Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis

 ,
,  ,
,  and
and 
Abstract

1. Introduction
2. In Vitro Studies
3. In Vivo Studies on Hepatic Steatosis Prevention
4. In Vivo Studies on Hepatic Steatosis Treatment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA-carboxylase |
| ACOX1 | Acyl-Coenzyme A oxidase 1 |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AKT | Protein kinase B |
| ALP | Alkaline phosphatase, |
| ALT | Alanine aminotransferase |
| AMP | Adenosine monophosphate |
| AMPK | 5′AMP-activated protein kinase |
| AST | Aspartate aminotransferase |
| ATGL | Adipose triglyceride lipase |
| ATP | Adenosine triphosphate |
| BW | Body weight |
| CaMKKβ | Ca2+/calmodulin-dependent protein kinase kinase β |
| cAMP | Cyclic adenosine monophosphate |
| CHREBP | Carbohydrate response element binding protein |
| CPT | Carnitine palmitoyl transferase |
| CS | Citrate synthase |
| FAS | Fatty acid synthase |
| FATP5 | Fatty acid transport protein 5 |
| FFA | Free fatty acid |
| FOXO 1 | Forkhead box protein O1 |
| GGT | Gamma glutamil transpeptidase |
| GPAT1 | Glycerol-3-phosphate acyltransferase 1 |
| G6PDH | Glucose-6P-dehydrogenase |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
| HBV | Hepatitis B virus |
| HFD | High-fat diet |
| HFHS | High-fat high-sucrose diet |
| HSL | Hormone sensitive lipase |
| IkBα | Inhibitor of nuclear factor kappa subunit α |
| IL | Interleukin |
| LDH | Lactate dehydrogenase |
| LFD | Low fat diet |
| LKB1 | Liver kinase B1 |
| LXR | Liver X receptor |
| MCAD | Mitochondrial medium-chain acyl-CoA dehydrogenase |
| MDA | Malonaldehyde |
| ME | Malic enzyme |
| MTP | Microsomal triglyceride transfer protein |
| NAD+ | Oxidized nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NAFLD | Non-alcoholic fatty liver disease |
| NF-kB p65 | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| O+A | Oleic acid plus alcohol |
| PDE | Phosphodiesterase |
| pIkBα | Phospho-inhibitory subunit of NF-KBα |
| PPAR | Peroxisome proliferator-activated receptor |
| PRKA | Protein kinase A |
| ROS | Reactive oxygen species |
| RSV | Resveratrol |
| si RNA | Small interfering RNA |
| SIRT1 | Silent information regulator 1; Surtuin-1 |
| SCD | Stearoyl-Coenzyme A desaturase 1 |
| SQSTM1 | Sequestosome 1 |
| SOD | Superoxide dismutase |
| SREBF | Sterol regulatory element-binding transcription factor |
| SREBP | Sterol regulatory element-binding protein |
| STD | Standard diet |
| TG | Triglyceride |
| TNF-α | Tumor necrosis factor- α |
| UCP | Uncoupling protein |
References
- Milton-Laskibar, I.; Aguirre, L.; Fernández-Quintela, A.; Rolo, A.P.; Soeiro Teodoro, J.; Palmeira, C.M.; Portillo, M.P. Lack of additive effects of resveratrol and energy restriction in the treatment of hepatic steatosis in rats. Nutrients 2017, 9, 737. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.; Portillo, M.P.; Hijona, E.; Bujanda, L. Effects of resveratrol and other polyphenols in hepatic steatosis. World J. Gastroenterol. 2014, 20, 7366–7380. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. Ampk as a therapeutic target for treating metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. Ampk signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. Ampk: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. The amp-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Minireview: The amp-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule ampk activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-specific activation of ampk prevents steatosis on a high-fructose diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Ampk: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintela, A.; Milton-Laskibar, I.; González, M.; Portillo, M.P. Antiobesity effects of resveratrol: Which tissues are involved? Ann. N. Y. Acad. Sci. 2017, 1403, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Heebøll, S.; Thomsen, K.L.; Pedersen, S.B.; Vilstrup, H.; George, J.; Grønbæk, H. Effects of resveratrol in experimental and clinical non-alcoholic fatty liver disease. World J. Hepatol. 2014, 6, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Heebøll, S.; Thomsen, K.L.; Clouston, A.; Sundelin, E.I.; Radko, Y.; Christensen, L.P.; Ramezani-Moghadam, M.; Kreutzfeldt, M.; Pedersen, S.B.; Jessen, N.; et al. Effect of resveratrol on experimental non-alcoholic steatohepatitis. Pharmacol. Res. 2015, 95–96, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Heebøll, S.; El-Houri, R.B.; Hellberg, Y.E.; Haldrup, D.; Pedersen, S.B.; Jessen, N.; Christensen, L.P.; Grønbaek, H. Effect of resveratrol on experimental non-alcoholic fatty liver disease depends on severity of pathology and timing of treatment. J. Gastroenterol. Hepatol. 2016, 31, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of sirt1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. Pgc-1alpha, sirt1 and ampk, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. Ampk and sirt1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. Sirt1 regulates hepatocyte lipid metabolism through activating amp-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. Sirt1 modulation of the acetylation status, cytosolic localization, and activity of lkb1. Possible role in amp-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. Amp-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting camp phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Xu, S.; Maitland-Toolan, K.A.; Zuccollo, A.; Hou, X.; Jiang, B.; Wierzbicki, M.; Verbeuren, T.J.; Cohen, R.A. Polyphenols stimulate amp-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 2006, 55, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Chen, L.L.; Xiao, F.X.; Sun, H.; Ding, H.C.; Xiao, H. Resveratrol improves non-alcoholic fatty liver disease by activating amp-activated protein kinase. Acta Pharmacol. Sin. 2008, 29, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Liu, D. Resveratrol suppresses t0901317-induced hepatic fat accumulation in mice. AAPS J. 2013, 15, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Suh, H.R.; Yoon, Y.; Lee, K.J.; Kim, D.G.; Kim, S.; Lee, B.H. Protective effect of resveratrol derivatives on high-fat diet induced fatty liver by activating amp-activated protein kinase. Arch. Pharm. Res. 2014, 37, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Chen, Y.; Rui, B.B.; Hu, C.M. Resveratrol ameliorates lipid accumulation in HepG2 cells, associated with down-regulation of lipin1 expression. Can. J. Physiol. Pharmacol. 2016, 94, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.L.; Zhou, Y.; Yi, L.; Gao, Y.X.; Ran, L.; Chen, S.H.; Zhang, T.; Zhou, X.; Zou, D.; et al. Resveratrol improves hepatic steatosis by inducing autophagy through the camp signaling pathway. Mol. Nutr. Food Res. 2015, 59, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ma, J.; Wang, W.; Zhang, L.; Xu, J.; Wang, K.; Li, D. Resveratrol supplement inhibited the NF-κB inflammation pathway through activating AMPKα-sirt1 pathway in mice with fatty liver. Mol. Cell. Biochem. 2016, 422, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Zuccollo, A.; Hou, X.; Nagata, D.; Walsh, K.; Herscovitz, H.; Brecher, P.; Ruderman, N.B.; Cohen, R.A. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 2004, 279, 47898–47905. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Repa, J.J. The liver x receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Ajmo, J.M.; Liang, X.; Rogers, C.Q.; Pennock, B.; You, M. Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G833–G842. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, G.; Rodríguez, V.M.; Macarulla, M.T.; Miranda, J.; Churruca, I.; Portillo, M.P. Hepatic lipid metabolic pathways modified by resveratrol in rats fed an obesogenic diet. Nutrition 2013, 29, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.C.; Lin, Y.H.; Chen, H.J.; Chou, S.C.; Cheng, A.C.; Kalyanam, N.; Ho, C.T.; Pan, M.H. Piceatannol exerts anti-obesity effects in c57bl/6 mice through modulating adipogenic proteins and gut microbiota. Molecules 2016, 21, 1419. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kita, T.; Yamasaki, S.; Kawahara, T.; Ueno, Y.; Yamada, M.; Mukai, Y.; Sato, S.; Kurasaki, M.; Saito, T. Maternal resveratrol intake during lactation attenuates hepatic triglyceride and fatty acid synthesis in adult male rat offspring. Biochem. Biophys. Rep. 2017, 9, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.; Morón, R.; Zarzuelo, A.; Galisteo, M. Long-term resveratrol administration reduces metabolic disturbances and lowers blood pressure in obese Zucker rats. Biochem. Pharmacol. 2009, 77, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Do, G.M.; Jung, U.J.; Park, H.J.; Kwon, E.Y.; Jeon, S.M.; McGregor, R.A.; Choi, M.S. Resveratrol ameliorates diabetes-related metabolic changes via activation of amp-activated protein kinase and its downstream targets in db/db mice. Mol. Nutr. Food Res. 2012, 56, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Hong, H.J.; Guan, J.; Kim, D.G.; Yang, E.J.; Koh, G.; Park, D.; Han, C.H.; Lee, Y.J.; Lee, D.H. Resveratrol improves insulin signaling in a tissue-specific manner under insulin-resistant conditions only: In vitro and in vivo experiments in rodents. Metabolism 2012, 61, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Chen, Y.F.; Hsu, W.H.; Yang, C.W.; Kao, C.H.; Tsai, T.F. Resveratrol helps recovery from fatty liver and protects against hepatocellular carcinoma induced by hepatitis b virus × protein in a mouse model. Cancer Prev. Res. 2012, 5, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, S.; Li, Z.; Zhao, X.; Li, W.; Sun, Y.; Zhang, Z.; Ling, W.; Feng, X. Effects and mechanisms of resveratrol on the amelioration of oxidative stress and hepatic steatosis in KKAy mice. Nutr. Metab. (Lond.) 2014, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lim, Y.; Yang, S.J. Involvement of resveratrol in crosstalk between adipokine adiponectin and hepatokine fetuin-A in vivo and in vitro. J. Nutr. Biochem. 2015, 26, 1254–1260. [Google Scholar] [CrossRef] [PubMed]

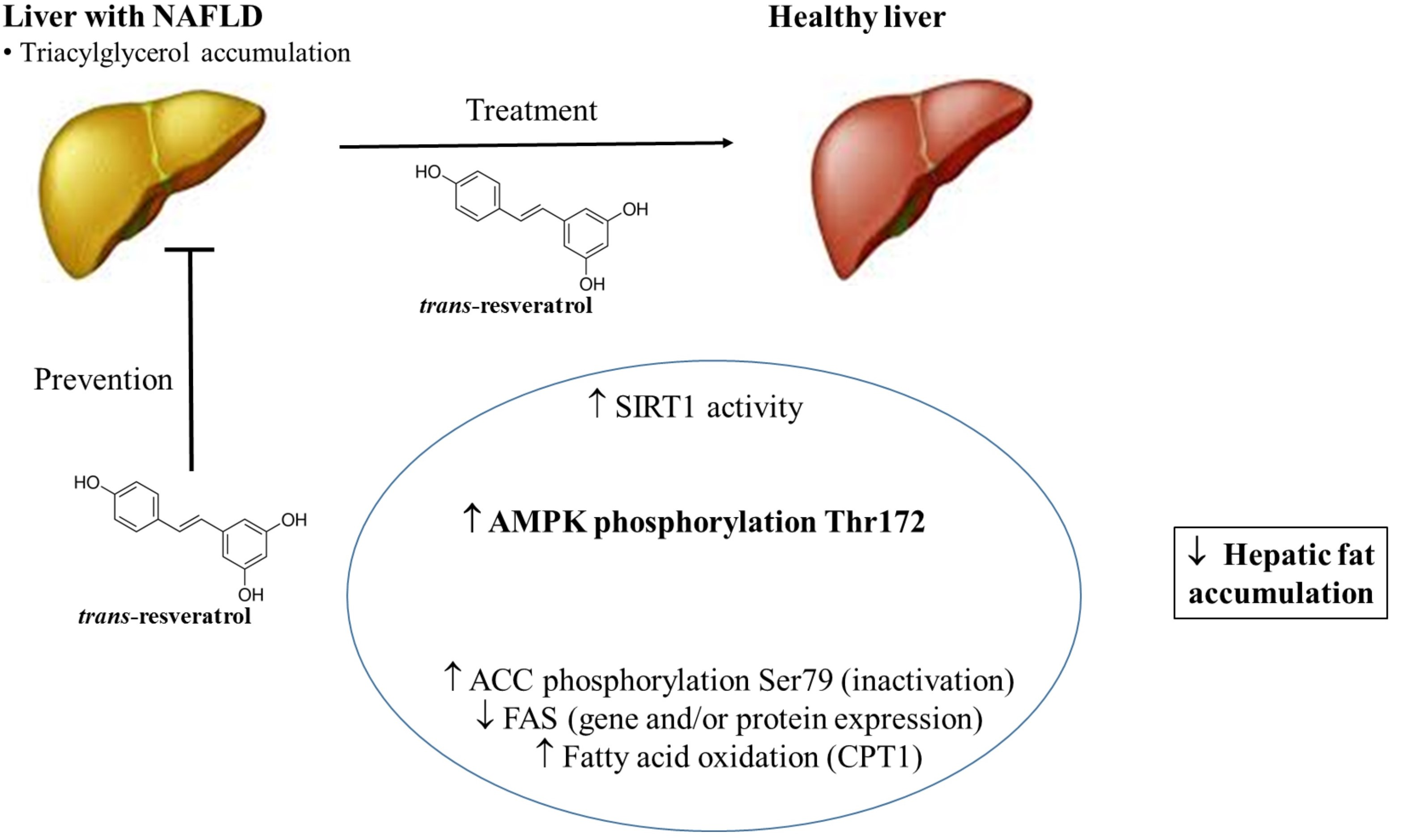
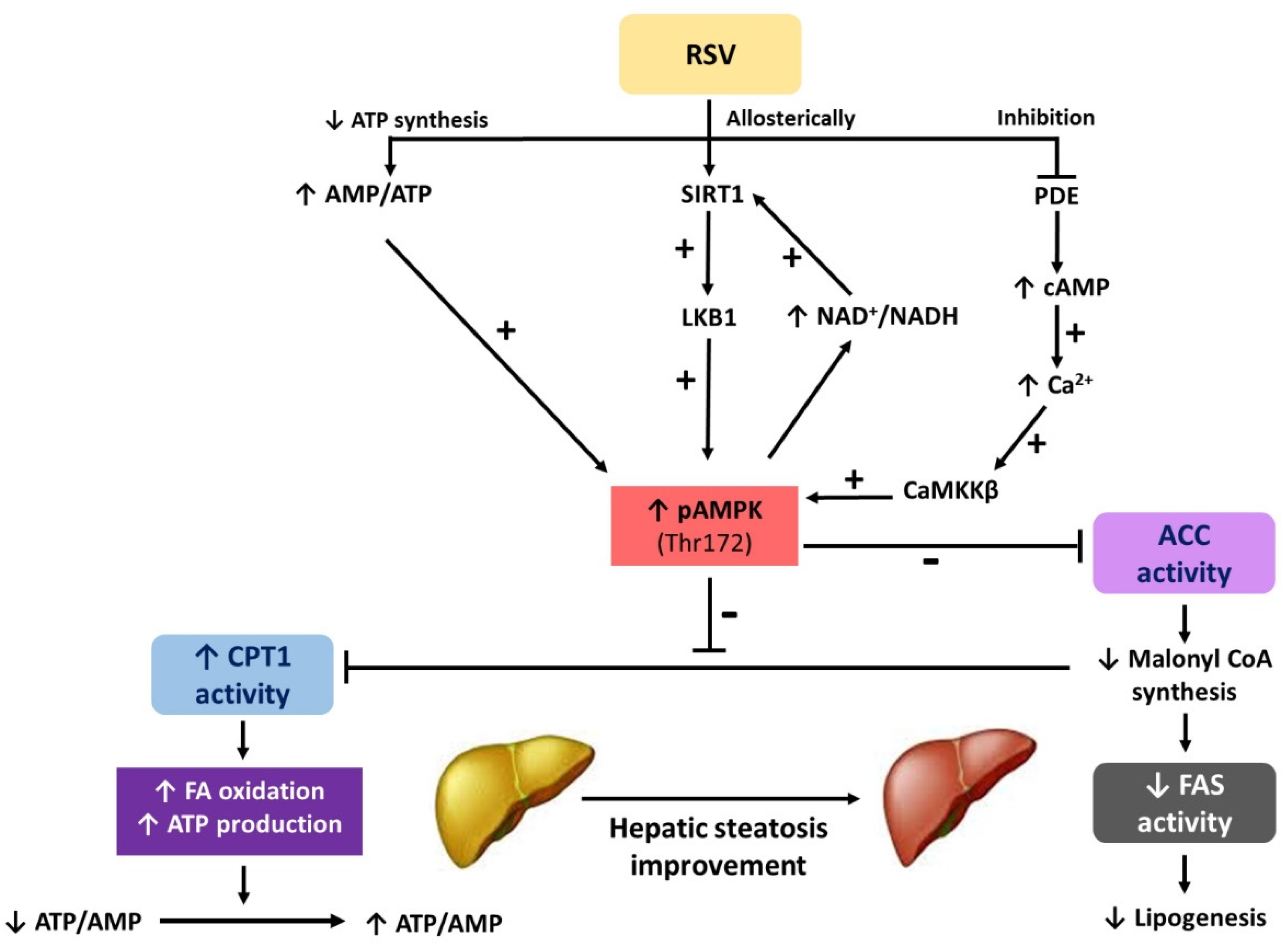
 : regulation direction;
: regulation direction;  : regulation inhibition).
: regulation direction; : regulation inhibition).
: regulation inhibition).
: regulation direction; : regulation inhibition).

{kind=link}
{kind=link}
{kind=link}
| Reference | Cell Line | Experimental Design | Effect of Resveratrol | Mechanism of Action |
|---|---|---|---|---|
| [23] | HepG2 | 24 h culture with RSV (10 µmol/L) 1 h culture with RSV (50 µmol/L) (AMPKα1 activity determination) | Prevention of high-glucose-induced lipid accumulation (in HepG2 cells) | ↑ AMPK (Thr-172) and ACC (Ser-79) phosphorylation (10 µmol/L RSV) ↑ AMPKα1 activity |
| [18] | HepG2 HEK293 | HepG2 24 h culture with RSV (1–100 μM) HEK293 cells pretreated with splitomicin (100 μM) for 24 h and incubated with RSV (50 μM) for an additional 1 h | ↓ High glucose-induced TG accumulation: RSV (10–50 μM) | ↑ SIRT1 activity (dose dependent (10–100 μM RSV) ↓ FAS protein expression ↑ AMPKα phosphorylation (Thr172) ↑ ACC phosphorylation (Ser79) |
| [24] | HepG2 | 6 or 24 h culture with 10, 25, and 50 μM of RSV | ↓ TG accumulation | ↑ AMPKα phosphorylation (Thr172) ↓ srebf1 and fasn gene expressions |
| [25] | Hepa 1–6 cell line (murine hepatocytes) | 24 h culture with T0901317 (1 μM, LXR activator: ↑ liver fat accumulation) + RSV (40 μM), with or without compound C (10 μM, AMPK inhibitor) | ↓ T0901317-induced fat accumulation | ↓ T0901317-induced fat accumulation (via AMPK activation) |
| [26] | H4IIEC3 rat hepatoma cells | 24 h culture with FFA [oleic acid and palmitic acid 2:1, 0.5 mM] or T0901317 (10 μM, LXR activator) + RSV or SY-102 (RSV derivative, 3.3–50 μM) | ↓ FFA-induced lipid accumulation by RSV (50 mΜ) and SY-102 (30 μM) ↓ T0901317-induced SREBP-1 maturation by RSV (30 μM) and SY-102 (10 μM) | ↓ srebf1 and fasn gene expression by SY-102 (50 μM) via AMPK/LXR pathway |
| [27] | HepG2 | 48 h treatment Control, oleic acid + alcohol (O + A), O + A-RSV (5, 15, 45, 13 5 µM), O + A-AICAR-RSV 45 µM, O + A-Compound C-RSV 45 µM | ↓ Lipid accumulation (15, 45, 135 μM) ↓ Hepatocyte TG content (45 and 135 μM) Attenuated hepatic steatosis | ↑ AMPKα phosphorylation * ↑ ACC phosphorylation * ↓ SREBP1c and lipin protein expression |
| [28] | HepG2 | 24 h culture with 0.2 mM palmitate Additional 24 h treated with RSV (20–80 μM) Also exposed to 3-MA autophagy inhibitor for 1 h or siRNAs before the addition of RSV | ↓ Lipid content | ↑ LC3-II protein expression and SQSTM1 protein degradation (3-MA pre-treatment inhibited this effect) ↑ SIRT1 protein expression/activity and cyclic AMP levels ↑ AMPK (Thr-172) and PRKA (Ser-96) phosphorylation |
| [29] | Primary hepatocytes from C57BL/6 mice | Treatment with NEFA, NEFA + RSV (50 and 100 μM), NEFA + Nicotinamide, NEFA + Compound C, NEFA + RSV + Nicotinamide, NEFA + RSV + Compound C Treatment length not specified | ↓ NEFA increased expression of several inflammatory markers | ↑ AMPK phosphorylation * ↑ sirt1 gene and SIRT1 protein expressions ↓ phosphorylation IκBα and NF-κB p65 ↓ il-1β, il-6, and tnf-α gene expression |
| Reference | Animal Model | Experimental Design | Effect of Resveratrol | Mechanism of Action |
|---|---|---|---|---|
| [33] | One-year-old male C57BL/6NIA mice | HFD (60% of calories as fat) RSV dose: 22.4 mg/kg bw/day Length: 6 months | Fatty liver development prevention (organ size) Prevention of cellular integrity loss and large lipid droplet accumulation | ↑AMPK phosphorylation (Thr172) ↑ACC phosphorylation (Ser79) |
| [35] | 6–8 week-old male C57BL/6J mice | LFD (10% of calories as fat) RSV dose: 200 and 400 mg/kg bw/day 3 groups: LF diet+ethanol, LF diet + ethanol + RSV200, LF diet + ethanol + RSV400 Length: 2 weeks | Prevention of liver weight, liver lipid droplets, hepatic TG content and serum ALT level increase | ↑sirt1 gene and SIRT1 protein expressions ↑ AMPKα and β phosphorylation * ↑ total AMPK levels ↑ ACC phosphorylation * ↓ SREBP1c protein expression ↓ fasn, gpat1, scd1, accα, me ↑acox1, mcad and cpt1a gene expression ↓ pparγ gene expression |
| [36] | Male Sprague Dawley rats | Obesogenic diet (45% of calories as fat) RSV dose: 30 mg/kg bw/day Length: 6 weeks | ↓ Hepatic fat content | ↑ AMPK phosphorylation (Thr172) ↑ ACC phosphorylation (Ser79) |
| [25] | Male C57BL/6 mice induced by LXR receptor | Groups: Control, T0901317 (LXR activator) and T0901317+ RSV RSV dose: 200 mg/kg bw/day Length: 5 days | Prevention of the increase in liver size, fat accumulation and TG content (induced by LXR activator) | ↑ AMPK phosphorylation (Thr172) ↑ACC phosphorylation (Ser79) ↓ srebf1, chrebp and acc expression (RSV alone) |
| [29] | 4 week-old C57BL/6 mice | HFD (60% of calories as fat) RSV dose: 30 mg/kg bw/day Length: 60 day | ↓ Liver weight ↓ GGT, AST, ALT, ALP, LDH plasma levels ↓ IL-1β, IL-6, and TNF-α plasma levels | ↑ AMPK phosphorylation (Thr172) ↑ SIRT1 protein expression ↓ IkBα and NF-kB p65 phosphorylation * ↓ il-1β, il-6, and tnf-α gene expression |
| [37] | 5 week-old male C57BL/6 mice | HFD (45% of energy as fat) RSV dose: 0.1% resveratrol (w/w) Length: 18 weeks | No changes in liver weight and serum AST and ALT levels | ↑ AMPK phosphorylation (Thr172) ↑ACC phosphorylation (Ser79) ↓ FAS protein expression ↓ Hepatic adipogenic protein expression |
| [38] | Pups from female Wistar rats | Control diet RSV dose: 20 mg/kg bw/day Length: 3 weeks (lactation period) | ↓ Hepatic lipid accumulation | ↑ AMPK phosphorylation (Ser403) ↑ SIRT1 protein expression ↓ Active/precursor SREBP-1c protein ratio ↓ ACC protein expression ↓ FAS protein expression ↓Hepatic adipogenic protein expression |
| Reference | Animal Model | Experimental Design | Effect of Resveratrol | Mechanism of Action |
|---|---|---|---|---|
| [24] | Male Wistar rats (180–200 g) | Acute treatment: Fed stated rats RSV dose: 100 mg/kg bw/day Length: 4 h | ↓ Hepatic fat content | ↑ AMPK phosphorylation (Thr172) ↓ srebf1 and fasn gene expressions |
| Chronic treatment: High-fat diet (59% of calories as fat) RSV dose: 100 mg/kg bw/day Length: 10 weeks | ||||
| [39] | Obese male Zucker rats and lean heterozygous littermates | STD RSV dose: 10 mg/kg bw/day Length: 8 weeks | No change in liver weight ↓ Liver TG and cholesterol content | ↑ AMPK phosphorylation (Thr172) ↑ ACC phosphorylation * |
| [40] | 4 week-old male C57BL/KsJ-db/db mice | RSV dose: 0.005% and 0.02% (w/w) Length: 6 weeks | ↓ Hepatic fat content (only in 0.02% RSV group) | ↓ ACC phosphorylation * ↑ srebf1 gene expression (0.02% RSV) ↑ PPARα protein expression (0.02% RSV) ↑ UCP2 protein expression ↑ AMPK phosphorylation * |
| [41] | 5 week-old male C57BL/6N mice | HFD RSV dose: 30 mg/kg bw/day Length: 2 weeks | ↓ Hepatic fat content | ↑ AKT phosphorylation (Ser473 and Thr308) ↓ AMPKα phosphorylation (Thr172) |
| [42] | 4 week-old male C57BL/6 mice expressing HBV X protein | RSV dose: 30 mg/kg bw/day Length: 2, 3, 7, and 14 days | Histopatology alteration reversion ↓ Serum ALT levels | ↓ srebf1 and lxrα gene expressions (from day 2 in advance) ↑ AMPK phosphorylation (Thr172) (from day 3 in advance) ↓ pparγ and acc gene expressions. ↑ SIRT1 protein expression and activity (from day 7 in advance) ↓ fasn gene expression |
| [26] | Male ICR mice (20–25 g) | HFD. RSV dose: 15 or 45 mg/kg bw/day. (same doses of SY-102, a RSV derivative). Length: 2 days | ↓ Hepatic TG levels (by SY-102 and RSV) | ↑ AMPK phosphorylation (Thr172) (by SY-102) ↓ srebf1 and fasn mRNA levels (by SY-102 and RSV) |
| [43] | 8 week-old male KKAy mice (genetic model of obesity) 8 week-old male C57BL/6J mice (control) | Chow diet (AIN93G) RSV dose: 2 or 4 g/kg diet Length: 12 weeks | ↓ Hepatic fat content (Oil Red) and TG levels Hepatic steatosis attenuation (histological study) ↓ MDA levels | ↑ AMPK phosphorylation (Thr172) ↑ SIRT1 protein expression ↑ FOXO1 phosphorylation (Thr24) ↓ ROS levels ↑ GSH levels, GPx and SOD activities ↑ hsl gene expression and HSL phosphorylation (Ser660) ↑ atgl gene expression and ATGL protein expression |
| [44] | 6 week-old male C57BL/6J mice | HFD RSV dose: 8 mg/kg bw/day Length: 4 weeks | ↓ Liver weight ↓ Plasma levels of Fetuin-A and ALT ↓ Hepatic index | ↑ ampk gene expression ↓ fetuin-A gene expression ↓ Fetuin-A protein expression ↓ nfκβ gene expression |
| [28] | 8 week-old 129/SvJ mice (male) | HFD (60% of energy as fat) RSV dose: 0.4% (w/w) Length: 8 weeks | ↓ Hepatic fat content | ↑ cyclic AMP levels ↑ PRKA phosphorylation (Ser96) ↑ AMPK phosphorylation (Thr172) ↑ SIRT1 protein expression |
| [1] | 6 week-old male Wistar rats | HFHS RSV dose: 30 mg/kg bw/day Length: 6 weeks | ↓ Hepatic TG content ↑ Plasma TG release (from liver) ↓ Liver fatty acid uptake | ↑ AMPK phosphorylation (Thr172) ↑ CPT1a activity ↑ CS activity ↑ MTP activity ↓ FATP5 protein expression |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trepiana, J.; Milton-Laskibar, I.; Gómez-Zorita, S.; Eseberri, I.; González, M.; Fernández-Quintela, A.; Portillo, M.P. Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis. Int. J. Mol. Sci. 2018, 19, 3473. https://doi.org/10.3390/ijms19113473
Trepiana J, Milton-Laskibar I, Gómez-Zorita S, Eseberri I, González M, Fernández-Quintela A, Portillo MP. Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis. International Journal of Molecular Sciences. 2018; 19(11):3473. https://doi.org/10.3390/ijms19113473
Chicago/Turabian StyleTrepiana, Jenifer, Iñaki Milton-Laskibar, Saioa Gómez-Zorita, Itziar Eseberri, Marcela González, Alfredo Fernández-Quintela, and María P. Portillo. 2018. "Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis" International Journal of Molecular Sciences 19, no. 11: 3473. https://doi.org/10.3390/ijms19113473
APA StyleTrepiana, J., Milton-Laskibar, I., Gómez-Zorita, S., Eseberri, I., González, M., Fernández-Quintela, A., & Portillo, M. P. (2018). Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis. International Journal of Molecular Sciences, 19(11), 3473. https://doi.org/10.3390/ijms19113473