Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
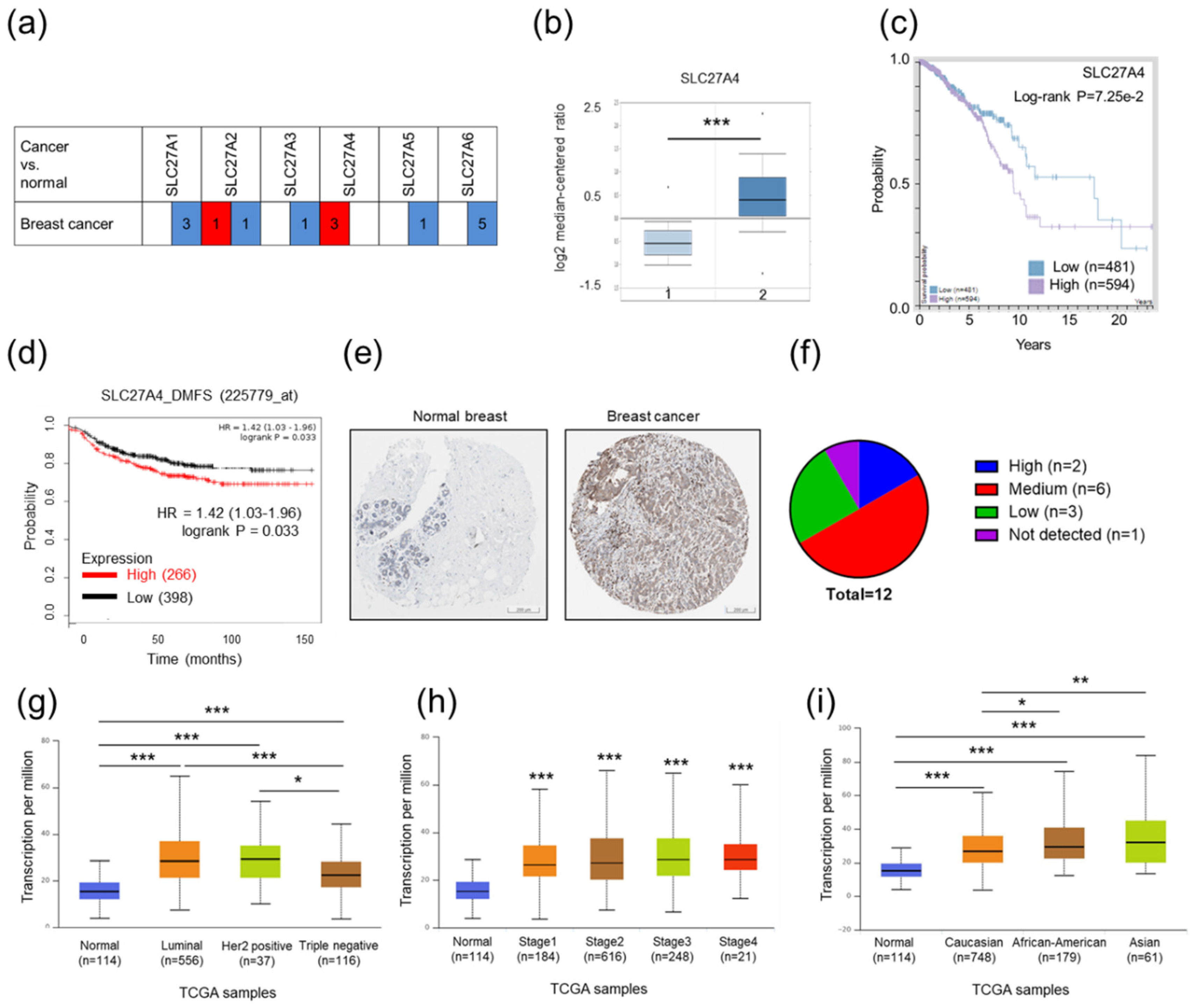
2.1. Relatively High SLC27A4 Expression Was Detected in Breast Cancer Tissue
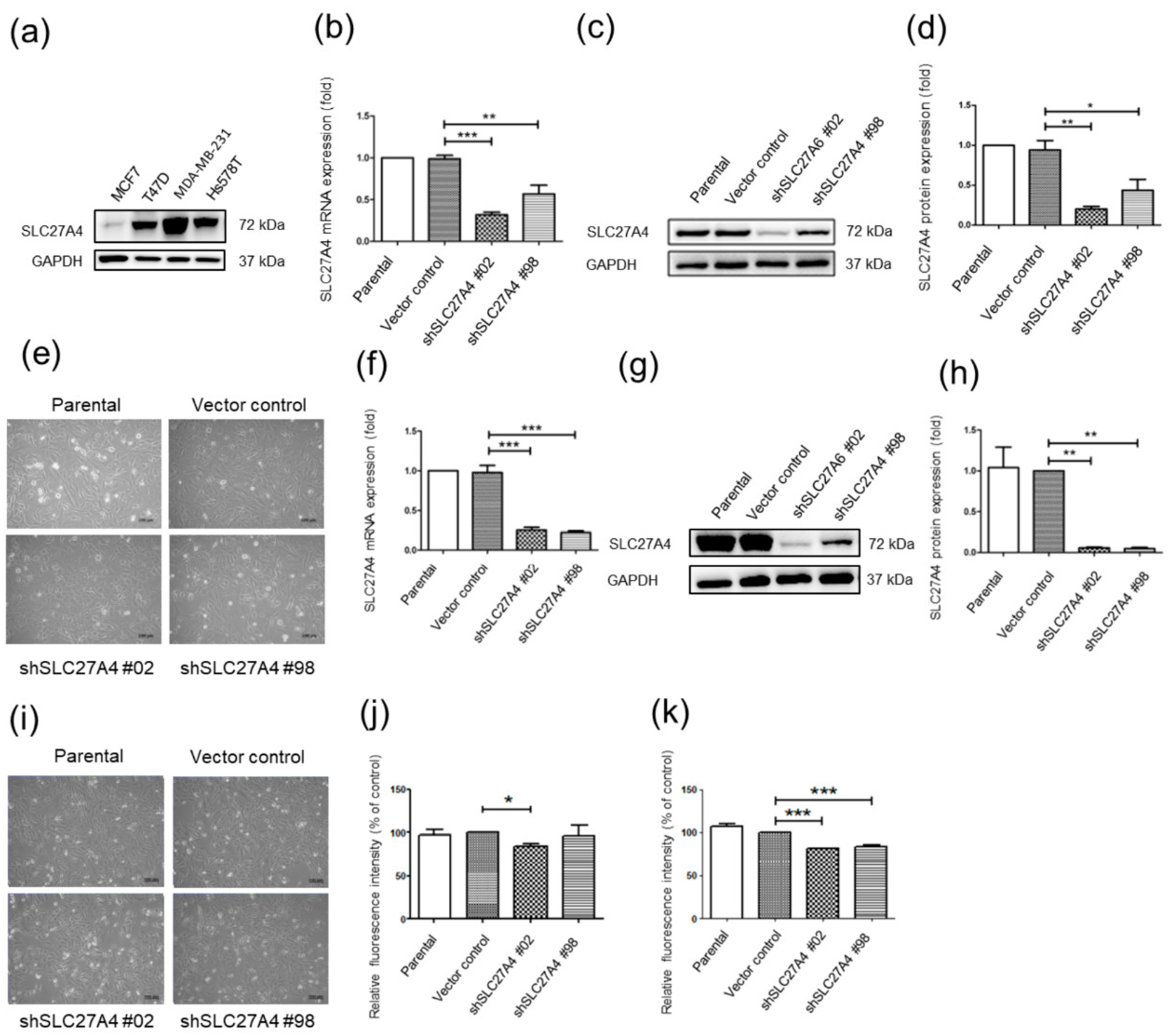
2.2. Silencing SLC27A4 in Breast Cancer Cell LINES Results in Decreasing Fatty Acids Uptake Capacity
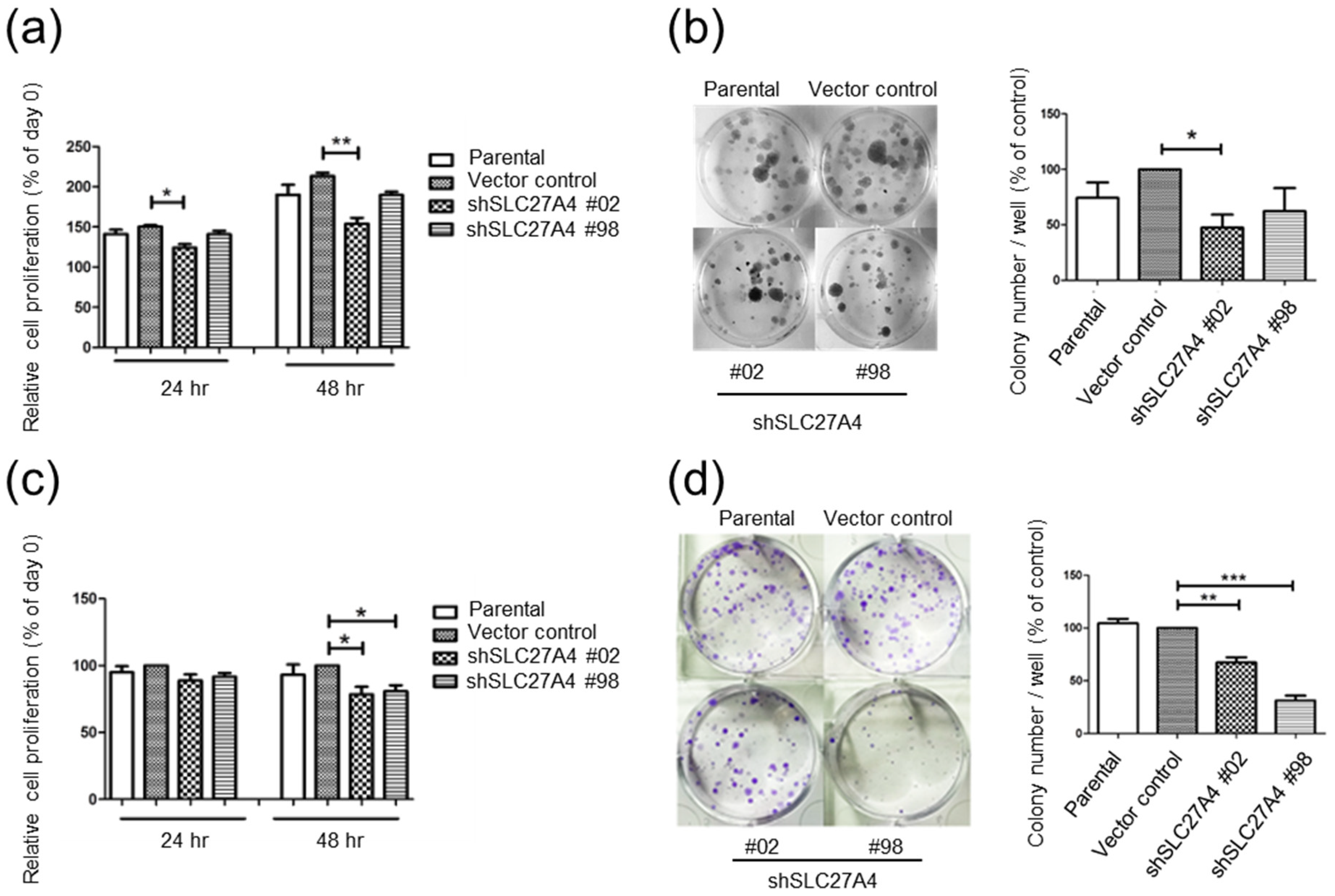
2.3. Silencing SLC27A4 in Breast Cancer Cell Lines Inhibited Cell Growth
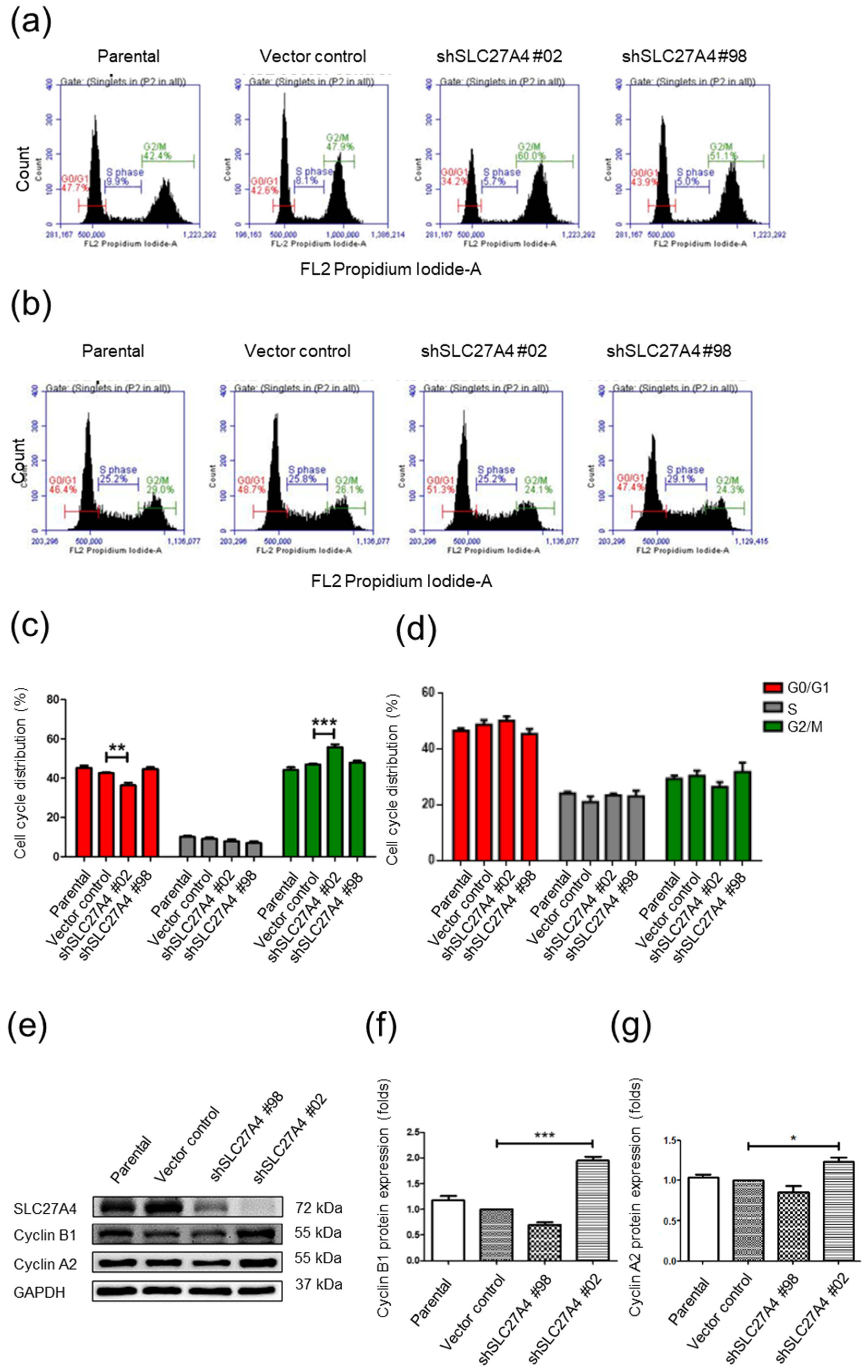
2.4. Silencing SLC27A4 in Breast Cancer Cell Lines Affected G0/G1 and G2/M Cell Cycle
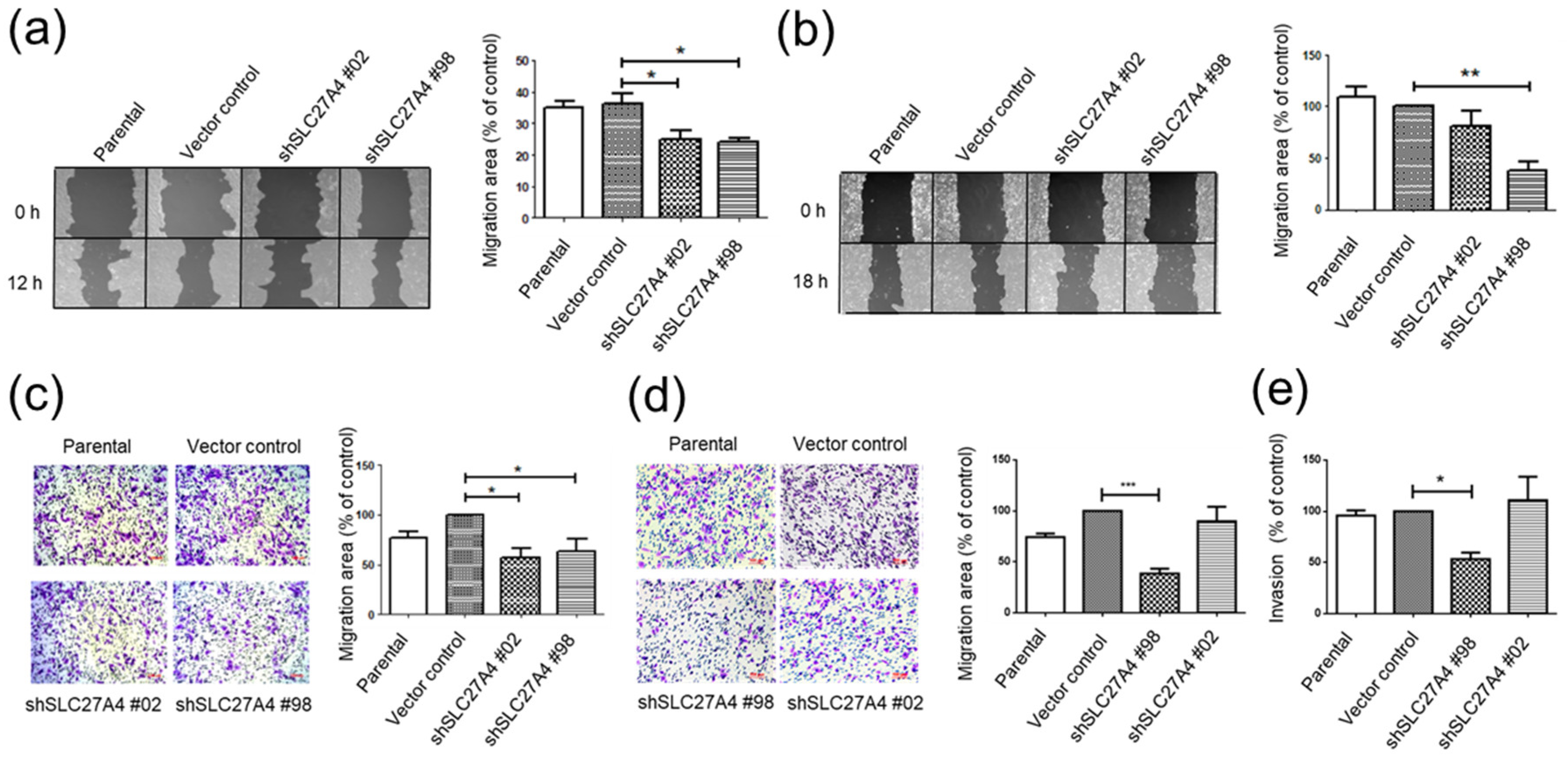
2.5. Silencing SLC27A4 in Breast Cancer Cell Lines Inhibited Capacity of Migration and Invasion
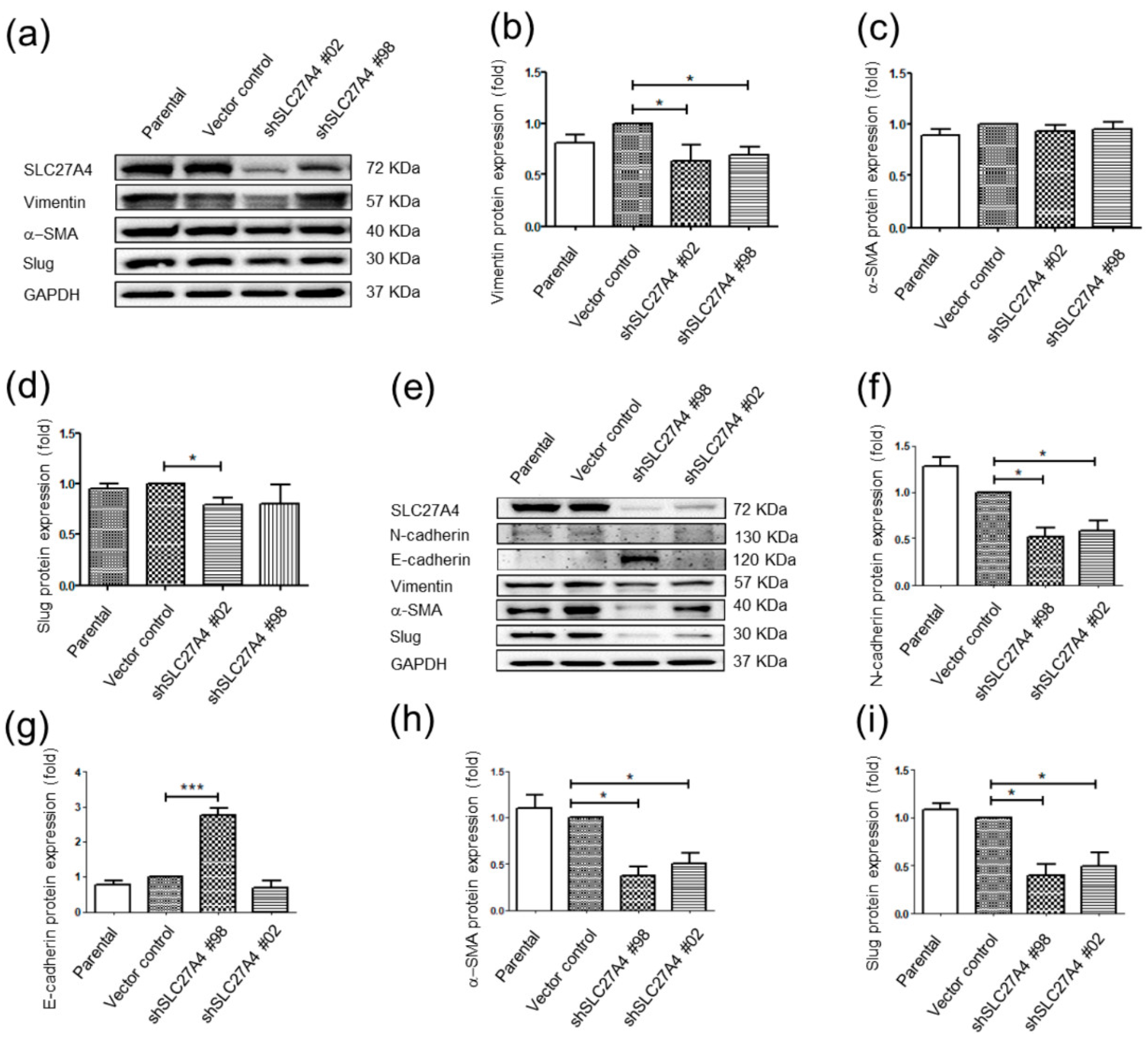
2.6. Silencing SLC27A4 in Breast Cancer Cell Lines Affected Regulatory Molecules of Epithelial Mesenchymal Transition Signaling Pathways
2.7. Potential Interacting Networks in SLC27A4-Silencing Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Bioinformatic Analysis
4.3. Western Blot Assay
4.4. Knockdown of SLC27A4
4.5. Fatty Acid Uptake Assay
4.6. Real-Time Quantitative PCR
4.7. WST-1 Assay
4.8. Colony Formation Assay
4.9. Cell Cycle Analysis
4.10. Wound Healing Assay
4.11. Transwell Migration and Invasion Assay
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Blucher, C.; Stadler, S.C. Obesity and breast cancer: Current insights on the role of fatty acids and lipid metabolism in promoting breast cancer growth and progression. Front. Endocrinol. (Lausanne) 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Germain, D.R.; Mackey, J.R.; Godbout, R. Association of fabp5 expression with poor survival in triple-negative breast cancer: Implication for retinoic acid therapy. Am. J. Pathol. 2011, 178, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.Y.; Umemura, S.; Tsukamoto, H.; Kumaki, N.; Tokuda, Y.; Osamura, R.Y. Overexpression of fatty acid binding protein-7 correlates with basal-like subtype of breast cancer. Pathol. Res. Pract. 2010, 206, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Lai, R.; Mackey, J.R.; Godbout, R. A fatty acid-binding protein 7/rxrbeta pathway enhances survival and proliferation in triple-negative breast cancer. J. Pathol. 2012, 228, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Wang, J.; Wen, X.; Marcus, M.T.; Daniels, G.; Zhang, D.Y.; Ye, F.; Wang, L.H.; Du, X.; et al. Long chain fatty acyl-coa synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS ONE 2013, 8, e77060. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G.; Li, L.O.; Coleman, R.A. Long-chain acyl-coa synthetases and fatty acid channeling. Future Lipidol. 2007, 2, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.C.; Kan, J.Y.; Hsieh, C.J.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. Association of long-chain acyl-coenzyme a synthetase 5 expression in human breast cancer by estrogen receptor status and its clinical significance. Oncol. Rep. 2017, 37, 3253–3260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via cd36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor cd36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Stahl, A. Slc27 fatty acid transport proteins. Mol. Aspects Med. 2013, 34, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A. Very-long-chain acyl-coa synthetases. J. Biol. Chem. 2008, 283, 1773–1777. [Google Scholar] [CrossRef] [PubMed]
- Black, P.N.; DiRusso, C.C. Yeast acyl-coa synthetases at the crossroads of fatty acid metabolism and regulation. Biochim. Biophys. Acta 2007, 1771, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, L.; Wei, D.; Wang, X.; Zhang, S.; Li, H. Fatty acid synthase mediates the epithelial-mesenchymal transition of breast cancer cells. Int. J. Biol. Sci. 2014, 10, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhang, C.; Arif, M.; Liu, Z.; Benfeitas, R.; Bidkhori, G.; Deshmukh, S.; Al Shobky, M.; Lovric, A.; Boren, J.; et al. Tcsbn: A database of tissue and cancer specific biological networks. Nucleic Acids Res. 2018, 46, D595–D600. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Pomerening, J.R.; Myers, J.W.; Gustavsson, C.; Jones, J.T.; Hahn, A.T.; Meyer, T.; Ferrell, J.E., Jr. Cyclin a2 regulates nuclear-envelope breakdown and the nuclear accumulation of cyclin b1. Curr. Biol. 2007, 17, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, N.; Tyekucheva, S.; Zadra, G.; Photopoulos, C.; Loda, M. De novo fatty acid synthesis at the mitotic exit is required to complete cellular division. Cell Cycle 2014, 13, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Mellor, P.; Ward, A.K.; Kendall, S.; McDonald, M.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Napper, S.; Anderson, D.H. Molecular characterization of breast cancer cell lines through multiple omic approaches. Breast Cancer Res. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the g2/m transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.H. The role of p53 in cell cycle regulation. Pathol. Res. Pract. 1996, 192, 669–675. [Google Scholar] [CrossRef]
- Son, H.; Moon, A. Epithelial-mesenchymal transition and cell invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in tgf-beta-induced epithelial to mesenchymal transition is regulated by activation of the mtor pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Gu, L.N.; Shan, B.E.; Geng, C.Z.; Sang, M.X. Biomarkers for emt and met in breast cancer: An update. Oncol. Lett. 2016, 12, 4869–4876. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jay, A.; Brunaldi, K.; Huang, N.; Hamilton, J.A. Cd36 enhances fatty acid uptake by increasing the rate of intracellular esterification but not transport across the plasma membrane. Biochemistry 2013, 52, 7254–7261. [Google Scholar] [CrossRef] [PubMed]
- Kuch, E.M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brugger, B.; Sreemmel, W.; Ehehalt, R.; Poppelreuther, M.; Fullekrug, J. Differentially localized acyl-coa synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 2014, 1841, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Borin, T.F.; Angara, K.; Rashid, M.H.; Achyut, B.R.; Arbab, A.S. Arachidonic acid metabolite as a novel therapeutic target in breast cancer metastasis. Int. J. Mol. Sci. 2017, 18, 2661. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, C.; Cavia Mdel, M.; Alonso-Torre, S.R. Antitumor effect of oleic acid; mechanisms of action: A review. Nutr. Hosp. 2012, 27, 1860–1865. [Google Scholar] [PubMed]
- Wu, S.; Su, J.; Qian, H.; Guo, T. Slc27a4 regulate atg4b activity and control reactions to chemotherapeutics-induced autophagy in human lung cancer cells. Tumour. Biol. 2016, 37, 6943–6952. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, S.; Xing, Z.; Lin, A.; Liang, K.; Song, J.; Hu, Q.; Yao, J.; Chen, Z.; Park, P.K.; et al. A ror1-her3-lncrna signalling axis modulates the hippo-yap pathway to regulate bone metastasis. Nat. Cell Biol. 2017, 19, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. Ualcan: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yen, M.-C.; Chou, S.-K.; Kan, J.-Y.; Kuo, P.-L.; Hou, M.-F.; Hsu, Y.-L. Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3434. https://doi.org/10.3390/ijms19113434
Yen M-C, Chou S-K, Kan J-Y, Kuo P-L, Hou M-F, Hsu Y-L. Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells. International Journal of Molecular Sciences. 2018; 19(11):3434. https://doi.org/10.3390/ijms19113434
Chicago/Turabian StyleYen, Meng-Chi, Shih-Kai Chou, Jung-Yu Kan, Po-Lin Kuo, Ming-Feng Hou, and Ya-Ling Hsu. 2018. "Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells" International Journal of Molecular Sciences 19, no. 11: 3434. https://doi.org/10.3390/ijms19113434
APA StyleYen, M.-C., Chou, S.-K., Kan, J.-Y., Kuo, P.-L., Hou, M.-F., & Hsu, Y.-L. (2018). Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells. International Journal of Molecular Sciences, 19(11), 3434. https://doi.org/10.3390/ijms19113434