Combinations of Small RNA, RNA, and Degradome Sequencing Uncovers the Expression Pattern of microRNA–mRNA Pairs Adapting to Drought Stress in Leaf and Root of Dactylis glomerata L.


 ,
, 
Abstract
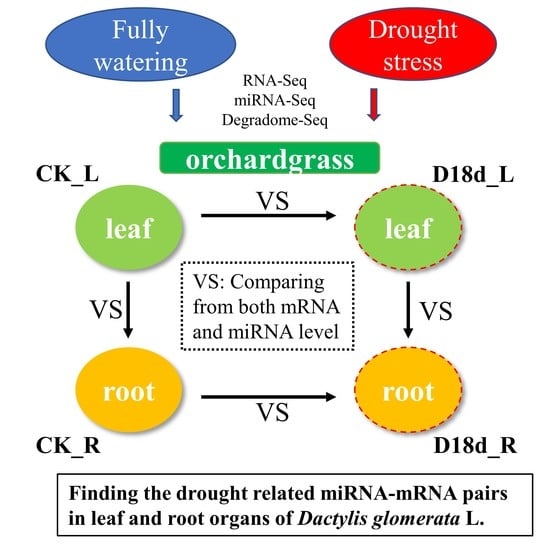
1. Introduction
2. Results
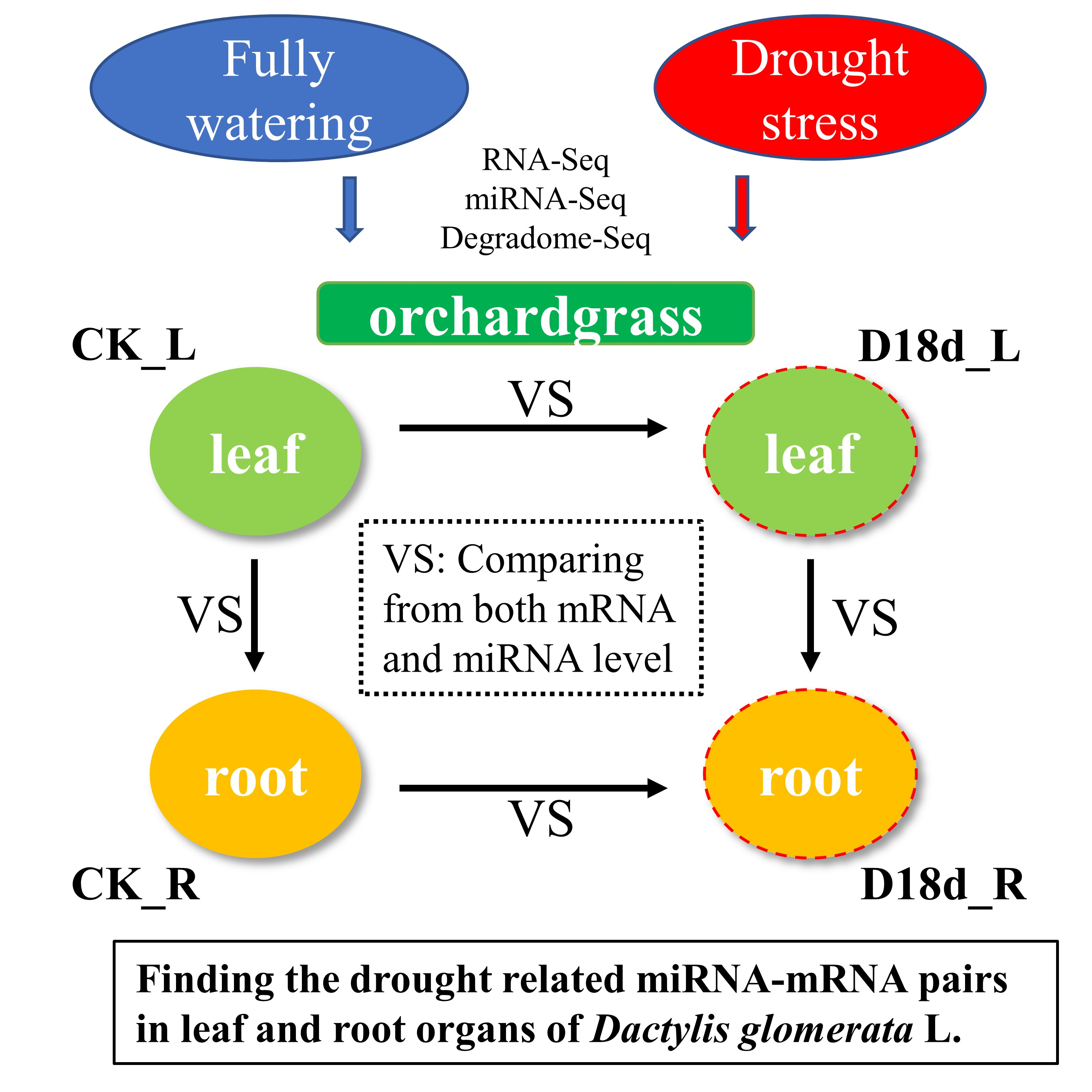
2.1. Transcriptome Sequencing of Orchardgrass under Drought Treatment
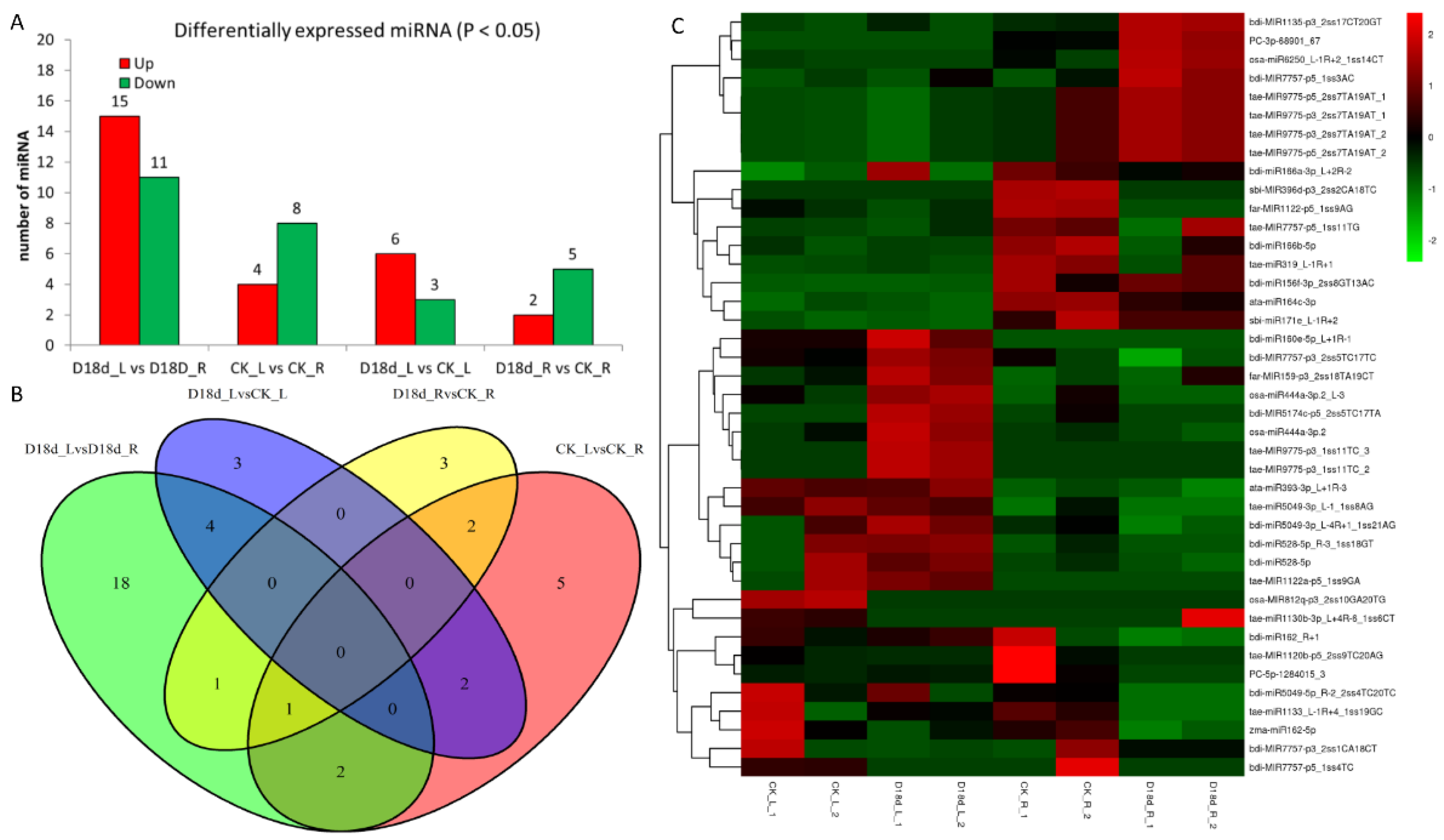
2.2. Construction of Global Small RNAs Library
2.3. Degradome Sequencing Analysis
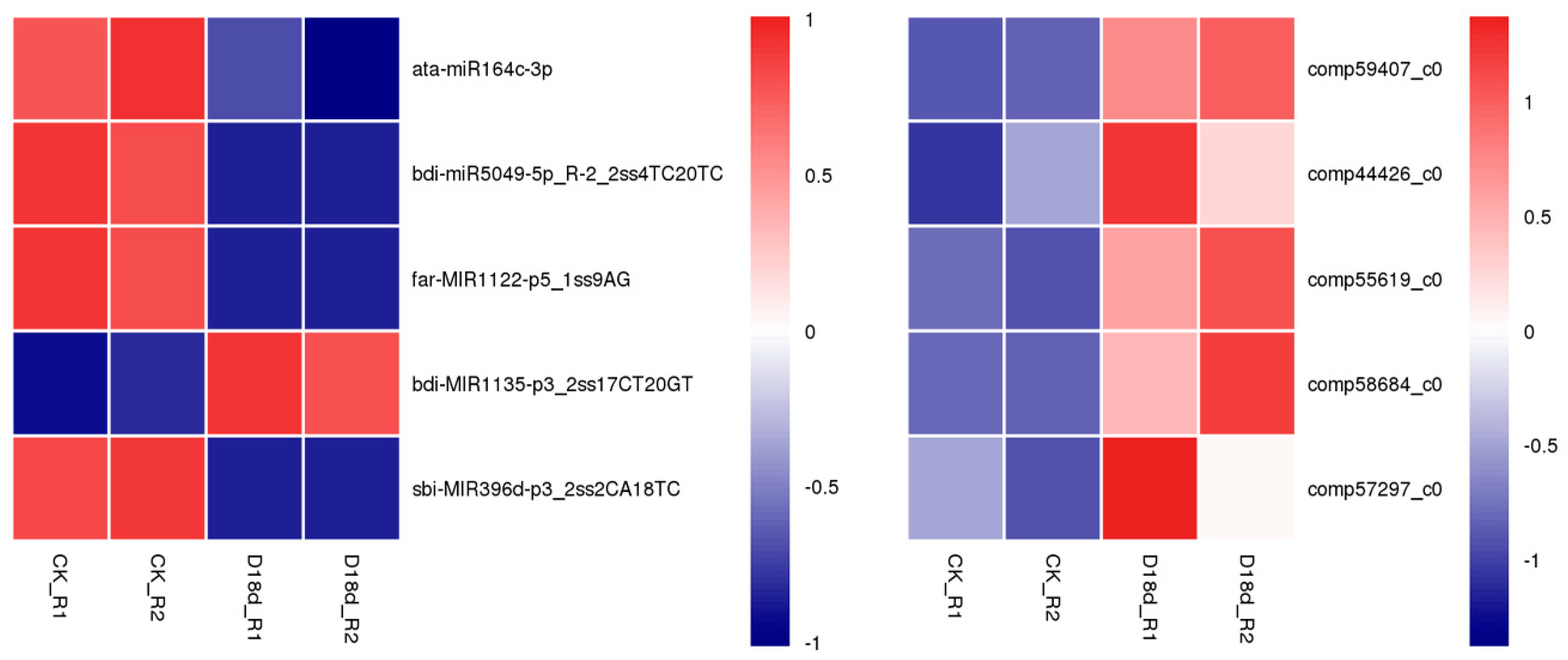
2.4. Correlation Analysis of miRNA–Target Pairs
3. Discussion
3.1. Small RNAs and Their Expression Pattern in Plants under Drought Stress
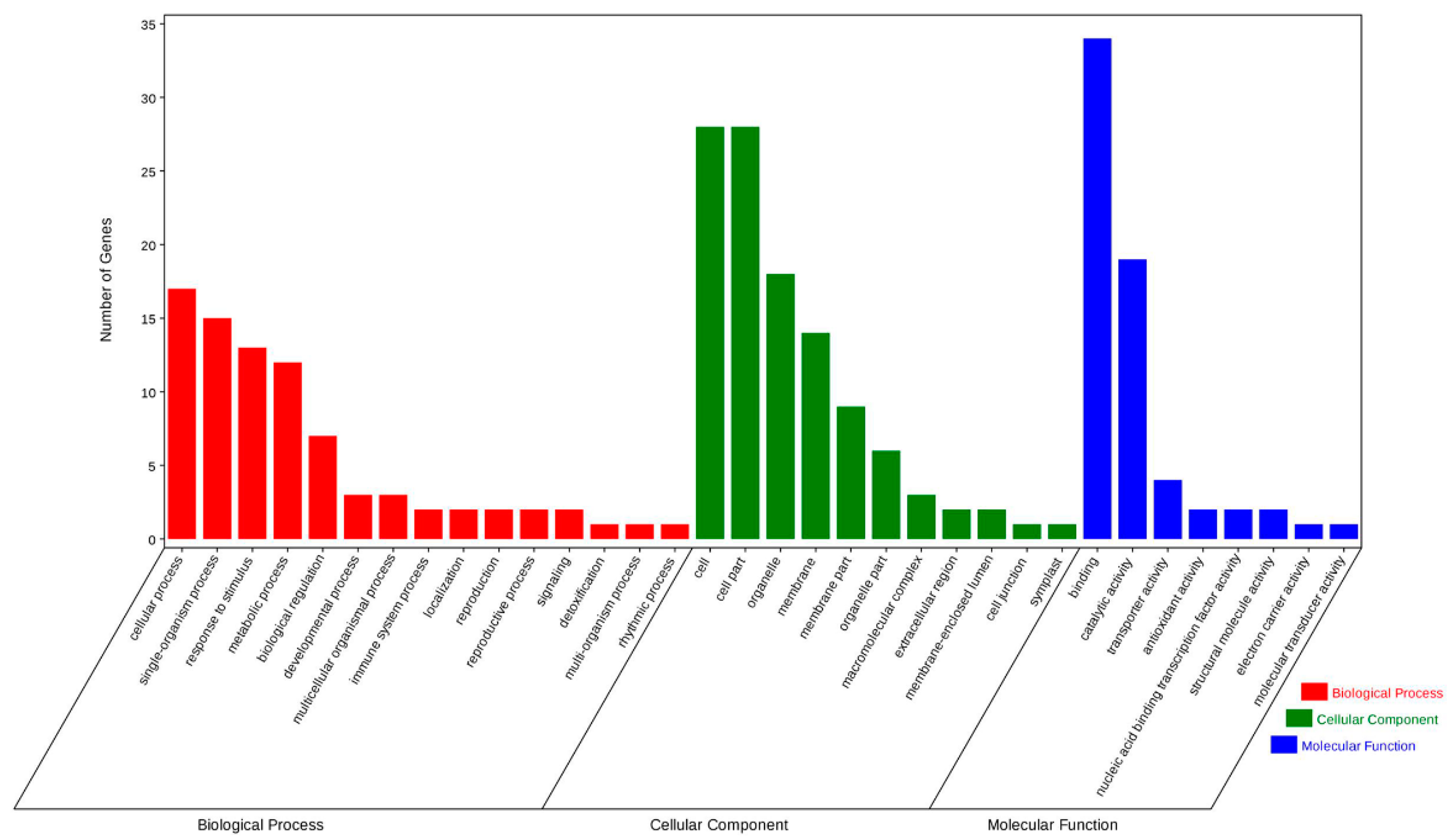
3.2. Target Parsing of Small RNAs in Orchardgrass
3.3. Insights to the Correlation of miRNAs and their Targets
4. Materials and Methods
4.1. Plant Material and Water Deficit Treatment
4.2. Transcriptome Sequencing and De Novo Assembly Analysis
4.3. Small RNA Sequencing and miRNAs Identification
4.4. Degradome Sequencing and Target Identification
4.5. Verification by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GO | Gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| MAPK | Mitogen-activated protein kinase |
| CDPK | Calcium-dependent protein kinase |
| miRNA | microRNA |
| SPL | Squamosa promoter binding protein-like |
| PARE | Parallel analysis of RNA ends |
| RACE | 5′-rapid amplification of cDNA ends |
| DEG | Differently expressed gene |
| AGO | Protein argonaute 2 |
| MYBAS2 | MYB-related protein |
| NAC74 | NAC domain-containing protein 74 |
| PERK2 | Protein kinase R (PKR)-like endoplasmic reticulum kinase 2 |
| STP13 | Sugar transport protein 13 |
| BIPE3 | Luminal-binding protein 3 |
| DEmiR | DIFFERENTLY expressed miRNA |
References
- Haines, A.; Kovats, R.S.; Campbelllendrum, D.; Corvalan, C. Climate change and human health: Impacts, vulnerability, and mitigation. Public Health 2006, 120, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, C.R.; Williams, C.A.; Schaefer, K.; Baldocchi, D.; Black, T.A.; Goldstein, A.H.; Law, B.E.; Oechel, W.C.; Kyaw, T.P.U.; Scott, R.L. Reduction in carbon uptake during turn of the century drought in western North America. Nat. Geosci. 2012, 5, 551–556. [Google Scholar] [CrossRef]
- Touma, D.; Ashfaq, M.; Nayak, M.A.; Kao, S.C.; Diffenbaugh, N.S. A multi-model and multi-index evaluation of drought characteristics in the 21st century. J. Hydrol. 2015, 526, 196–207. [Google Scholar] [CrossRef]
- Schwalm, C.R.; Wrl, A.; Michalak, A.M.; Fisher, J.B.; Biondi, F.; Koch, G.; Litvak, M.; Ogle, K.; Shaw, J.D.; Wolf, A.; et al. Global patterns of drought recovery. Nature 2015, 548, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N.V.; Battisti, D.S.; Beachy, R.N.; Cooper, P.J.; Fischhoff, D.A.; Hodges, C.N.; Knauf, V.C.; Lobell, D.; Mazur, B.J.; Molden, D.; et al. Radically rethinking agriculture for the 21st century. Science 2010, 327, 833–834. [Google Scholar] [CrossRef] [PubMed]
- Trenberth, K.E.; Dai, A.; Schrier, G.V.D.; Jones, P.D.; Barichivich, J.; Briffa, K.R.; Sheffield, J. Global warming and changes in drought. Nat. Clim. Chang. 2014, 4, 17–22. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Nakano, T.; Miyagawa, N.; Ishihama, N.; Yoshioka, M.; Katou, Y.; Yaeno, T.; Shirasu, K.; Yoshioka, H. WRKY transcription factors phosphorylated by MAPK regulate a plant immune NADPH oxidase in Nicotiana benthamiana. Plant Cell 2015, 27, 2645–2663. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, G.; Wang, C.; He, Z.; Lan, X.; Zhang, S.; Lan, H. The bHLH transcription factor CgbHLH001 is a potential interaction partner of CDPK in halophyte Chenopodium glaucum. Sci. Rep. 2017, 7, 8441. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 2009, 2009, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Q. MicroRNA-based biotechnology for plant improvement. J. Cell. Physiol. 2015, 230, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. MiRNA-based drought regulation in wheat. Funct. Integr. Genomics 2016, 16, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Shaik, R.; Ramakrishna, W. Bioinformatic analysis of epigenetic and microRNA mediated regulation of drought responsive genes in rice. PLoS ONE 2012, 7, e49331. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Gao, J.; Liu, M.; Qin, C.; Zhang, W.; Yang, A.; Xia, M.; Zhang, Z.; Shen, Y.; Lin, H.; et al. Genome-wide analysis of water-stress-responsive microRNA expression profile in tobacco roots. Funct. Integr. Genom. 2014, 14, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xing, T.; Yuan, H.; Liu, Z.; Jin, Z.; Zhang, L.; Pei, Y. Hydrogen sulfide improves drought tolerance in Arabidopsis thaliana by microRNA expressions. PLoS ONE 2013, 8, e77047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Z.; Gong, P.; Zhang, J.; Ziaf, K.; Li, H.; Xiao, F.; Ye, Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 2011, 33, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Grativol, C.; Tanurdzic, M.; Carnavalebottino, M.; Vieira, T.; Motta, M.R.; Rojas, C.; Vincentini, R.; Chabregas, S.M.; Hemerly, A.S.; et al. Differential sRNA regulation in leaves and roots of sugarcane under water depletion. PLoS ONE 2014, 9, e93822. [Google Scholar] [CrossRef] [PubMed]
- Hamza, N.B.; Sharma, N.; Tripathi, A.; Sanan-Mishra, N. MicroRNA expression profiles in response to drought stress in Sorghum bicolor. Gene Expr. Patterns 2016, 20, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Gustafson, P.; Langridge, P.; Shi, B.J. Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol. J. 2015, 13, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, M.N.; Sathish, S.; Kavitha, P.; Dachinamoorthy, P.; Ravikesavan, R. Comparative miRNAome analysis revealed numerous conserved and novel drought responsive miRNAs in cotton (Gossypium spp.). Cotton Genom. Genet. 2016, 7, 1–23. [Google Scholar]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017, 258, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Srivastava, A.K.; Pan, Y.; Bai, J.; Fang, J.; Shi, H.; Zhu, J.K. Knockdown of rice microRNA166 confers drought resistance by causing leaf rolling and altering stem xylem development. Plant Physiol. 2018, 176, 2082–2094. [Google Scholar] [CrossRef] [PubMed]
- Pinzón, N.; Li, B.; Martinez, L.; Sergeeva, A.; Presumey, J.; Apparailly, F.; Seitz, H. MicroRNA target prediction programs predict many false positives. Genome Res. 2017, 27, 234–245. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Kan, N.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Addoquaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Lu, C.W.; Shen, B.N.; Guan-Zong, L.; Bowman, J.L.; Arteaga-Vazquez, M.A.; Liu, L.Y.D.; Hong, S.F.; Chu-Fang, L.; Su, G.M.; et al. Identification of miRNAs and their targets in the liverwort Marchantia polymorphaby integrating RNA-seq and degradome analyses. Plant Cell Physiol. 2016, 57, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Arsovski, A.A.; Yu, K.; Wang, A. Genome-Wide investigation using sRNA-seq, degradome-seq and transcriptome-seq reveals regulatory networks of microRNAs and their target genes in doybean during doybean mosaic virus infection. PLoS ONE 2016, 11, e0150582. [Google Scholar] [CrossRef] [PubMed]
- CandarCakir, B.; Arican, E.; Zhang, B. SmallRNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Kole, C. (Ed.) Millets and grasses. In Wild Crop Relatives: Genomic and Breeding Resources; Springer: Berlin/Heidelberg, Germany, 2011; Volume 70, pp. 135–151. [Google Scholar]
- Hulten, E. Flora of Alaska and Neighbouring Territories: A Manual of the Vascular Plants; Stanford University Press: Stanford, CA, USA, 1968. [Google Scholar]
- Feng, G.; Huang, L.; Li, J.; Wang, J.; Xu, L.; Pan, L.; Zhao, X.; Wang, X.; Huang, T.; Zhang, X. Comprehensive transcriptome analysis reveals distinct regulatory programs during vernalization and floral bud development of orchardgrass (Dactylis glomerata L.). BMC Plant Biol. 2017, 17, 216. [Google Scholar] [CrossRef] [PubMed]
- Last, L.; Widmer, F.; Fjellstad, W.; Stoyanova, S.; Kölliker, R. Genetic diversity of natural orchardgrass (Dactylis glomerata L.) populations in three regions in Europe. BMC Genet. 2013, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- Zhouri, L.; Kallida, R.; Shaimi, N.; Barre, P.; Volaire, F.; Gaboun, F.; Fakiri, M. Evaluation of Cocksfoot (Dactylis glomerata L.) Population for Drought Survival and Behavior. Saudi J. Biol. Sci. 2016, in press. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, X.Q.; Pen, Y.; Liang, X.Y.; Huang, L.K.; Chen, L.Z.; Li, Z.; Ma, Y.M. Effects of drought stress on the root growth and photosynthetic characters of Dactylis glomerata seedlings. Ying Yong Sheng Tai Xue Bao 2013, 24, 2763–2769. (In Chinese) [Google Scholar] [PubMed]
- Ji, Y.; Zhang, X.; Peng, Y.; Liang, X.; Huang, L.; Ma, X.; Ma, Y. Effects of drought stress on lipid peroxidation, osmotic adjustment and activities of protective enzymes in the roots and leaves of orchardgrass. Acta Prataculture Sin. 2014, 23, 144–151. (In Chinese) [Google Scholar]
- Ding, Y.; Tao, Y.; Zhu, C. Emerging roles of microRNAs in the mediation of drought stress response in plants. J. Exp. Bot. 2013, 64, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Kakimoto, M.; Sakuma, Y.; Maruyama, K.; Osakabe, Y.; Tran, L.S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J. 2007, 50, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, Z.; Liu, Y.; Zhang, H.; Zhang, M.; Liu, Q.; Hong, X.; Zhu, J.K.; Gong, Z. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. 2010, 63, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.N.; Chen, B.; Lu, G.J.; Han, B. Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem. Biophys. Res. Commun. 2009, 379, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.J.; Xu, S.C.; Liu, N.; Zhang, G.W.; Hu, Q.Z.; Gong, Y.M. Soybean TCP transcription factors: Evolution, classification, protein interaction and stress and hormone responsiveness. Plant Physiol. Biochem. 2018, 127, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xu, Y.; Ma, Q.; Xu, W.; Wang, T.; Xue, Y.; Chong, K. Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic Arabidopsis. Plant Physiol. 2007, 143, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.S.; Liang, D.; Shuai, P.; Xia, X.L.; Yin, W.L. The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J. Exp. Bot. 2010, 61, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Kantar, M.; Lucas, S.J.; Budak, H. MiRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 2011, 233, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Pasini, L.; Bergonti, M.; Fracasso, A.; Marocco, A.; Amaducci, S. Microarray analysis of differentially expressed mRNAs and miRNAs in young leaves of sorghum under dry-down conditions. J. Plant Physiol. 2014, 171, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, J.; Wang, Z.; Wen, Y.; Wang, J.; He, W.; Liu, B.; Si, H.; Wang, D. Identification of novel and conserved microRNAs related to drought stress in potato by deep sequencing. PLoS ONE 2014, 9, e95489. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.V.; Ellison, N.W. Dactylis. Wild Crop Relat. Genomic Breed. Resour. 2011, 73–87. [Google Scholar] [CrossRef]
- Chen, Z.H.; Bao, M.L.; Sun, Y.Z.; Yang, Y.J.; Xu, X.H.; Wang, J.H.; Han, N.; Bian, H.W.; Zhu, M.Y. Regulation of auxin response by miR393-targeted is involved in normal development in Arabidopsis. Plant Mol. Biol. 2011, 77, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Windels, D.; Vazquez, F. MiR393: Integrator of environmental cues in auxin signaling? Plant Signal. Behav. 2011, 6, 1672–1675. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Pan, X.; Li, Y.; Cai, H.; Ji, W.; Chen, Q.; Zhu, Y. Osa-MIR393: A salinity- and alkaline stress-related microRNA gene. Mol. Biol. Rep. 2011, 38, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, L.; Han, N.; Hu, J.; Yang, Y.; Xiang, T.; Zhang, X.; Wang, L. Overexpression of a miR393-resistant form of transport inhibitor response protein 1 (mTIR1) enhances salt tolerance by increased osmoregulation and Na+ exclusion in Arabidopsis thaliana. Plant Cell Physiol. 2015, 56, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Bian, H.; Zeng, Z.; Hou, N.; Shi, B.; Wang, J.; Zhu, M.; Han, N. MiR393-mediated auxin signaling regulation is involved in root elongation inhibition in response to toxic aluminum stress in barley. Plant Cell Physiol. 2017, 58, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xie, K.; Xiong, L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 2014, 65, 2119–2135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, L.; Sha, T.; Liu, J.; Zhang, H.; Hui, Z.; Jia, G.; Diao, X. Combined small RNA and degradome sequencing to identify miRNAs and their targets in response to drought in foxtail millet. BMC Genet. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Kansal, S.; Devi, R.M.; Balyan, S.C.; Arora, M.K.; Singh, A.K.; Mathur, S.; Raghuvanshi, S. Unique miRNome during anthesis in drought-tolerant indica rice var. Nagina 22. Planta 2015, 241, 1543–1559. [Google Scholar] [CrossRef] [PubMed]
- Phookaew, P.; Netrphan, S.; Sojikul, P.; Narangajavana, J. Involvement of miR164- and miR167-mediated target gene expressions in responses to water deficit in cassava. Biol. Plant. 2014, 58, 469–478. [Google Scholar] [CrossRef]
- Song, C.P.; Agarwal, M.; Ohta, M.; Guo, Y.; Halfter, U.; Zhu, J.K. Role of an Arabidopsis AP2/EREBP-type transcriptional repressor in abscisic acid and drought stress responses. Plant Cell 2005, 17, 2384–2396. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tang, N.; Du, H.; Ye, H.; Xiong, L. Characterization of OsbZIP23 as a key player of the basic leucine zipper transcription factor family for conferring abscisic acid sensitivity and salinity and drought tolerance in rice. Plant Physiol. 2008, 148, 1938–1952. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.M.; Ali, M.; Feng, X.; Li, X. The essence of NAC gene family to the cultivation of drought-resistant soybean (Glycine max L. Merr.) cultivars. BMC Plant Biol. 2017, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Yamaguchishinozaki, K.; Urao, T.; Iwasaki, T.; Hosokawa, D.; Shinozaki, K. Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 1997, 9, 1859–1868. [Google Scholar] [PubMed]
- Cho, E.K.; Hong, C.B. Over-expression of tobacco NtHSP70-1 contributes to drought-stress tolerance in plants. Plant Cell Rep. 2006, 25, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Maruyama, K.; Osakabe, Y.; Qin, F.; Seki, M.; Shinozaki, K.; Yamaguchishinozaki, K. Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 2006, 18, 1292–1309. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. MiRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.K.; Choi, Y.J. A nuclear-localized HSP70 confers thermoprotective activity and drought-stress tolerance on plants. Biotechnol. Lett. 2009, 31, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.Q.; Jiang, S.L.; Huang, C.M.; Deng, Z.N.; Wu, K.C.; Xu, L.; Luo, H.B.; Lu, Z.; Wei, Y.W. Cloning of ATP binding protein gene in sugarcane and its expression characteristics under drought stress. J. South. Agric. 2017, 48, 1537–1547. (In Chinese) [Google Scholar] [CrossRef]
- Buriani, G.; Mancini, C.; Benvenuto, E.; Baschieri, S. Plant heat shock protein 70 as carrier for immunization against a plant-expressed reporter antigen. Transgenic Res. 2011, 20, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Zhao, M.; Gao, W.; Sun, S.; Li, W.X. MicroRNA/microRNA* complementarity is important for the regulation pattern of NFYA5 by miR169 under dehydration shock in Arabidopsis. Plant J. 2017, 91, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.H.; Ponnuchamy, M.; Kumar, J.; Reddy, N.R.R. Exploring drought stress-regulated genes in senna (Cassia angustifolia Vahl.): A transcriptomic approach. Funct. Integr. Genomics 2017, 17, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, L.L.; Tanabe, N.; Lymperopoulos, P.; Khan, N.Z.; Rodermel, S.R.; Aronsson, H.; Clarke, A.K. Quantitative analysis of the chloroplast molecular chaperone ClpC/Hsp93 in Arabidopsis reveals new insights into its localization, interaction with the Clp proteolytic core, and functional importance. J. Biol. Chem. 2014, 289, 11318–11330. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Sun, D.; Wang, C.; Li, Y.; Guo, T. Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Physiol. Biochem. 2014, 80, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R.A.; Bi, Y.M.; Kant, S.; Rothstein, S.J. Over-expression of STP13, a hexose transporter, improves plant growth and nitrogen use in Arabidopsis thaliana seedlings. Plant Cell Environ. 2009, 32, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Nørholm, M.H.H.; Nour-Eldin, H.H.; Brodersen, P.; Mundy, J.; Halkier, B.A. Expression of the Arabidopsis high-affinity hexose transporter STP13 correlates with programmed cell death. FEBS Lett. 2006, 580, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, P.; Gaillard, C.; Veillet, F.; Verbeke, J.; Lemoine, R.; Coutosthévenot, P.; La, C.S. Expression of Arabidopsis sugar transport protein STP13 differentially affects glucose transport activity and basal resistance to Botrytis cinerea. Plant Mol. Biol. 2014, 85, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Mostajeran, A.; Rahimieichi, V. Effects of drought stress on growth and yield of rice (Oryza sativa L.) cultivars and accumulation of proline and soluble sugars in sheath and blades of their different ages leaves. Am. Eurasian J. Agric. Environ. Sci. 2009, 5, 264–272. [Google Scholar]
- Ji, Y.; Zhang, X.; Peng, Y.; Huang, L.; Liang, X.; Wang, K.; Yin, G.; Zhao, X. Osmolyte accumulation, antioxidant enzyme activities and gene expression patterns in leaves of orchardgrass during drought stress and recovery. Grassl. Sci. 2015, 60, 131–141. [Google Scholar] [CrossRef]
- Schachtman, D.P.; Goodger, J.Q.D. Chemical root to shoot signaling under drought. Trends Plant Sci. 2008, 13, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17. [Google Scholar] [CrossRef]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef] [PubMed]
- Addoquaye, C.; Snyder, J.A.; Yong, B.P.; Li, Y.F.; Sunkar, R.; Axtell, M.J. Sliced microRNA targets and precise loop-first processing of MIR319 hairpins revealed by analysis of the Physcomitrella patens degradome. RNA 2009, 15, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.R.; Coruh, C.; Axtell, M.J. Arabidopsis lyrata small RNAs: Transient miRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell 2010, 22, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yin, L.; Ying, Q.; Song, H.; Xue, D.; Lai, T.; Xu, M.; Shen, B.; Wang, H.; Shi, X. High-throughput sequencing and degradome analysis identify miRNAs and their targets involved in fruit senescence of Fragaria ananassa. PLoS ONE 2013, 8, e70959. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, L.; Yuan, D.; Lindsey, K.; Zhang, X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J. Exp. Bot. 2013, 64, 1521–1536. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of miRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.E.; Olsson, N.; Schlosser, J.; Peng, F.; Lund, S.T. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006, 6, 27. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | miR_Name | Regulation | Gene_ID | Gene Annotation | Regulation |
|---|---|---|---|---|---|
| CKL vs. CKR | ata-miR164c-3p | down | comp50213_c0 | - | down |
| ata-miR164c-3p | down | comp59407_c0 | Heat shock 70kda protein 5 | down | |
| PC-3p-68901_67 | down | comp50628_c0 | 11beta-hydroxysteroid dehydrogenase | down | |
| sbi-MIR396d-p3_2ss2CA18TC | down | comp57297_c0 | Proton-dependent oligopeptide transporter | down | |
| tae-miR319_L-1R+1 | down | comp59014_c0 | - | down | |
| ata-miR164c-3p | down | comp59034_c0 | - | down | |
| bdi-miR160e-5p_L+1R-1 | up | comp42625_c0 | Peroxidase | down | |
| far-MIR1122-p5_1ss9AG | down | comp58564_c0 | ATP-dependent Clp protease ATP-binding subunit clpc | Up | |
| tae-miR1130b-3p_L+4R-6_1ss6CT | up | comp59143_c0 | - | down | |
| D18dR vs. CKR | ata-miR164c-3p | down | comp59407_c0 | Heat shock 70kda protein 5 | up |
| bdi-miR5049-5p_R-2_2ss4TC20TC | down | comp44426_c0 | Naringenin 3-dioxygenase | up | |
| bdi-miR5049-5p_R-2_2ss4TC20TC | down | comp55619_c0 | Naringenin 3-dioxygenase | up | |
| far-MIR1122-p5_1ss9AG | down | comp55619_c0 | Proton-dependent oligopeptide transporter | up | |
| far-MIR1122-p5_1ss9AG | down | comp58684_c0 | - | up | |
| bdi-MIR1135-p3_2ss17CT20GT | up | comp58684_c0 | - | up | |
| sbi-MIR396d-p3_2ss2CA18TC | down | comp57297_c0 | - | up |
| Comparison | KEGG ID | Gene_ID | Regulation | KEGG_Name |
|---|---|---|---|---|
| CK_L vs. CK_R | ko00360 | comp42625_c0 | down | Phenylalanine metabolism |
| ko00680 | comp42625_c0 | down | Methane metabolism | |
| ko00940 | comp42625_c0 | down | Phenylpropanoid biosynthesis | |
| D18d_R vs. CK_R | ko03060 | comp59407_c0 | up | Protein export |
| ko00941 | comp55619_c0 | up | Flavonoid biosynthesis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Chen, P.; Chen, J.; Pennerman, K.K.; Liang, X.; Yan, H.; Zhou, S.; Feng, G.; Wang, C.; Yin, G.; et al. Combinations of Small RNA, RNA, and Degradome Sequencing Uncovers the Expression Pattern of microRNA–mRNA Pairs Adapting to Drought Stress in Leaf and Root of Dactylis glomerata L. Int. J. Mol. Sci. 2018, 19, 3114. https://doi.org/10.3390/ijms19103114
Ji Y, Chen P, Chen J, Pennerman KK, Liang X, Yan H, Zhou S, Feng G, Wang C, Yin G, et al. Combinations of Small RNA, RNA, and Degradome Sequencing Uncovers the Expression Pattern of microRNA–mRNA Pairs Adapting to Drought Stress in Leaf and Root of Dactylis glomerata L. International Journal of Molecular Sciences. 2018; 19(10):3114. https://doi.org/10.3390/ijms19103114
Chicago/Turabian StyleJi, Yang, Peilin Chen, Jing Chen, Kayla K. Pennerman, Xiaoyu Liang, Haidong Yan, Sifan Zhou, Guangyan Feng, Chengran Wang, Guohua Yin, and et al. 2018. "Combinations of Small RNA, RNA, and Degradome Sequencing Uncovers the Expression Pattern of microRNA–mRNA Pairs Adapting to Drought Stress in Leaf and Root of Dactylis glomerata L." International Journal of Molecular Sciences 19, no. 10: 3114. https://doi.org/10.3390/ijms19103114
APA StyleJi, Y., Chen, P., Chen, J., Pennerman, K. K., Liang, X., Yan, H., Zhou, S., Feng, G., Wang, C., Yin, G., Zhang, X., Hu, Y., & Huang, L. (2018). Combinations of Small RNA, RNA, and Degradome Sequencing Uncovers the Expression Pattern of microRNA–mRNA Pairs Adapting to Drought Stress in Leaf and Root of Dactylis glomerata L. International Journal of Molecular Sciences, 19(10), 3114. https://doi.org/10.3390/ijms19103114