Ubiquitination without e1 and e2 enzymes. Nature 2016, 533, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Lucas, M.; Evans, T.R.; Abascal-Palacios, G.; Doms, A.G.; Beauchene, N.A.; Rojas, A.L.; Hierro, A.; Machner, M.P. Ravn is a member of a previously unrecognized group of legionella pneumophila e3 ubiquitin ligases. PLoS Pathog. 2018, 14, e1006897. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M., Jr.; Zaika, A.I. Helicobacter pylori pathogen regulates p14arf tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, G.; Chavez, C.V.; Taieb, F.; Banfield, M.J.; Samba-Louaka, A.; Nobe, R.; Nougayrede, J.P.; Zumbihl, R.; Givaudan, A.; Escoubas, J.M.; et al. Cycle inhibiting factors (cifs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS ONE 2009, 4, e4855. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine deamidation and dysfunction of ubiquitin/nedd8 induced by a bacterial effector family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrede, J.P.; Watrin, C.; Jubelin, G.; Oswald, E.; Taieb, F. Bacterial cyclomodulin cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cell. Microbiol. 2008, 10, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.Y.; Gan, Y.H.; Hagen, T. Characterisation of cellular effects of burkholderia pseudomallei cycle inhibiting factor (cif). Biol. Open 2018, 7, bio-028225. [Google Scholar] [CrossRef] [PubMed]
- McCormack, R.M.; Lyapichev, K.; Olsson, M.L.; Podack, E.R.; Munson, G.P. Enteric pathogens deploy cell cycle inhibiting factors to block the bactericidal activity of perforin-2. eLife 2015, 4, e06505. [Google Scholar] [CrossRef] [PubMed]
- Malet, J.K.; Impens, F.; Carvalho, F.; Hamon, M.A.; Cossart, P.; Ribet, D. Rapid remodeling of the host epithelial cell proteome by the listeriolysin o (llo) pore-forming toxin. Mol. Cell. Proteom. 2018, 17, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Tcherpakov, M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010, 143, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Scully, I.L.; Swanson, K.; Green, L.; Jansen, K.U.; Anderson, A.S. Anti-infective vaccination in the 21st century-new horizons for personal and public health. Curr. Opin. Microbiol. 2015, 27, 96–102. [Google Scholar] [CrossRef] [PubMed]
- McAdow, M.; Kim, H. Preventing Staphylococcus aureus Sepsis through the inhibition of its agglutination in blood. PLoS Pathog. 2011, 7, e1002307. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Ito, E.; Nguyen, V.H.; Haight, M. Anti-pcrv antibody strategies against virulent pseudomonas aeruginosa. Hum. Vacc. Immunother. 2014, 10, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.E.; Hartland, E.L. Post-translational mechanisms of host subversion by bacterial effectors. Trends Mol. Med. 2017, 23, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Maculins, T.; Carter, N.; Dorval, T.; Hudson, K.; Nissink, J.W.; Hay, R.T.; Alwan, H. A generic platform for cellular screening against ubiquitin ligases. Sci. Rep. 2016, 6, 18940. [Google Scholar] [CrossRef] [PubMed]
- Perrett, C.A.; Lin, D.Y.; Zhou, D. Interactions of bacterial proteins with host eukaryotic ubiquitin pathways. Front. Microbiol. 2011, 2, 143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, Y. Diversity of bacterial manipulation of the host ubiquitin pathways. Cell. Microbiol. 2015, 17, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Polajnar, M.; Dietz, M.S.; Heilemann, M.; Behrends, C. Expanding the host cell ubiquitylation machinery targeting cytosolic salmonella. EMBO Rep. 2017, 18, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Alba, M.; Garcia-Gomez, J.J.; Aguilera-Herce, J.; Ramos-Morales, F. Proteomic insight into the effects of the salmonella ubiquitin ligase slrp on host cells. Biochem. Biophys. Res. Commun. 2016, 472, 539–544. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
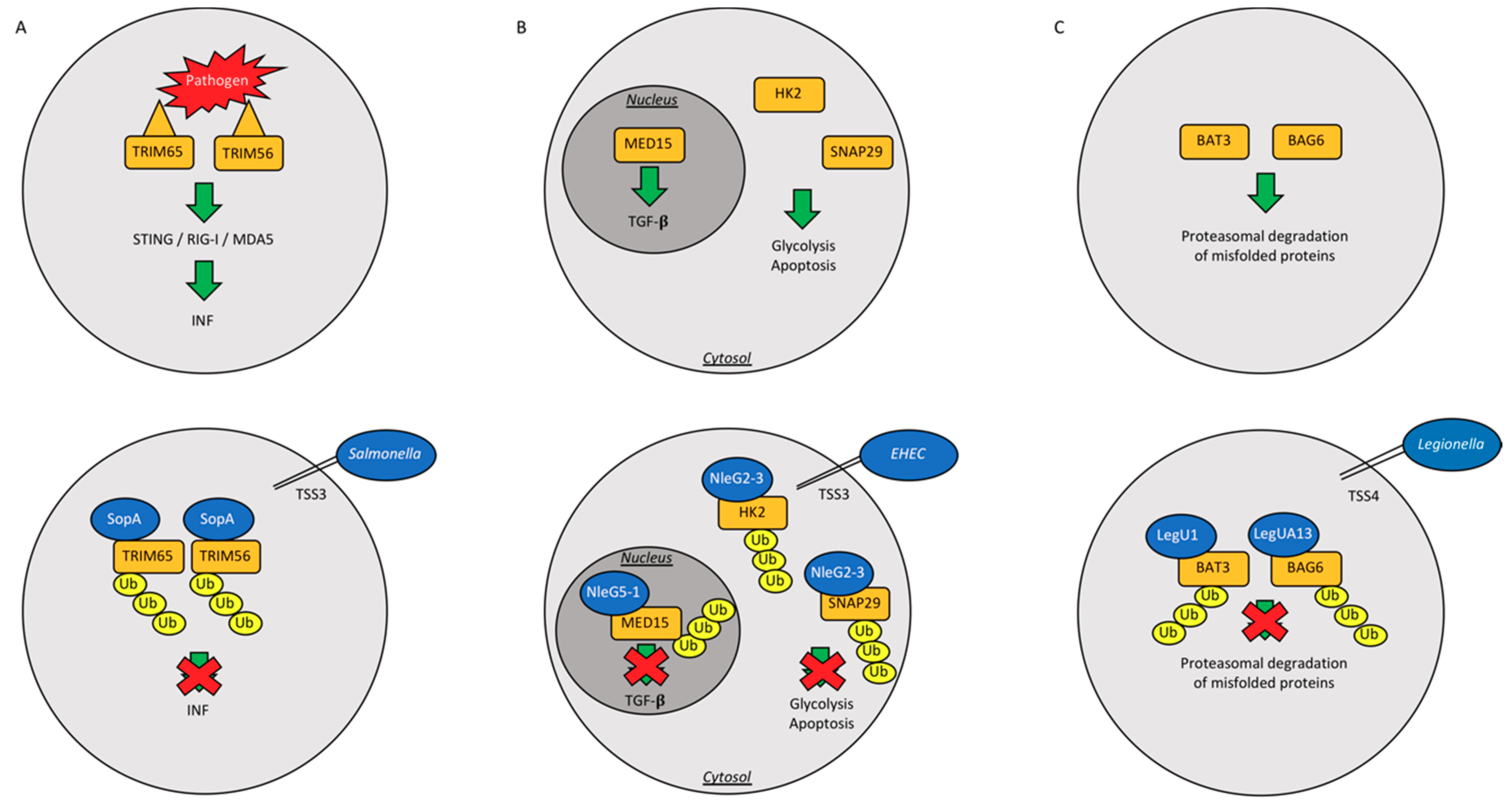{kind=link}
| Bacterial Strain | Bacterial Protein | Effector Type | Action | Reference |
|---|---|---|---|---|
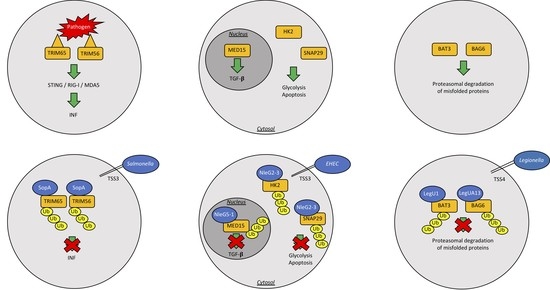
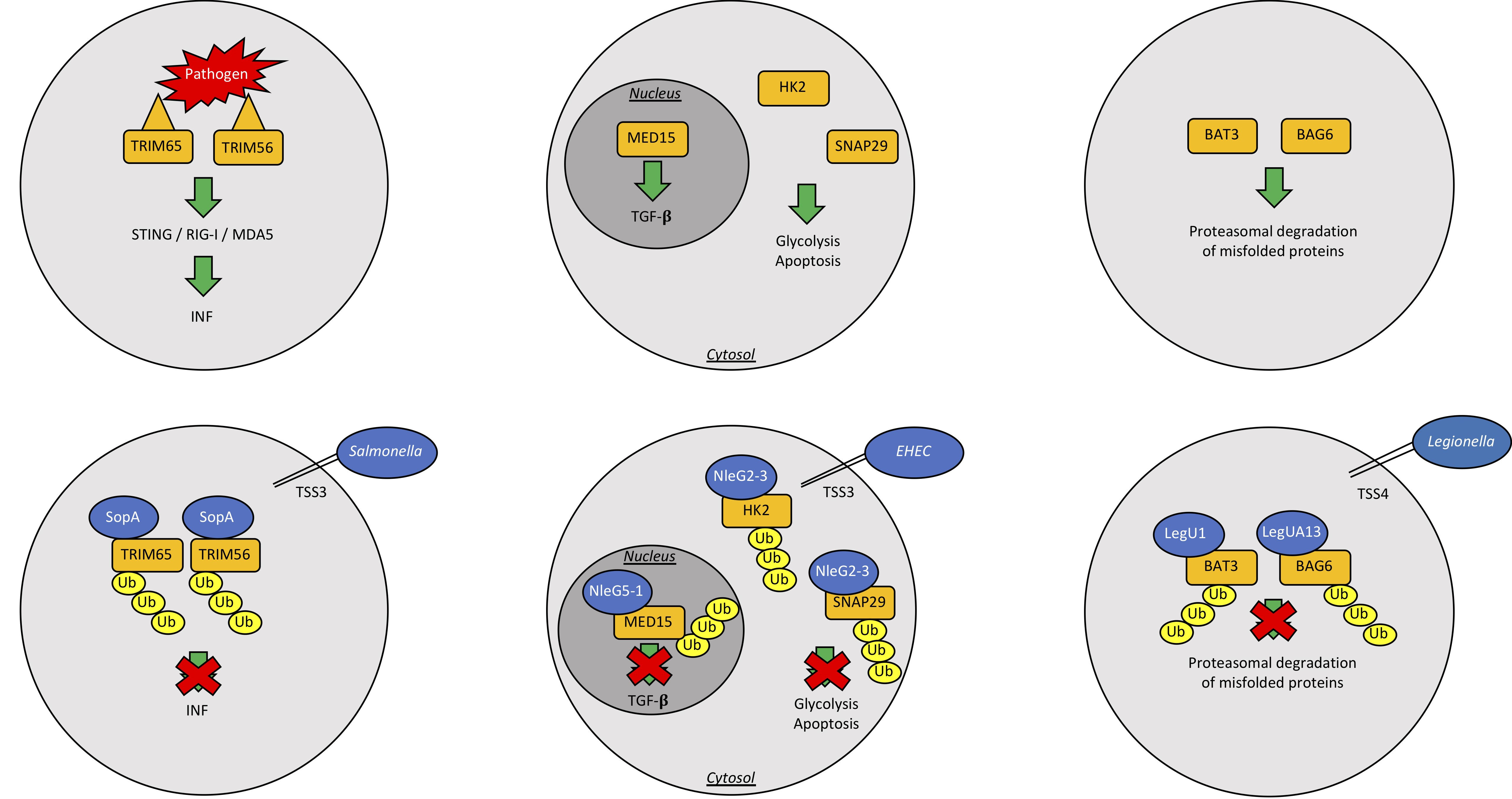
| Salmonella typhimurium | SopA | HECH TSS3 | Ubiquitinates host TRIM65 and TRIM56, blocking the production of interferon-α, helps machrophages and epithelial cell invasion | [52,53,54,55] |
| SidC | RING/U-Box E3 | Binds membrane PI4P and helps SCV generation | [59] | |
| Enterohemorrhagic Escherichia coli | NleL | HECH TSS3 | Ubiquitinates Tir into host intestinal cells; Ubiquitinates JNK | [56,57,58] |
| Escherichia coli | NleG | U-box Ubiquitin Ligase | Interacts with UBE2E2, UBE2E3 and UBE2D2 | [60] |
| Legionella pneumophila | LubX | U-box Ubiquitin Ligase | Ubiquitinates Clk1; ubiquitinates SidH to preserve host survival | [61,62,63] |
| GobX | U-box Ubiquitin Ligase | Exploites host ubiquitination and palmitoylation | [64] | |
| LegU1/LegUA13 | F-box proteins and TSS4 | Degrades host BAT3 or BAG6 | [65,66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisano, A.; Albano, F.; Vecchio, E.; Renna, M.; Scala, G.; Quinto, I.; Fiume, G. Revisiting Bacterial Ubiquitin Ligase Effectors: Weapons for Host Exploitation. Int. J. Mol. Sci. 2018, 19, 3576. https://doi.org/10.3390/ijms19113576
Pisano A, Albano F, Vecchio E, Renna M, Scala G, Quinto I, Fiume G. Revisiting Bacterial Ubiquitin Ligase Effectors: Weapons for Host Exploitation. International Journal of Molecular Sciences. 2018; 19(11):3576. https://doi.org/10.3390/ijms19113576
Chicago/Turabian StylePisano, Antonio, Francesco Albano, Eleonora Vecchio, Maurizio Renna, Giuseppe Scala, Ileana Quinto, and Giuseppe Fiume. 2018. "Revisiting Bacterial Ubiquitin Ligase Effectors: Weapons for Host Exploitation" International Journal of Molecular Sciences 19, no. 11: 3576. https://doi.org/10.3390/ijms19113576
APA StylePisano, A., Albano, F., Vecchio, E., Renna, M., Scala, G., Quinto, I., & Fiume, G. (2018). Revisiting Bacterial Ubiquitin Ligase Effectors: Weapons for Host Exploitation. International Journal of Molecular Sciences, 19(11), 3576. https://doi.org/10.3390/ijms19113576