Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
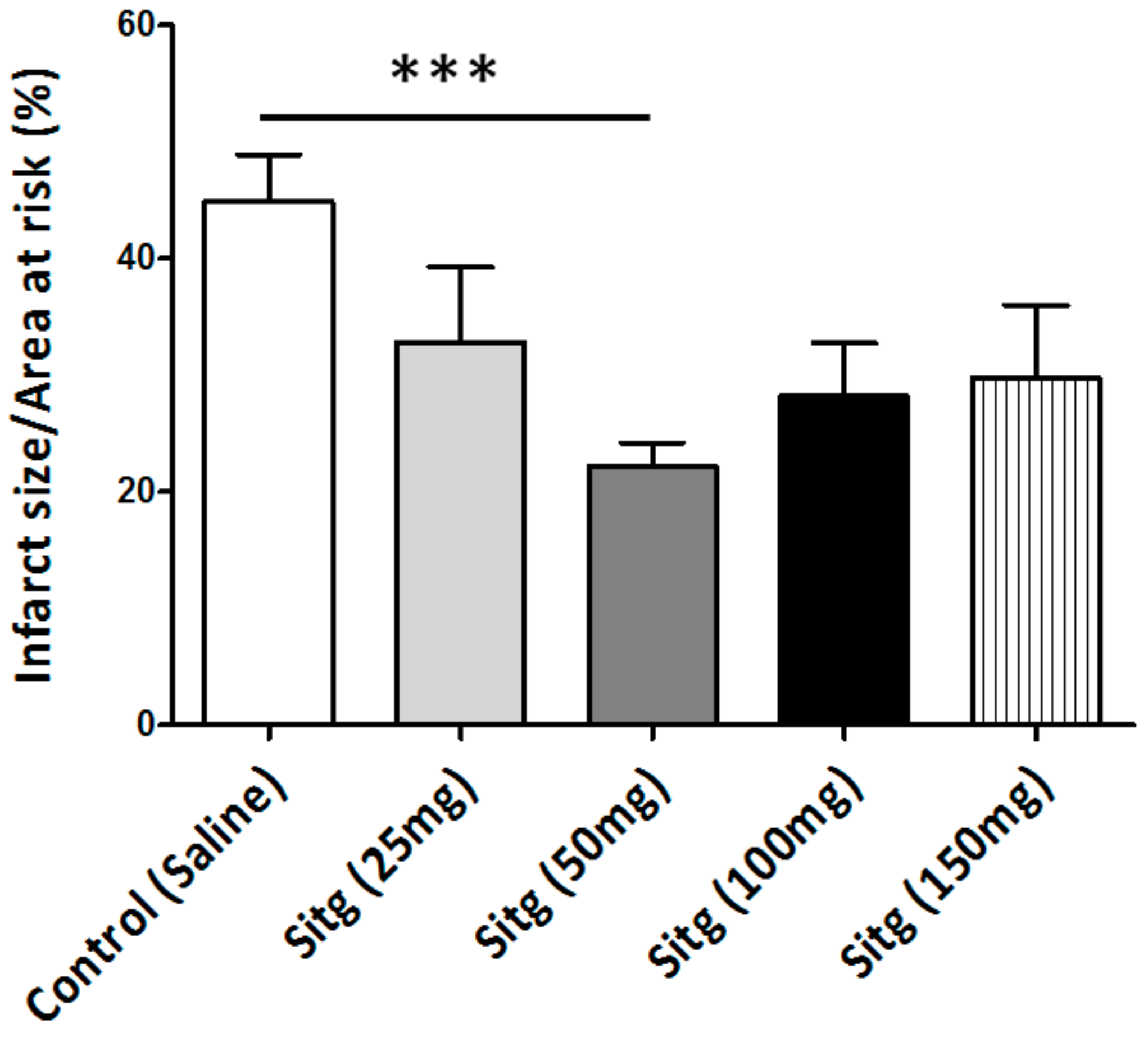
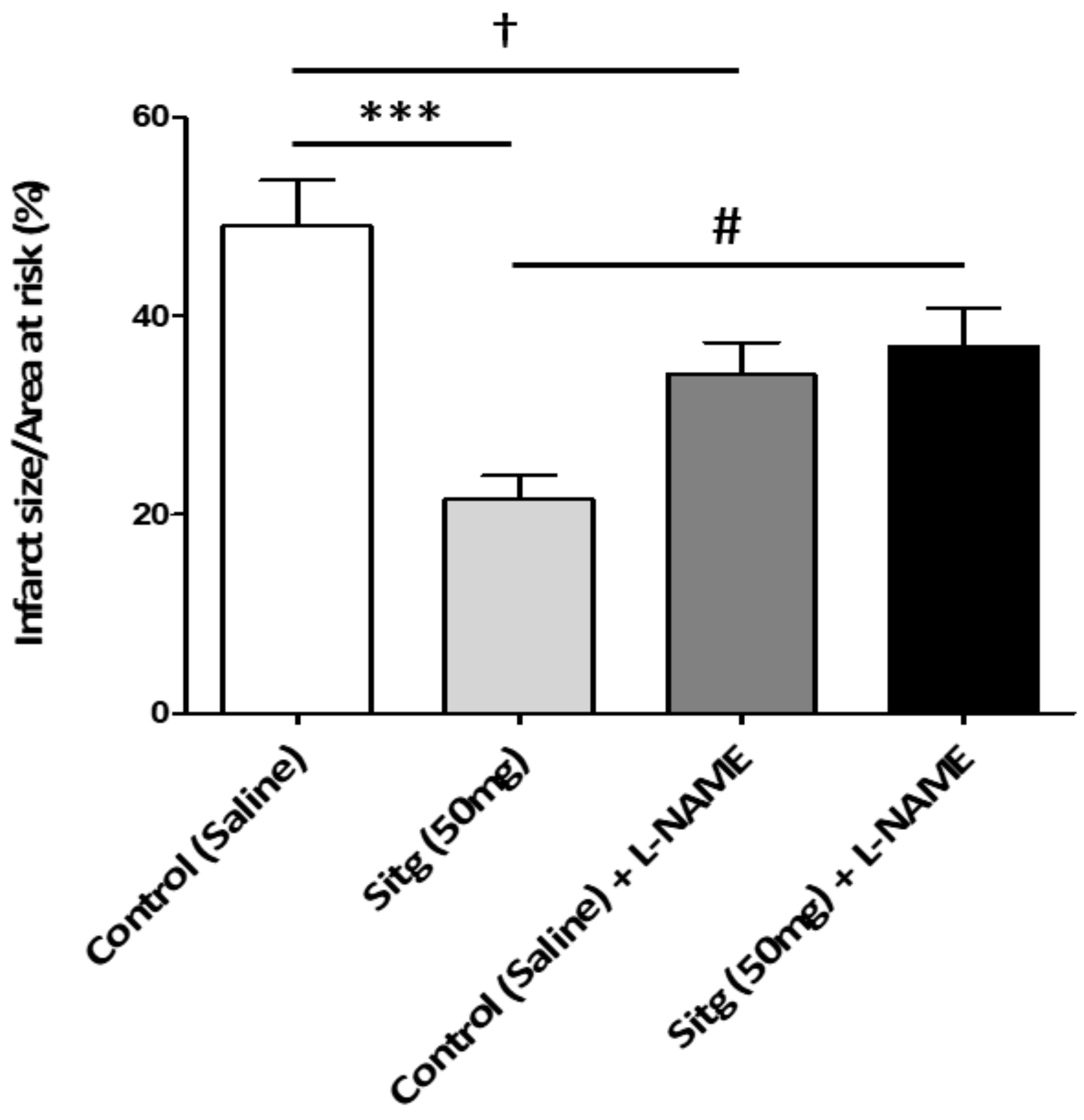
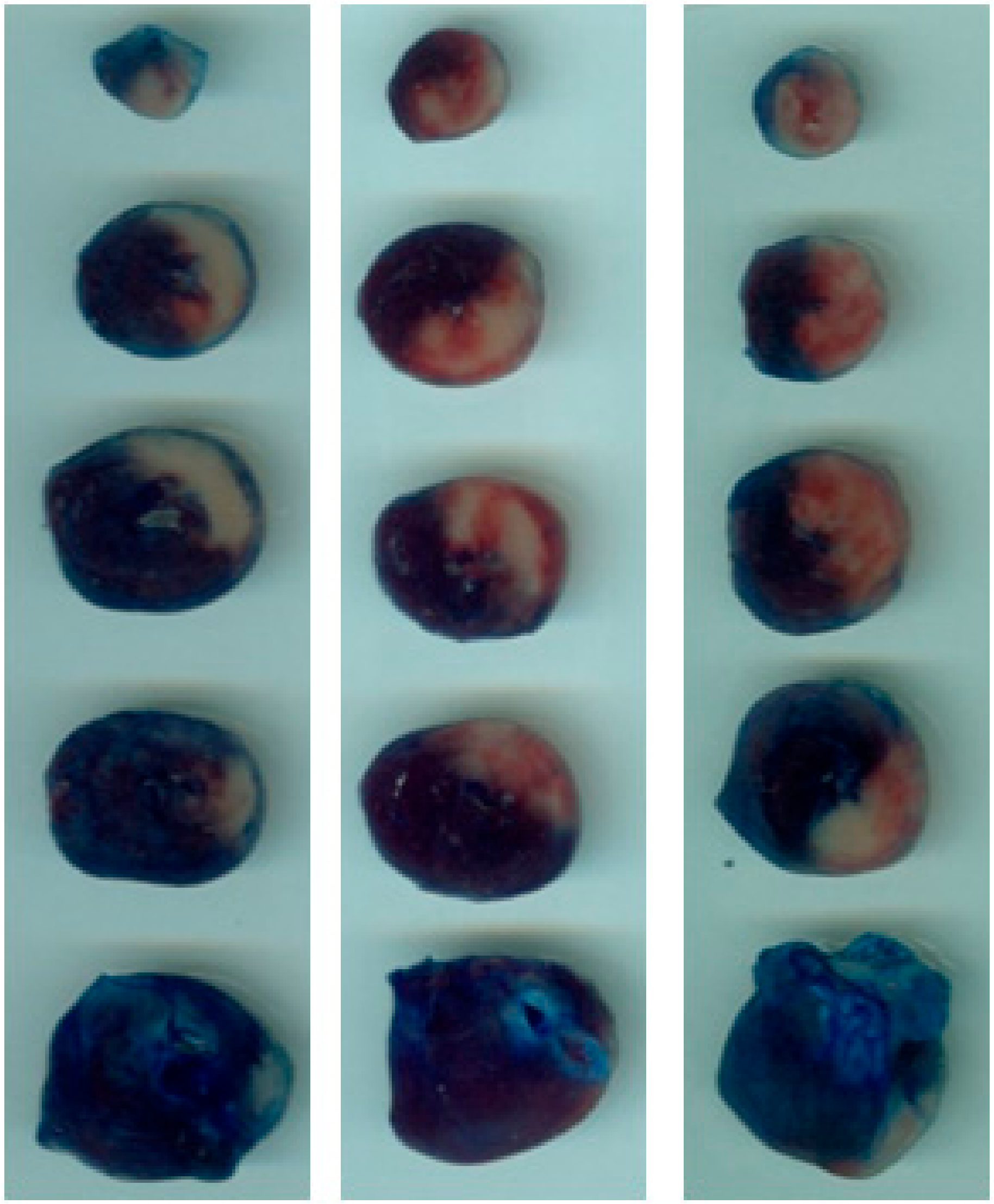
2.1. DPP-4i Decreased the Infarct Size in Heart Tissues of Sitg (50 mg) Group
2.2. DPP-4i Normalized DPP-4 Activity and Enhanced GLP-1 Level
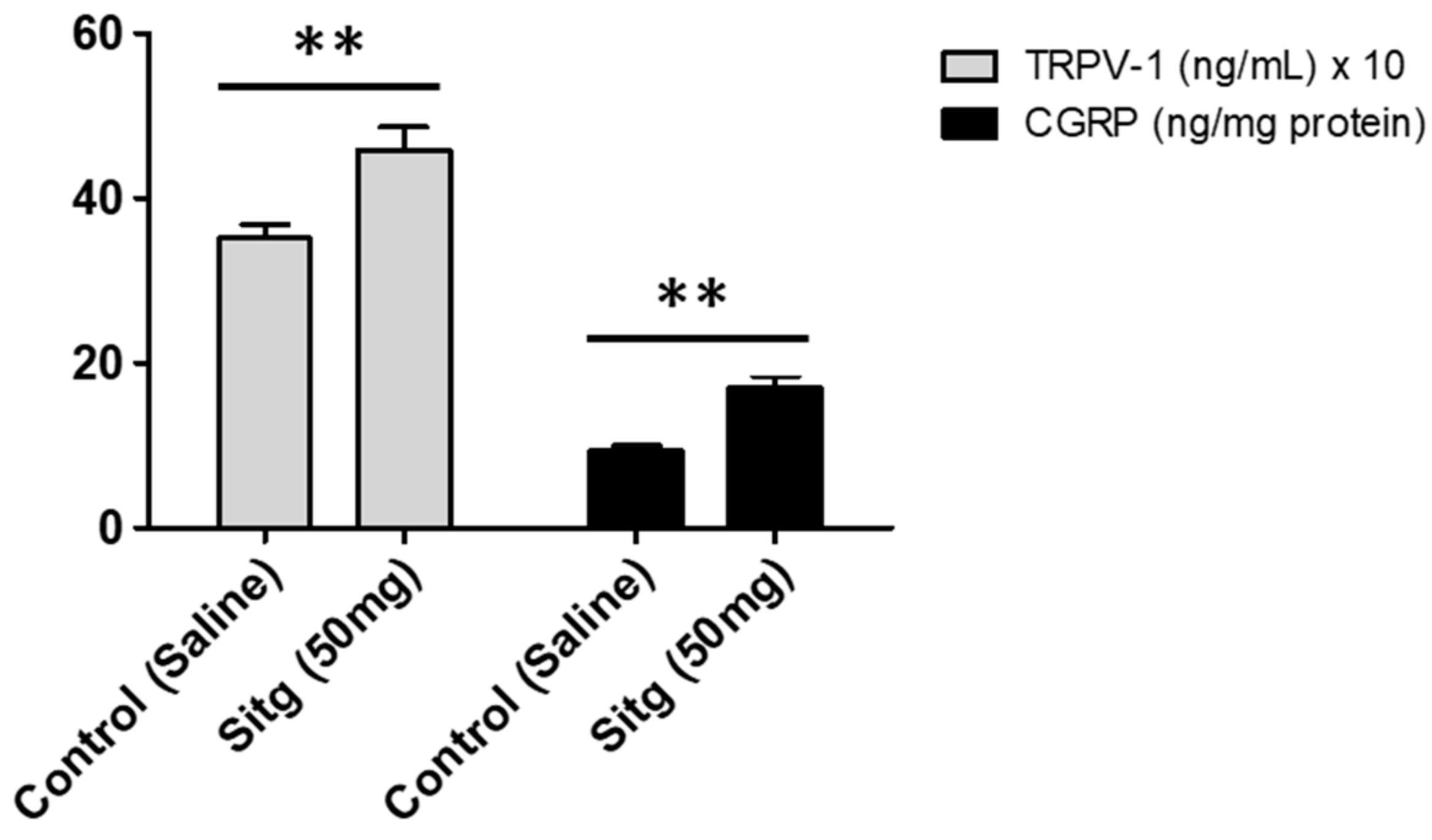
2.3. DPP-4i Increased TRPV-1 and CGRP Levels in Heart Tissues of Sitg (50 mg)
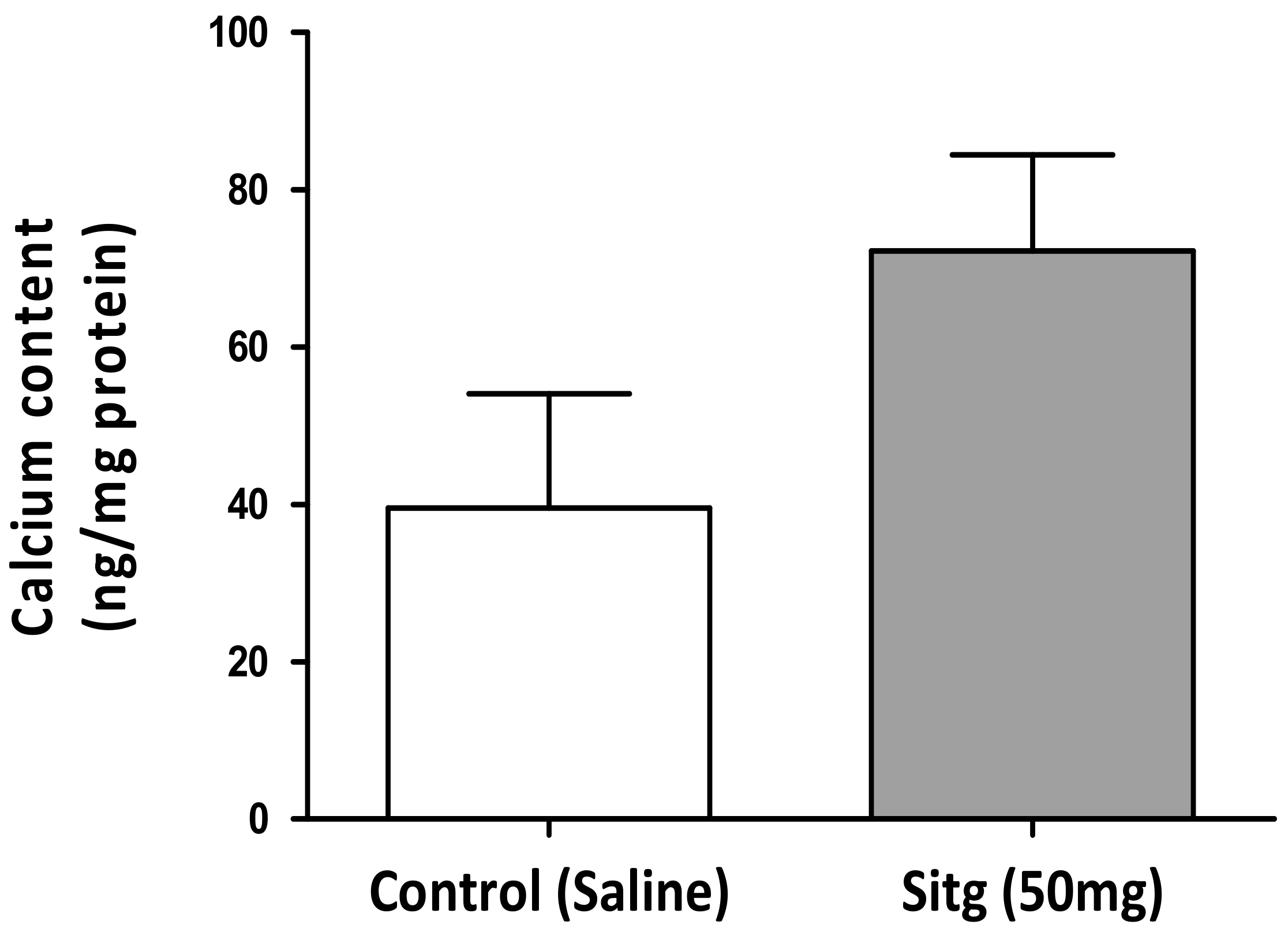
2.4. DPP-4i Augmented Cardiac Calcium (Ca2+) Content in Hearts of Sitg (50 mg) Group
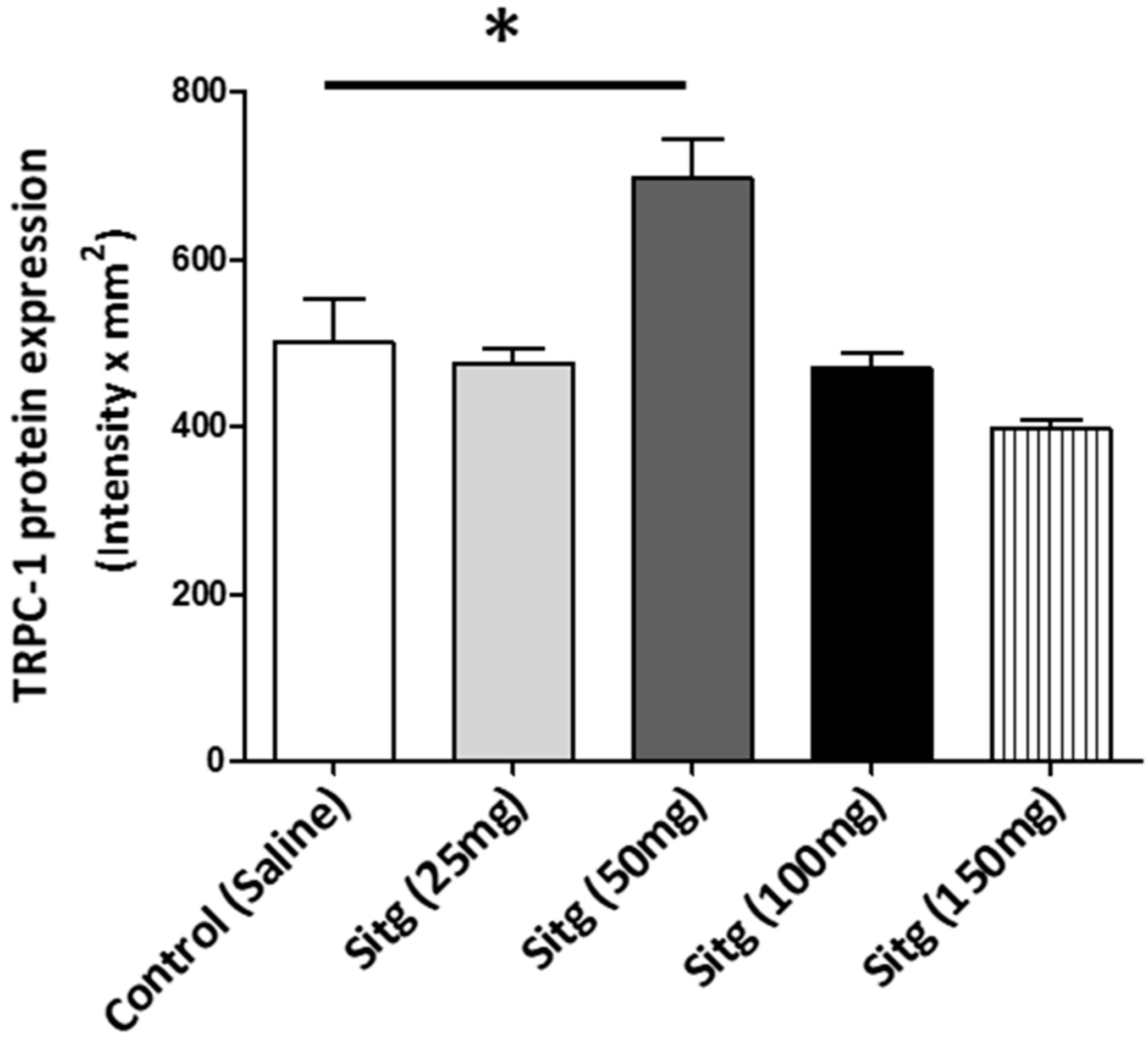
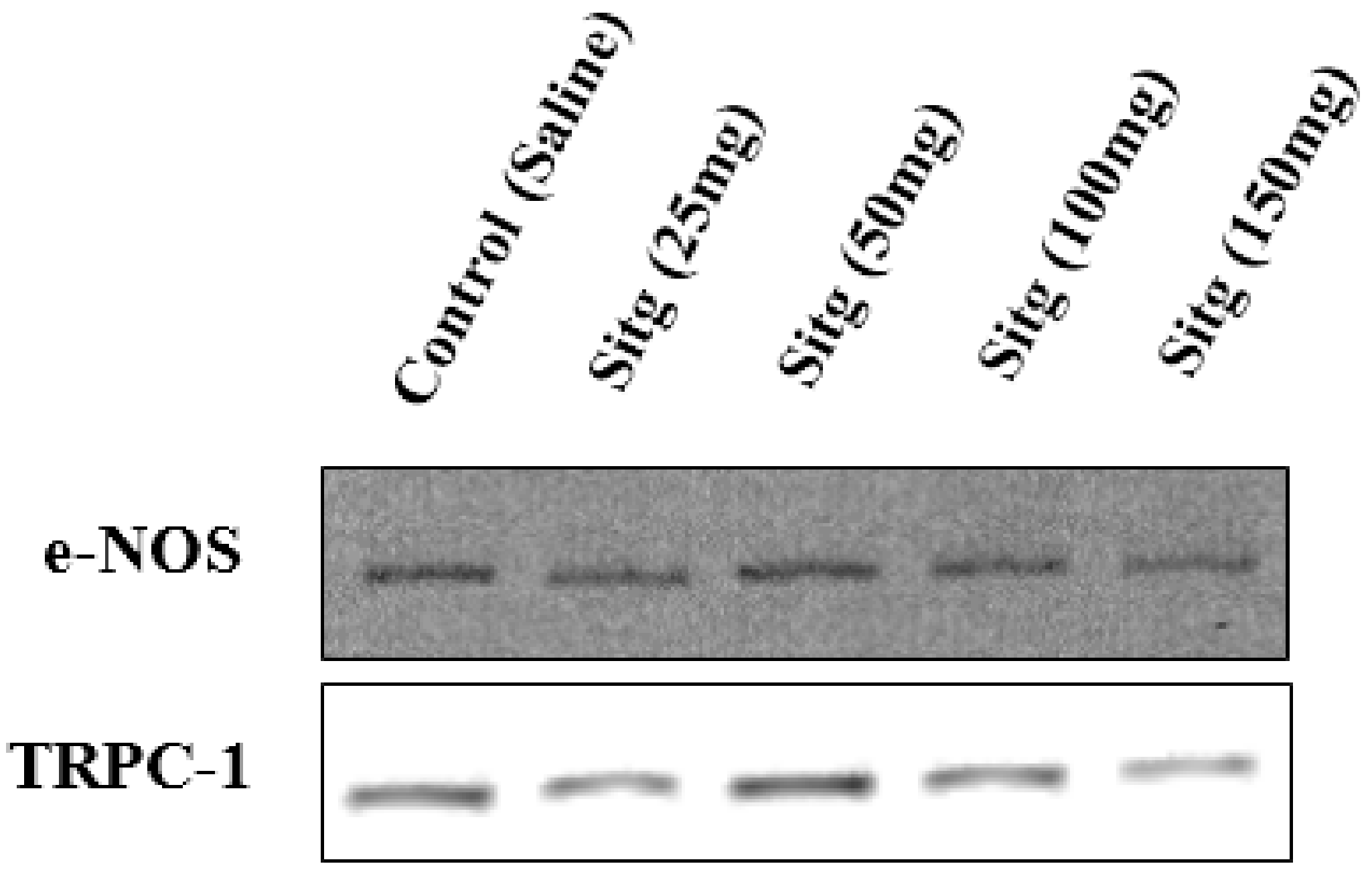
2.5. DPP-4i Positively Affected TRPC-1 Protein Expression
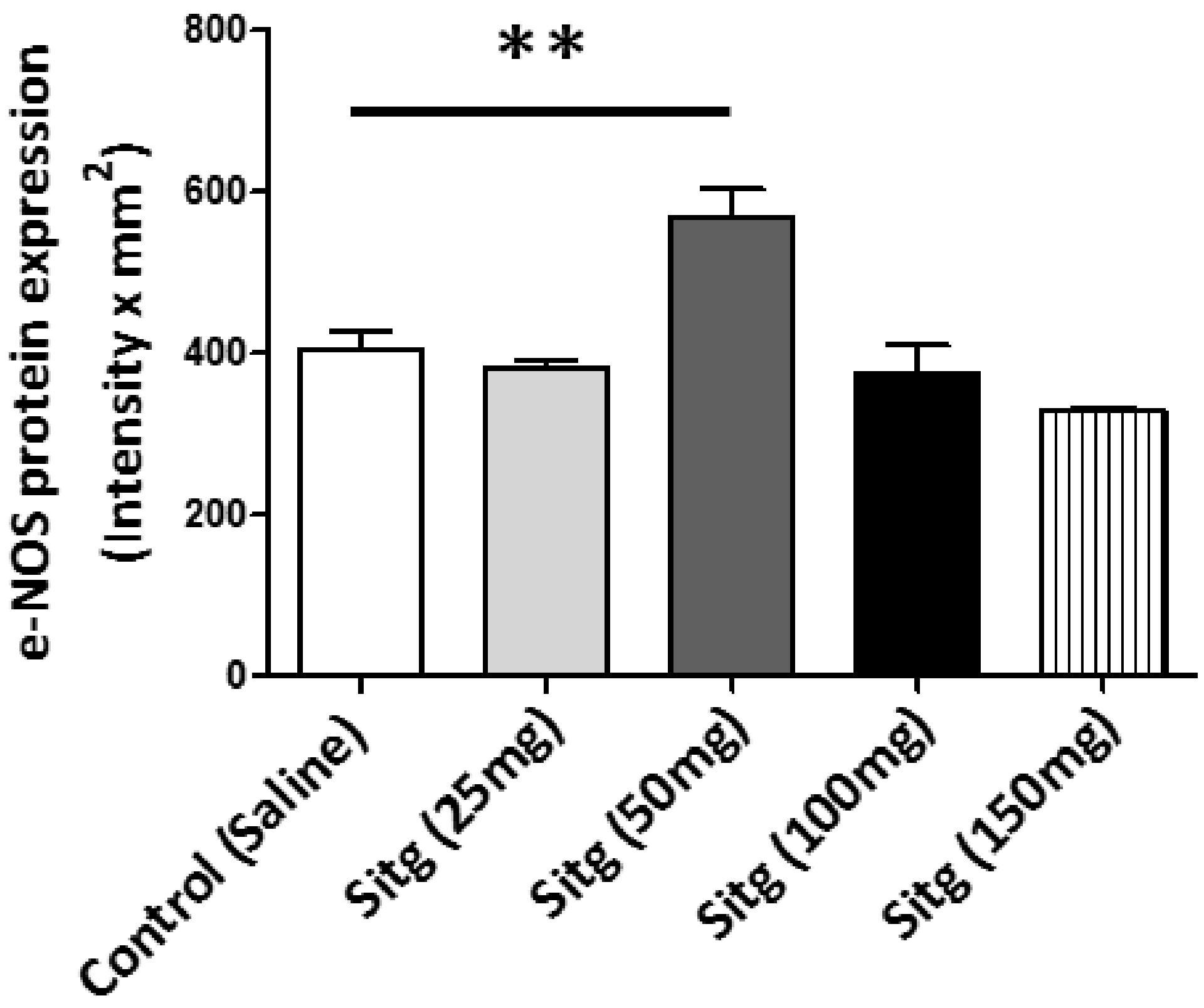
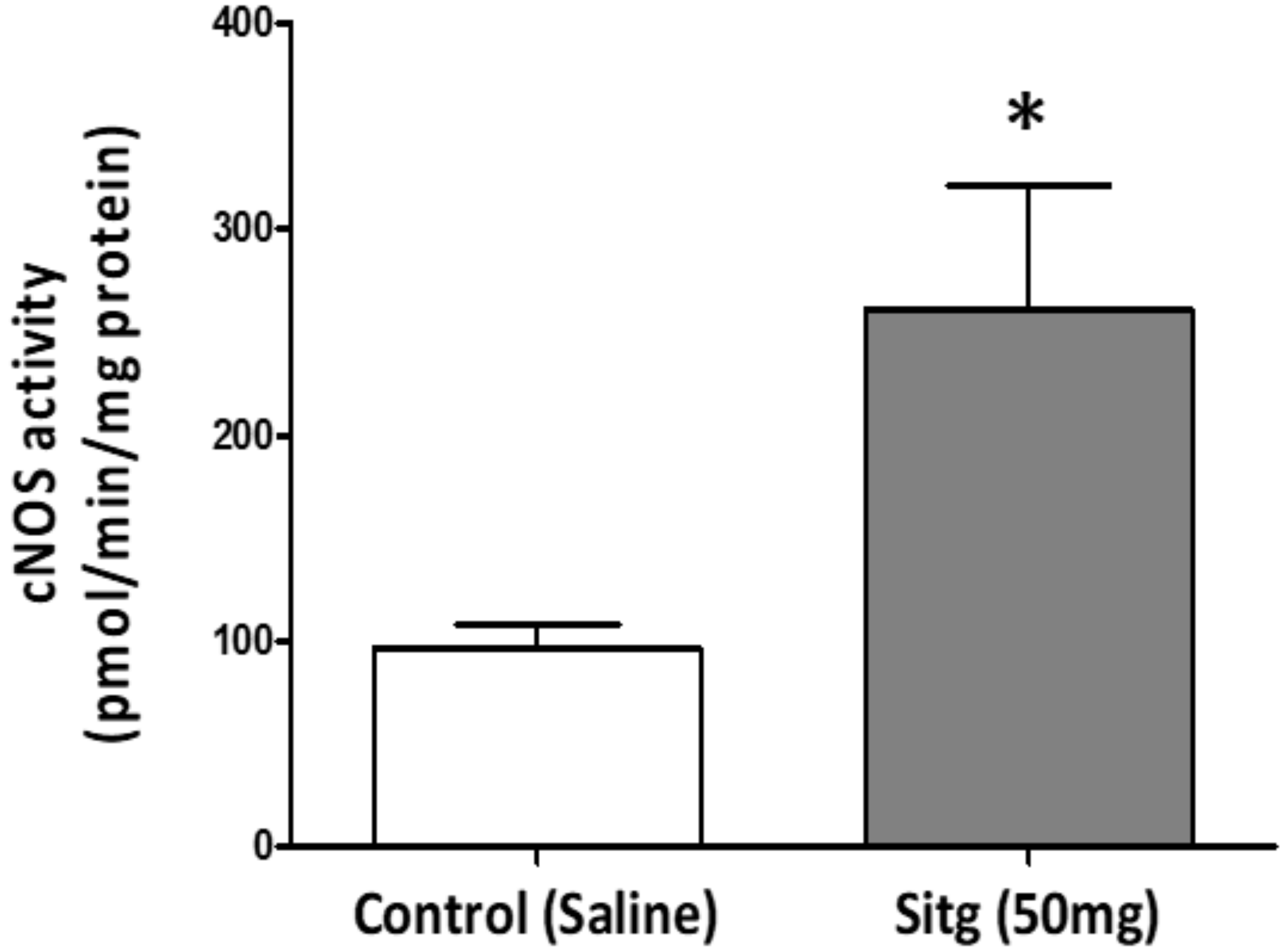
2.6. DPP-4i Upregulated e-NOS Protein Expression and cNOS Activity in Heart Tissues of Sitg (50 mg)
2.6.1. e-NOS Protein Expression
2.6.2. cNOS Activity
2.7. l-NAME Inhibited NOS-Mediated Cardioprotection against Infarct
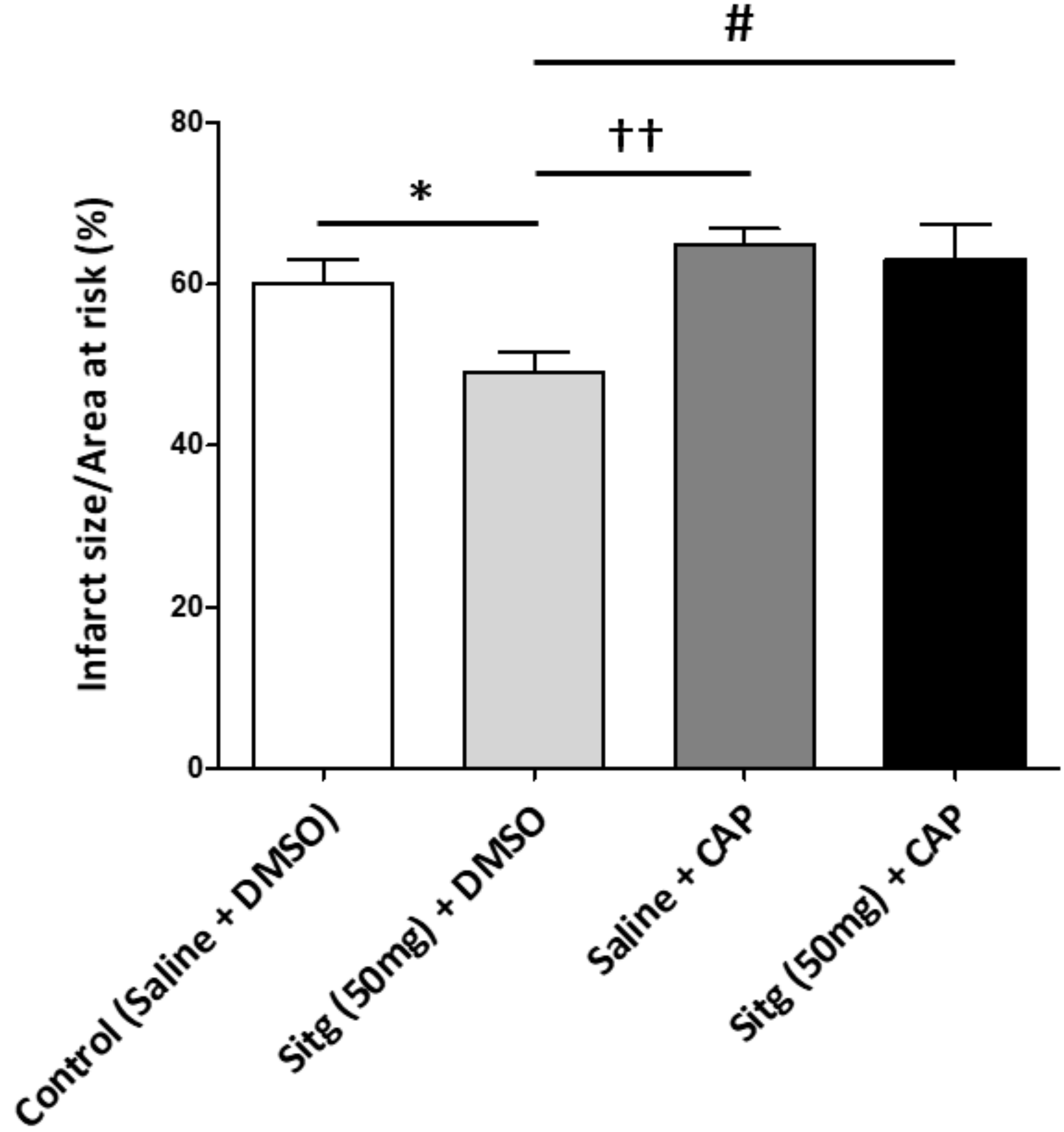
2.8. Capsazepine Inhibited TRPV-1-Mediated Cardioprotection against Infarct
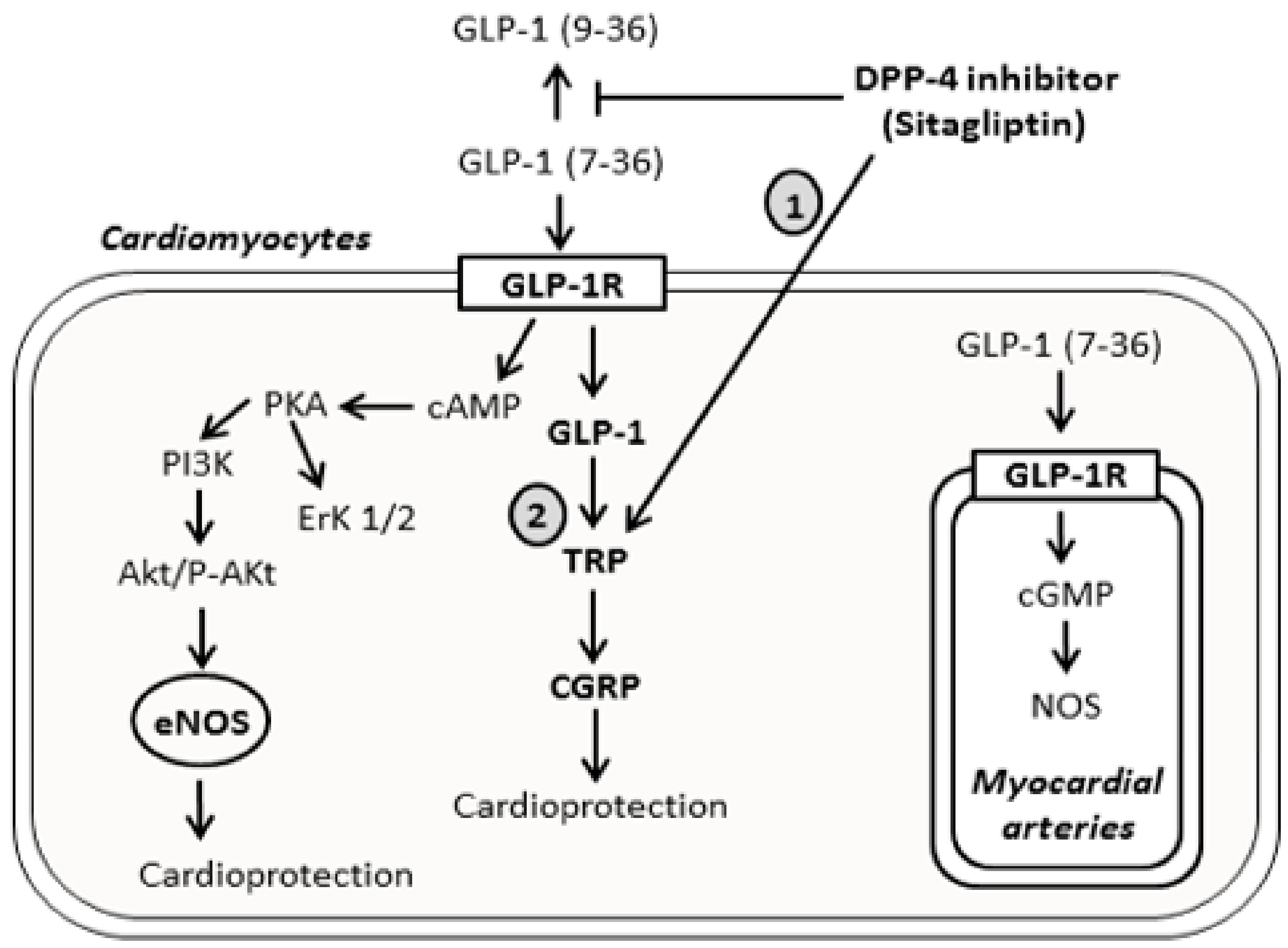
3. Discussion
Limitations
4. Materials and Methods
4.1. Drug Preparations
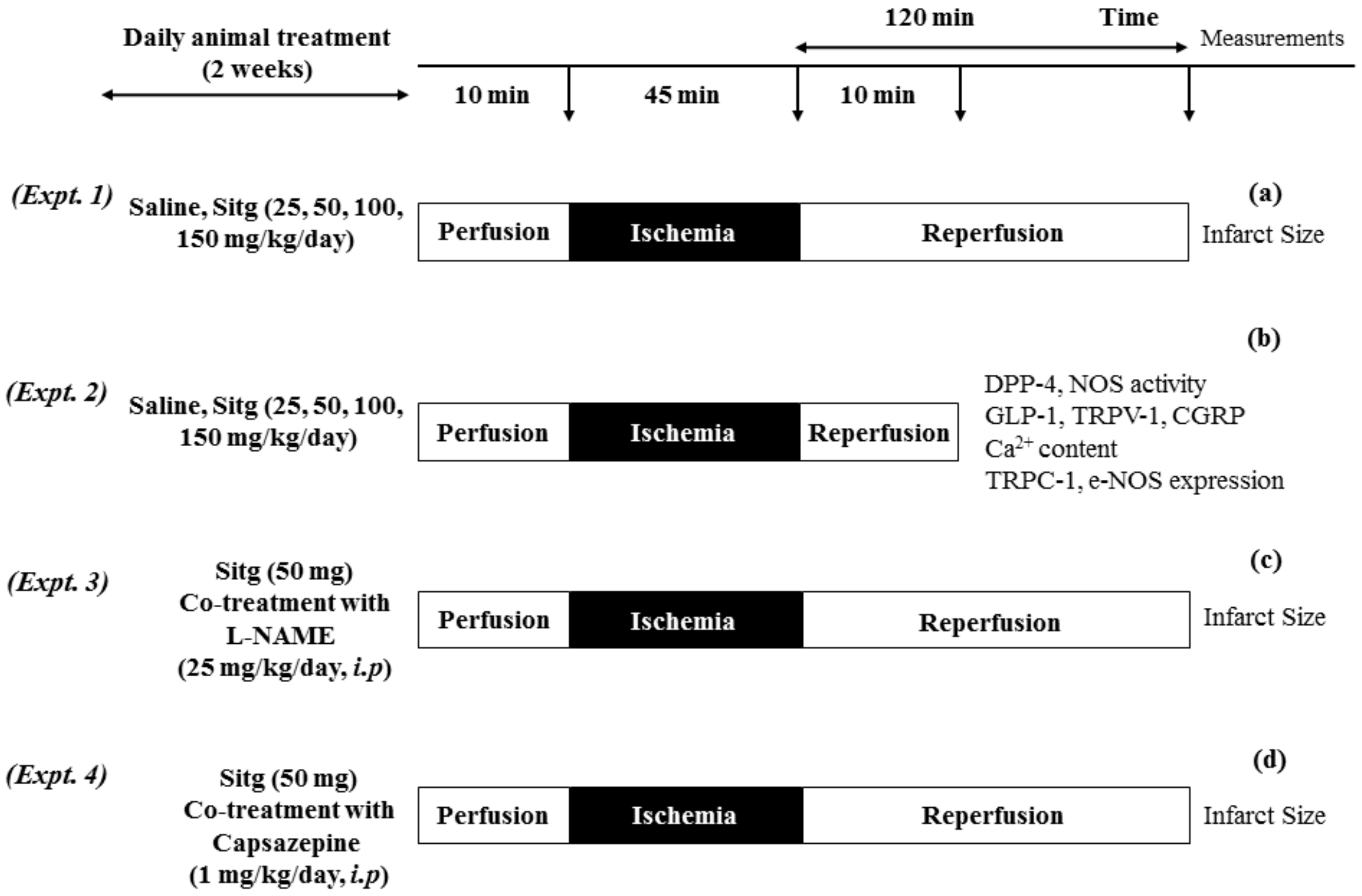
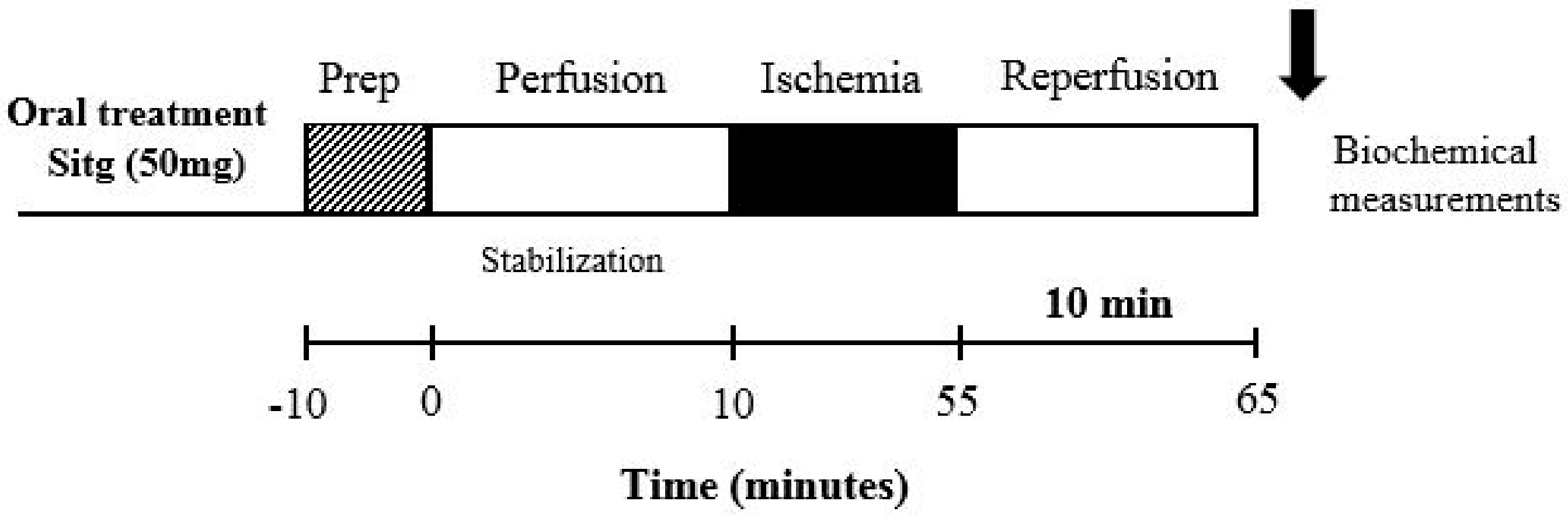
4.2. Animals and Experimental Design
4.3. Tissue Staining and Infarct Size Measurement
4.4. DPP-4 Activity Test
4.5. Nitric Oxide Synthase (NOS) Activity
4.6. ELISA Measurements (GLP-1, TRPV-1 and CGRP)
4.7. Calcium (Ca2+) Content Test
4.8. TRPC-1 and e-NOS Protein Expression by Western Blotting
4.9. Protein Determination
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MI | Myocardial infarction |
| IR | Ischemia-reperfusion |
| IS | Infarct size |
| DPP-4 | Dipeptidyl peptidase- 4 |
| DPP-4i | Dipeptidyl peptidase-4 inhibitor |
| GLP-1 | Glucagon-like peptide-1 |
| NOS | Nitric oxide synthase |
| e-NOS | Endothelial nitric oxide synthase |
| i-NOS | Inducible nitric oxide synthase |
| n-NOS | Neuronal nitric oxide synthase |
| TRP | Transient receptor potential |
| TRPC | Transient receptor potential canonical |
| TRPV | Transient receptor potential vanilloid |
| CGRP | Calcitonin gene-related peptide |
| l-NAME | Nω-nitro-l-arginine methyl ester |
| CAP | Capsazepine |
| TTC | Triphenyltetrazolium chloride |
| LAD | Left anterior descending |
| AAR | Area at risk |
| Sitg | Sitagliptin |
| PBS | Phosphate buffer saline |
| DMSO | Dimethyl sulfoxide |
References
- Acar, E.; Ural, D.; Bildirici, U.; Sahin, T.; Yilmaz, I. Diabetic cardiomyopathy. Anadolu Kardiyol. Derg. 2011, 11, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Zhang, P.; Ye, L.; Lu, K.; Wang, Y.; Duan, Q.; Zheng, A.; Qin, S.; Zhang, D. Protective effects of sitagliptin on myocardial injury and cardiac function in an ischemia/reperfusion rat model. Eur. J. Pharmacol. 2013, 718, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Ferreira, R.M.; Tavares, J.G.P.; Paula, L.; Araujo, E.A.; Govato, T.C.P.; Tikazawa, E.H.; Reis, M.; Luna-Filho, B.; et al. Cardioprotective effect of lipstatin derivative orlistat on normotensive rats submitted to cardiac ischemia and reperfusion. Acta Cir. Bras. 2018, 33, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.M.; Keyes, K.T.; Zhang, C.F.; Perez-Polo, J.R.; Lin, Y.; Birnbaum, Y. The myocardial infarct size-limiting effect of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1454–H1465. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Asakura, M.; Ito, S.; Min, K.D.; Shindo, K.; Yan, Y.; Liao, Y.; Yamazaki, S.; Sanada, S.; Asano, Y.; et al. Dipeptidyl-peptidase IV inhibition improves pathophysiology of heart failure and increases survival rate in pressure-overloaded mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1361–H1369. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Varin, E.M.; Ussher, J.R.; Campbell, J.E.; Bang, K.W.; Abdullah, T.; Baggio, L.L.; Drucker, D.J. Inhibition of Dipeptidyl Peptidase-4 Impairs Ventricular Function and Promotes Cardiac Fibrosis in High Fat-Fed Diabetic Mice. Diabetes 2016, 65, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Maiseyeu, A.; Davis, S.N.; Rajagopalan, S. DPP4 in cardiometabolic disease: Recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ. Res. 2015, 116, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Schulz, R.; Kelm, M.; Heusch, G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Pabla, R.; Curtis, M.J. Endogenous protection against reperfusion-induced ventricular fibrillation: Role of neuronal versus non-neuronal sources of nitric oxide and species dependence in the rat versus rabbit isolated heart. J. Mol. Cell. Cardiol. 1996, 28, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, R.R.; Panda, A.; Xia, Y.; Sanders, S.P.; Zweier, J.L. Decreased nitric-oxide synthase activity causes impaired endothelium-dependent relaxation in the postischemic heart. J. Biol. Chem. 1997, 272, 21420–21426. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, re3. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef] [PubMed]
- Craelius, W.; Chen, V.; El-Sherif, N. Stretch activated ion channels in ventricular myocytes. Biosci. Rep. 1988, 8, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Shenton, F.C.; Pyner, S. Expression of transient receptor potential channels TRPC1 and TRPV4 in venoatrial endocardium of the rat heart. Neuroscience 2014, 267, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Minke, B. TRP channels and Ca2+ signaling. Cell Calcium 2006, 40, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, F.; Zhang, J.C.; Wang, T.T.; Wei, X.; Wu, J.; Feng, Y.L.; Dai, Z.L.; Wu, Q.P. Neuroprotective Effect of Resveratrol on Ischemia/Reperfusion Injury in Rats through TRPC6/CREB Pathways. J. Mol. Neurosci. 2013, 50, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Kim, H.S.; Choi, W.; Ha, T.S.; Ahn, H.Y.; Kim, C.H. Mechanical Stretch-Induced Protection against Myocardial Ischemia-Reperfusion Injury Involves AMP-Activated Protein Kinase. Korean J. Physiol. Pharmacol. 2010, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Li, Y.J.; Deng, H.W. Evidence for calcitonin gene-related peptide-mediated ischemic preconditioning in the rat heart. Regul. Pept. 1999, 82, 53–57. [Google Scholar] [CrossRef]
- Chai, W.; Mehrotra, S.; Jan Danser, A.H.; Schoemaker, R.G. The role of calcitonin gene-related peptide (CGRP) in ischemic preconditioning in isolated rat hearts. Eur. J. Pharmacol. 2006, 531, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Aslan, C.; Melikoglu, C.; Ocal, I.; Saglam, G.; Sutcu, R.; Hosnuter, M. Effect of epigallocatechin gallate on ischemia-reperfusion injury: An experimental study in a rat epigastric island flap. Int. J. Clin. Exp. Med. 2014, 7, 57–66. [Google Scholar] [PubMed]
- Kloner, R.A.; Rezkalla, S.H. Cardiac protection during myocardial infarction: Where acute do we stand in 2004? J. Am. Coll. Cardiol. 2004, 44, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Sokos, G.G.; Nikolaidis, L.A.; Mankad, S.; Elahi, D.; Shannon, R.P. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J. Card. Fail. 2006, 12, 694–699. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, C.H.S. Dipeptidyl peptidase iv inhibitors and diabetes therapy. Front. Biosci. 2008, 13, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; de Kreutzenberg, S.; Fadini, G. Dipeptidyl-peptidase 4 inhibition: Linking metabolic control to cardiovascular protection. Curr. Pharm. Des. 2014, 20, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Chinda, K.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Dipeptidyl peptidase-4 inhibitor reduces infarct size and preserves cardiac function via mitochondrial protection in ischaemia-reperfusion rat heart. Diabetes Vasc. Dis. Res. 2014, 11, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.Y.; Woo, J.S.; Seo, J.W.; Lee, A.; Kim, D.J.; Kim, Y.G.; Kim, S.Y.; Lee, K.H.; Lim, S.J.; Cheng, X.W.; et al. The Dose-Dependent Organ-Specific Effects of a Dipeptidyl Peptidase-4 Inhibitor on Cardiovascular Complications in a Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0150745. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhao, M.; Bi, X.Y.; Yu, X.J.; Zang, W.J. Delayed preconditioning prevents ischemia/reperfusion-induced endothelial injury in rats: Role of ROS and eNOS. Lab. Investig. 2013, 93, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, X.; Belmadani, S.; Park, Y.; Tang, Z.; Feldman, A.M.; Chilian, W.M.; Zhang, C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.V.; Birdsey, G.M.; Randi, A.M. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vasc. Pharmacol. 2016, 86, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Tamaki, K.; Ito, K.; Yan, X.; Yamamoto, T.; Katsumata, Y.; Matsuhashi, T.; Sano, M.; Fukuda, K.; Suematsu, M.; et al. Indispensable role of endothelial nitric oxide synthase in caloric restriction-induced cardioprotection against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H894–H903. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zweier, J.L. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J. Biol. Chem. 1996, 271, 29223–29230. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.; Koitabashi, N.; Kass, D.A. TRP-ing up Heart and Vessels: Canonical Transient Receptor Potential Channels and Cardiovascular Disease. J. Cardiovasc. Transl. 2010, 3, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Murakami, M.; Ohba, T.; Takahashi, Y.; Ito, H. TRP channel and cardiovascular disease. Pharmacol. Ther. 2008, 118, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.D.; Alessandri-Haber, N. TRP channels: Targets for the relief of pain. Biochim. Biophys. Acta 2007, 1772, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, L.Z.; Han, Y.; Guo, Z. Differential effects of the calcitonin gene-related peptide on cardiac performance in acute myocardial ischemia and reperfusion in isolated rat hearts. Minerva Anestesiol. 2011, 77, 789–796. [Google Scholar] [PubMed]
- Jaarin, K.; Foong, W.D.; Yeoh, M.H.; Kamarul, Z.Y.; Qodriyah, H.M.; Azman, A.; Zuhair, J.S.; Juliana, A.H.; Kamisah, Y. Mechanisms of the antihypertensive effects of Nigella sativa oil in l-NAME-induced hypertensive rats. Clinics 2015, 70, 751–757. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Z.L.; Du, Y.H.; Fu, M.; Han, H.L.; Wang, Y.T.; Dong, Z.F. Capsaicin Attenuates Amyloid-beta-Induced Synapse Loss and Cognitive Impairments in Mice. J. Alzheimers Dis. 2017, 59, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Boughton-Smith, N.K.; Evans, S.M.; Laszlo, F.; Whittle, B.J.; Moncada, S. The induction of nitric oxide synthase and intestinal vascular permeability by endotoxin in the rat. Br. J. Pharmacol. 1993, 110, 1189–1195. [Google Scholar] [CrossRef] [PubMed]










 ) indicates mechanism’s activation, and the T bar sign (
) indicates mechanism’s activation, and the T bar sign ( ) indicates the inhibitory effect.
) indicates mechanism’s activation, and the T bar sign () indicates the inhibitory effect.
) indicates the inhibitory effect.
) indicates mechanism’s activation, and the T bar sign () indicates the inhibitory effect.




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-awar, A.; Almási, N.; Szabó, R.; Takacs, I.; Murlasits, Z.; Szűcs, G.; Török, S.; Pósa, A.; Varga, C.; Kupai, K. Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model. Int. J. Mol. Sci. 2018, 19, 3226. https://doi.org/10.3390/ijms19103226
Al-awar A, Almási N, Szabó R, Takacs I, Murlasits Z, Szűcs G, Török S, Pósa A, Varga C, Kupai K. Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model. International Journal of Molecular Sciences. 2018; 19(10):3226. https://doi.org/10.3390/ijms19103226
Chicago/Turabian StyleAl-awar, Amin, Nikoletta Almási, Renáta Szabó, Istvan Takacs, Zsolt Murlasits, Gergő Szűcs, Szilvia Török, Anikó Pósa, Csaba Varga, and Krisztina Kupai. 2018. "Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model" International Journal of Molecular Sciences 19, no. 10: 3226. https://doi.org/10.3390/ijms19103226
APA StyleAl-awar, A., Almási, N., Szabó, R., Takacs, I., Murlasits, Z., Szűcs, G., Török, S., Pósa, A., Varga, C., & Kupai, K. (2018). Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model. International Journal of Molecular Sciences, 19(10), 3226. https://doi.org/10.3390/ijms19103226