Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity


Abstract
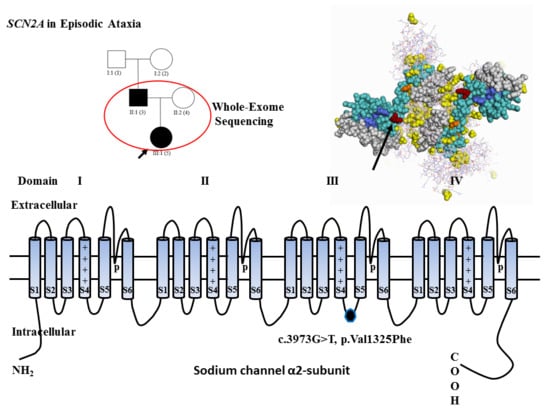
1. Introduction
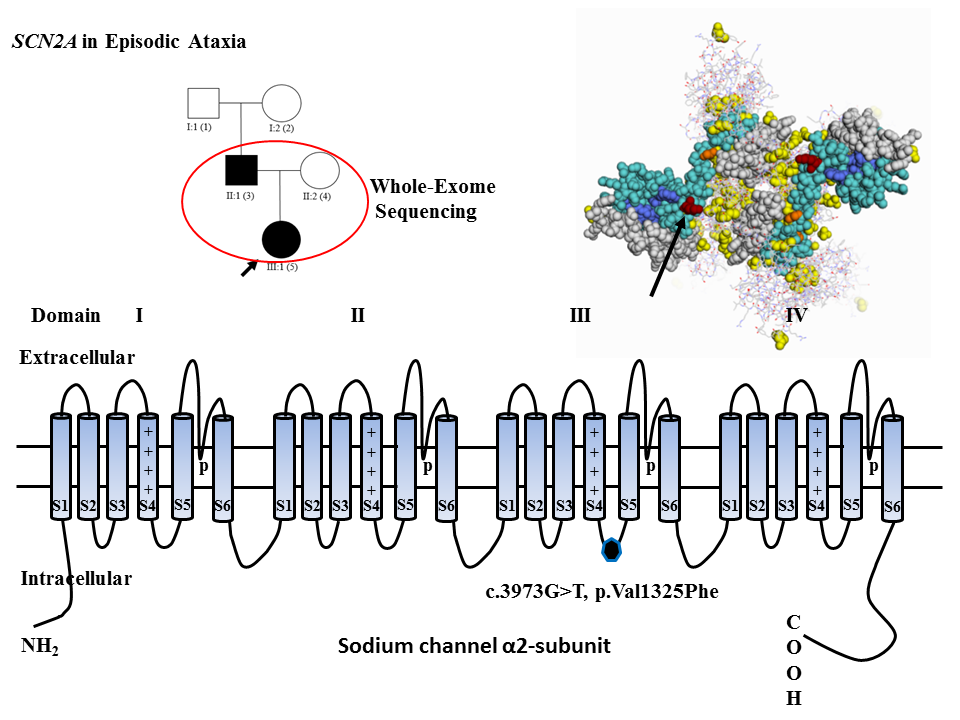
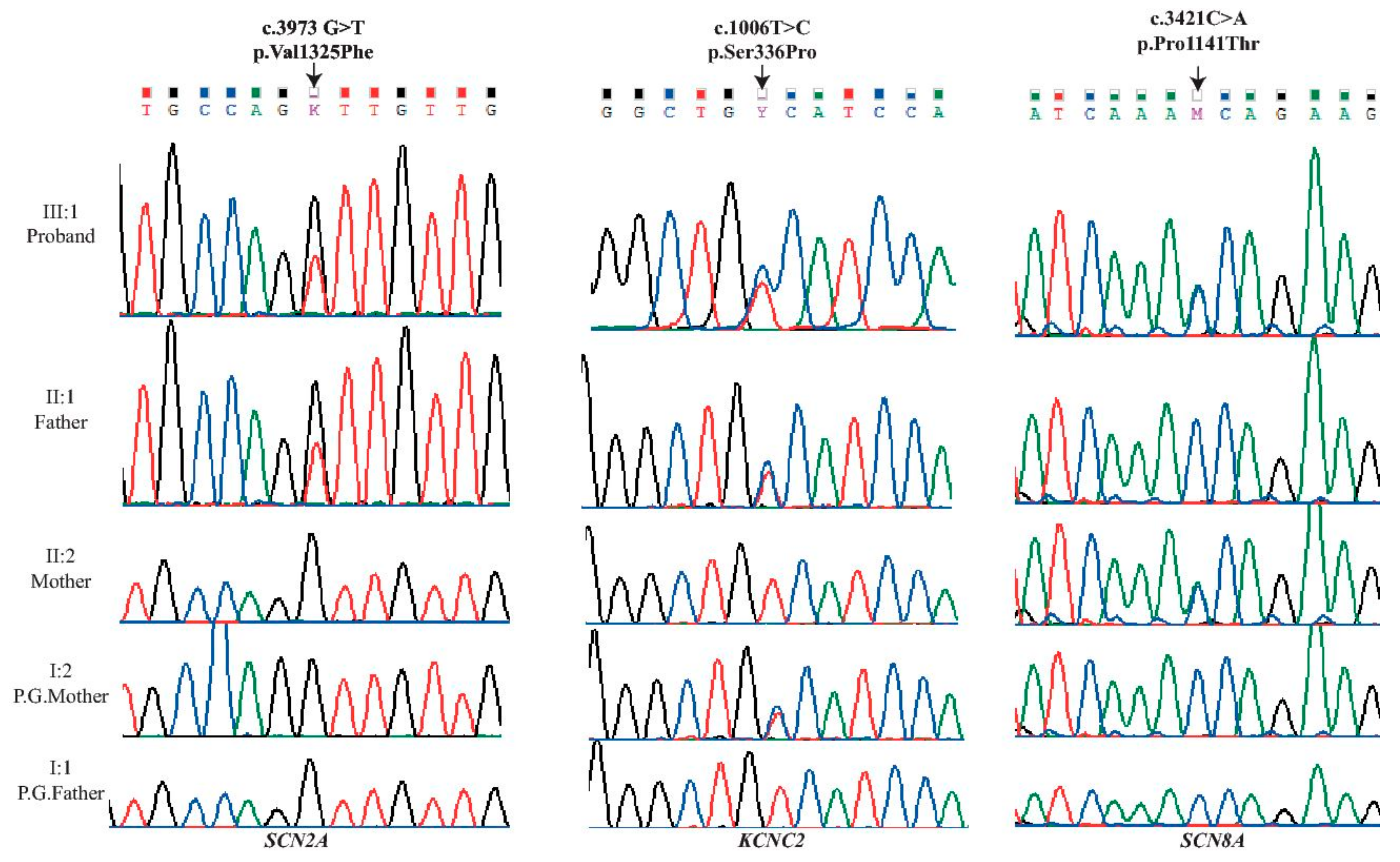
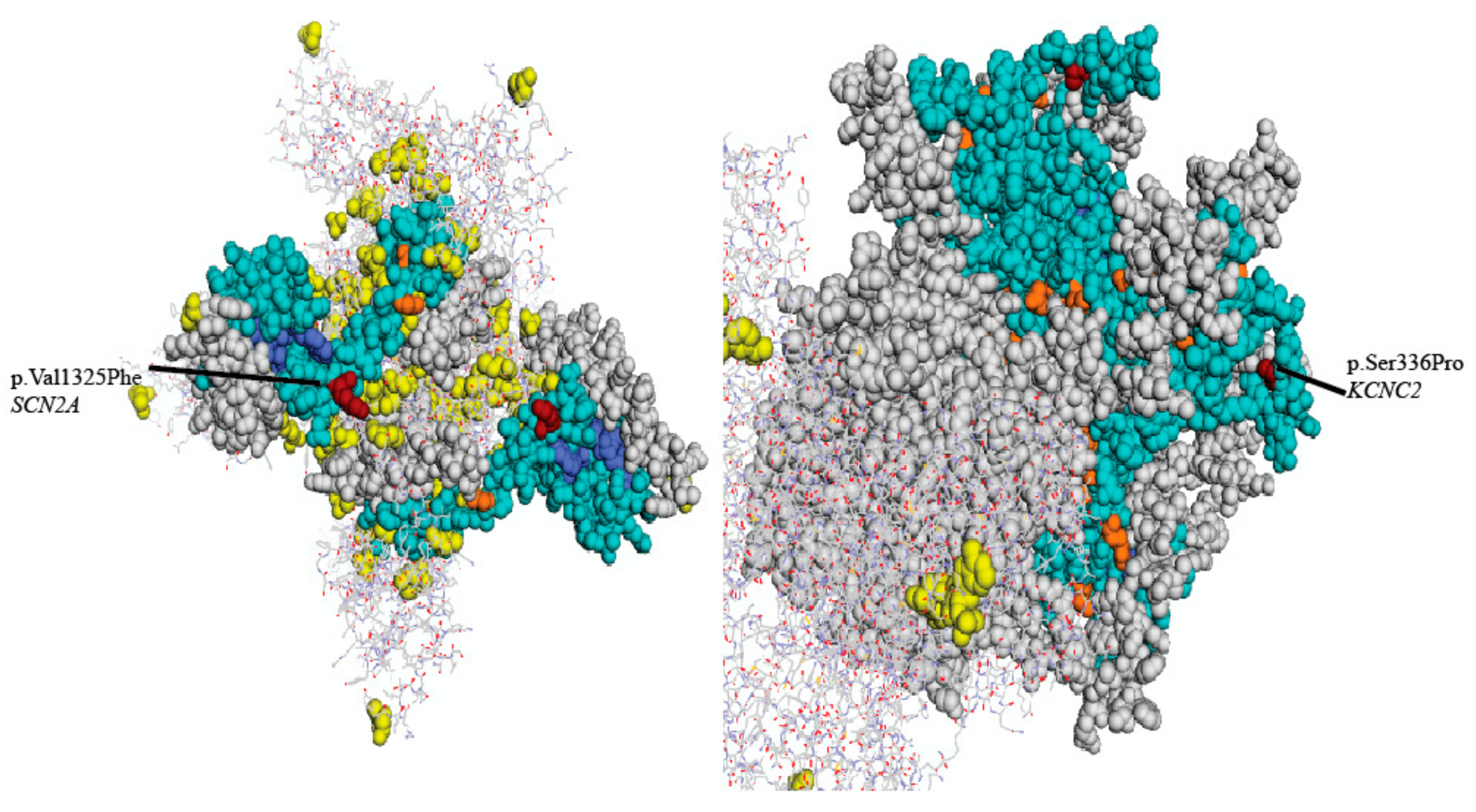
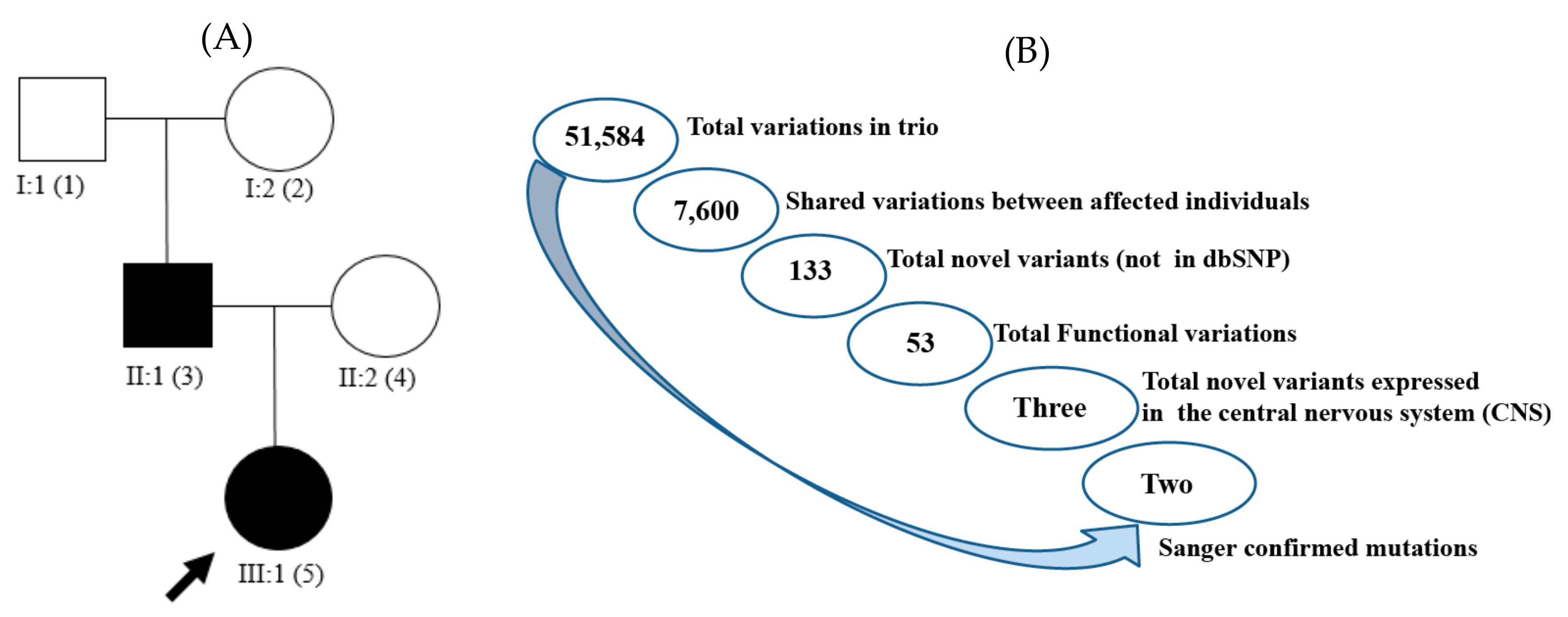
2. Results
3. Discussion
4. Materials and Methods
4.1. Subject
4.2. Clinical Context
4.3. Whole-Exome Sequencing
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jen, J.C.; Graves, T.D.; Hess, E.J.; Hanna, M.G.; Griggs, R.C.; Baloh, R.W.; The CINCH Investigators. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain 2007, 130, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Maksemous, N.; Roy, B.; Smith, R.A.; Griffiths, L.R. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol. Genet. Genomic Med. 2016, 4, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.; Luty, J.; Kwak, M.; Allen, L.; Magenis, R.E.; Mandel, G. Localization of a human brain sodium channel gene (SCN2A) to chromosome 2. Genomics 1989, 5, 204–208. [Google Scholar] [CrossRef]
- Ben-Shalom, R.; Caroline, M.K.; Kiara, N.B.; Joon, Y.A.; Stephan, J.S.; Kevin, J.B. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 2017, 82, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Kubota, T.; Negoro, T.; Saitoh, M.; Mizuguchi, M.; Ihara, Y.; Ishii, A.; Hirose, S. A case of recurrent encephalopathy with SCN2A missense mutation. Brain Dev. 2015, 37, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Leach, E.L.; van Karnebeek, C.D.; Townsend, K.N.; Tarailo-Graovac, M.; Hukin, J.; Gibson, W.T. Episodic ataxia associated with a de novo SCN2A mutation. Eur. J. Paediatr. Neurol. 2016, 20, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Anttonen, A.K.; Liukkonen, E.; Gaily, E.; Maljevic, S.; Schubert, S.; Bellan-Koch, A.; Petrou, S.; Ahonen, V.E.; Lerche, H.; et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 2010, 75, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Hahn, A.; Bast, T.; Müller, S.; Löffler, H.; Maljevic, S.; Gaily, E.; Prehl, I.; Biskup, S.; Joensuu, T.; et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 2016, 263, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Friedland, D.R.; Eernisse, R.; Popper, P. Potassium channel gene expression in the rat cochlear nucleus. Hear. Res. 2007, 228, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, S.; Roberts, J.; Koltzenburg, M.; Hanna, M.G.; Stewart, H. Deletion of chromosome 12q21 affecting KCNC2 and ATXN7L3B in a family with neurodevelopmental delay and ataxia. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Rea, R.; Zuberi, S.M.; Youroukos, S.; Panayiotopoulos, C.P.; Liguori, R.; Avoni, P.; McWilliam, R.C.; Stephenson, J.B.; Hanna, M.G.; et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 2000, 48, 647–656. [Google Scholar] [CrossRef]
- Lubbers, W.J.; Brunt, E.R.; Scheffer, H.; Litt, M.; Stulp, R.; Browne, D.L.; van Weerden, T.W. Hereditary myokymia and paroxysmal ataxia linked to chromosome 12 is responsive to acetazolamide. J. Neurol. Neurosurg. Psychiatry 1995, 59, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Moxley, R.T., III; Lafrance, R.A.; McQuillen, J. Hereditary paroxysmal ataxia: Response to acetazolamide. Neurology 1978, 28, 1259–1264. [Google Scholar]
- Larsen, J.; Carvill, G.L.; Gardella, E.; Kluger, G.; Schmiedel, G.; Barisic, N.; Depienne, C.; Brilstra, E.; Mang, Y.; Nielsen, J.E.; et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015, 84, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bagnasco, I.; Dassi, P.; Blé, R.; Vigliano, P. A relatively mild phenotype associated with mutation of SCN8A. Seizure 2018, 56, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Becker, F.; Møller, R.S.; Schubert, J.; Lemke, J.R.; Larsen, L.H.; Eiberg, H.; Nothnagel, M.; Thiele, H.; Altmüller, J.; et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann. Neurol. 2016, 79, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Wagnon, J.L.; Meisler, M.H. Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Front. Neurol. 2015, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.E.; Meisler, M.H. Sodium channel SCN8A (Nav1.6): Properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 2013, 4, 213. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.G.; Fann, Y.C.; et al. StemCellDB: The human pluripotent stem cell database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, F.; Jurkat-Rott, K. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 1999, 79, 1317–1372. [Google Scholar] [CrossRef] [PubMed]
- Klassen, T.; Davis, C.; Goldman, A.; Burgess, D.; Chen, T.; Wheeler, D.; McPherson, J.; Bourquin, T.; Lewis, L.; Villasana, D.; et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011, 145, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Gene | Exon | Protein | Coding | SIFT | Polyphen | Mutation Taster | PROVEAN |
|---|---|---|---|---|---|---|---|---|
| chr2:166231195 | SCN2A | 21 | p.Val1325Phe | c.3973G>T | D (0.0) | D (1) | D | D (−4.5) |
| chr12:75444779 | KCNC2 | 3 | p.Ser336Pro | c.1006T>C | D (0.023) | D (0.994) | D | D (−3.65) |
| chr12:52163700 | SCN8A | 18 | p.Pro1141Thr | c.3421C>A | D (0.046) | D(0.957) | D | D (−2.68) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksemous, N.; Smith, R.A.; Sutherland, H.G.; Sampaio, H.; Griffiths, L.R. Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. Int. J. Mol. Sci. 2018, 19, 3113. https://doi.org/10.3390/ijms19103113
Maksemous N, Smith RA, Sutherland HG, Sampaio H, Griffiths LR. Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. International Journal of Molecular Sciences. 2018; 19(10):3113. https://doi.org/10.3390/ijms19103113
Chicago/Turabian StyleMaksemous, Neven, Robert A. Smith, Heidi G. Sutherland, Hugo Sampaio, and Lyn R. Griffiths. 2018. "Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity" International Journal of Molecular Sciences 19, no. 10: 3113. https://doi.org/10.3390/ijms19103113
APA StyleMaksemous, N., Smith, R. A., Sutherland, H. G., Sampaio, H., & Griffiths, L. R. (2018). Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. International Journal of Molecular Sciences, 19(10), 3113. https://doi.org/10.3390/ijms19103113