A Protein Intrinsic Disorder Approach for Characterising Differentially Expressed Genes in Transcriptome Data: Analysis of Cell-Adhesion Regulated Gene Expression in Lymphoma Cells

Abstract
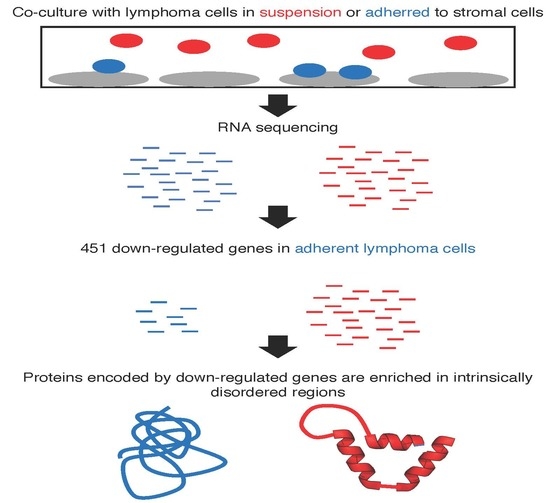
1. Introduction
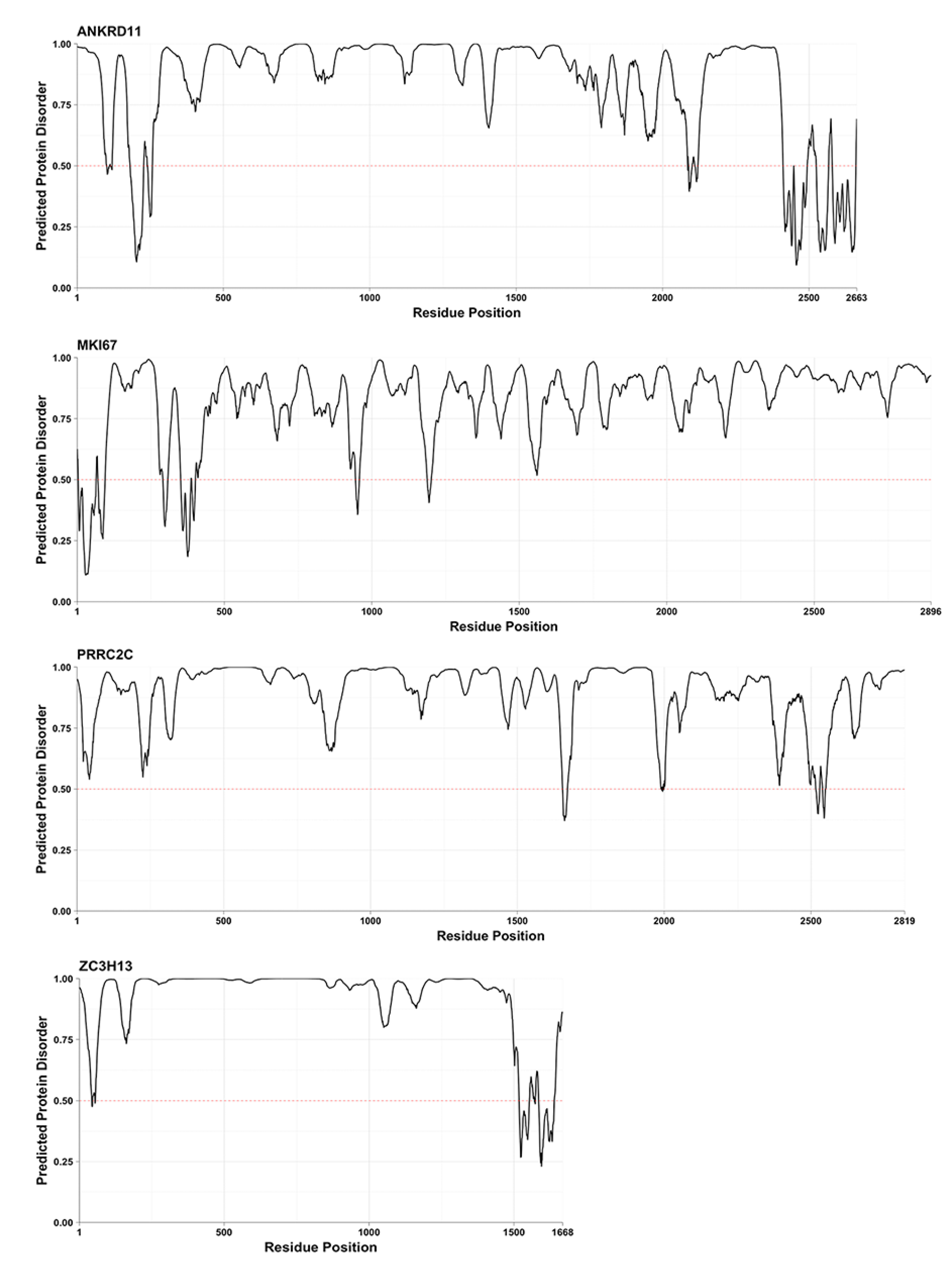
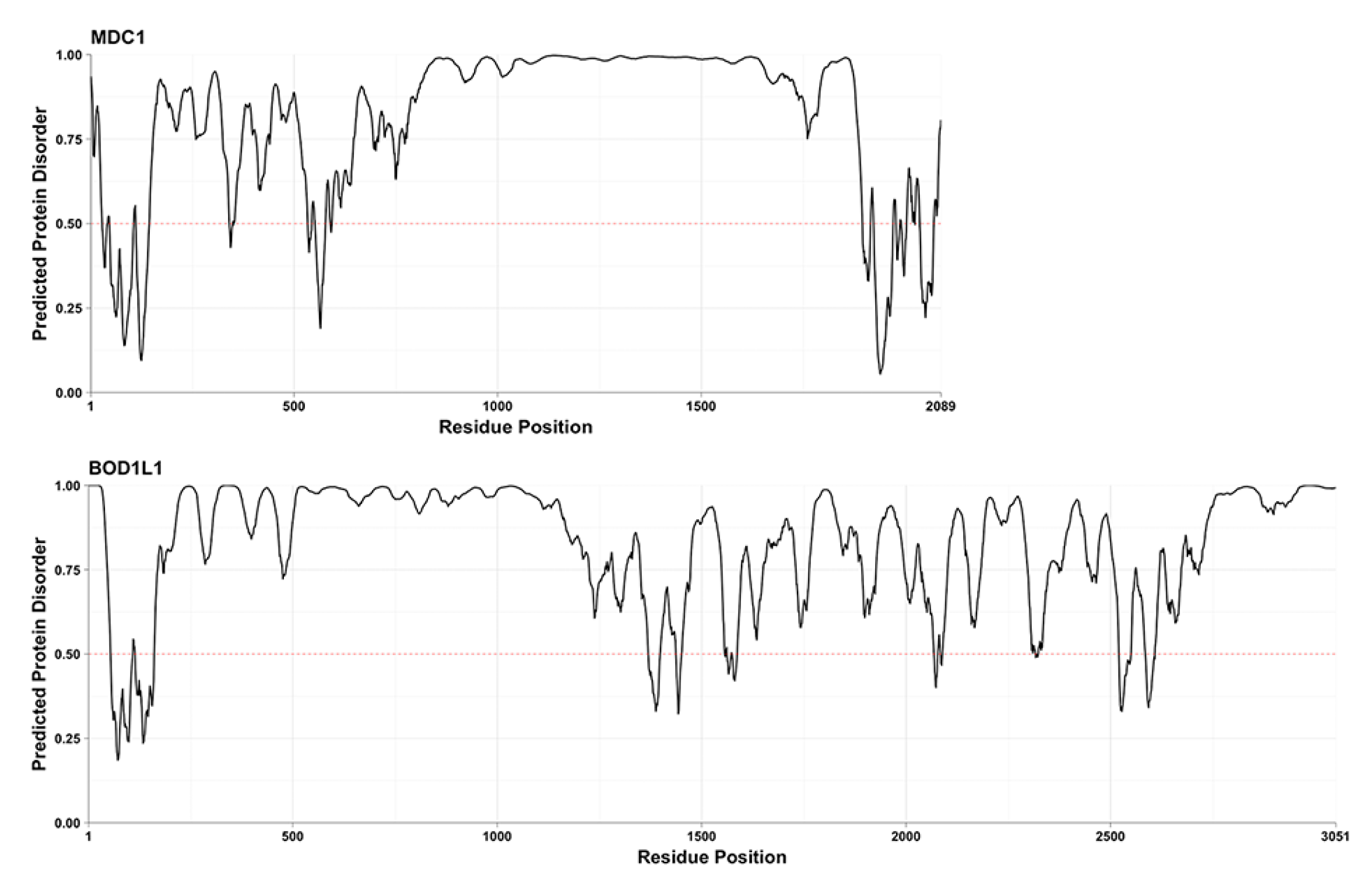
2. Results
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Data Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Sugase, K.; Dyson, H.J.; Wright, P.E. Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc. Natl. Acad. Sci. USA 2015, 112, 9614–9619. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the d2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Marasco, D.; Scognamiglio, P.L. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int. J. Mol. Sci. 2015, 16, 7394–7412. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, G.; Henriksson, J.; Sander, B.; Wright, A.P. Mixed-species rnaseq analysis of human lymphoma cells adhering to mouse stromal cells identifies a core gene set that is also differentially expressed in the lymph node microenvironment of mantle cell lymphoma and chronic lymphocytic leukemia patients. Haematologica 2018, 103, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of b-cell activating factor and activation of the canonical and non-canonical nuclear factor kappab pathways. Haematologica 2012, 97, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Rossi, P.; Saio, T.; Kalodimos, C.G. Structural basis for the antifolding activity of a molecular chaperone. Nature 2016, 537, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Poletto, M.; Vitagliano, L.; Tell, G.; Marasco, D. G-quadruplex DNA recognition by nucleophosmin: New insights from protein dissection. Biochim. Biophys. Acta 2014, 1840, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, K.; Lebrun, P.; Tompa, P. To be disordered or not to be disordered: Is that still a question for proteins in the cell? Cell. Mol. Life Sci. 2017, 74, 3185–3204. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.A.; Forman-Kay, J.D. Liquid-liquid phase separation in cellular signaling systems. Curr. Opin. Struct. Biol. 2016, 41, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically disordered proteome of human membrane-less organelles. Proteomics 2018, 18, e1700193. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell. Biol. 2014, 206, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian wtap is a regulatory subunit of the rna n6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Kalousi, A.; Furst, A.; Koch, M.; Fischer, B.; Soutoglou, E. Tankyrases promote homologous recombination and check point activation in response to dsbs. PLoS Genet. 2016, 12, e1005791. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.G.; Earnshaw, W.C. Ki-67 and the chromosome periphery compartment in mitosis. Trends Cell. Biol. 2017, 27, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting liquid phases underlie nucleolar subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Katzenberger, T.; Petzoldt, C.; Holler, S.; Mader, U.; Kalla, J.; Adam, P.; Ott, M.M.; Muller-Hermelink, H.K.; Rosenwald, A.; Ott, G. The ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood 2006, 107, 3407. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: A more stochastic view of protein function. Adv. Exp. Med. Biol. 2012, 725, 1–14. [Google Scholar] [PubMed]
- Lee, S.H.; Kim, D.H.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Lee, C.; Han, K.H. Understanding pre-structured motifs (presmos) in intrinsically unfolded proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Li, S.; Chan, E.K. Function of gw182 and gw bodies in sirna and mirna pathways. Adv. Exp. Med. Biol. 2013, 768, 71–96. [Google Scholar] [PubMed]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse marrow stromal cells protect cll cells from spontaneous and drug-induced apoptosis: Development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Lwin, T.; Hazlehurst, L.A.; Dessureault, S.; Lai, R.; Bai, W.; Sotomayor, E.; Moscinski, L.C.; Dalton, W.S.; Tao, J. Cell adhesion induces p27kip1-associated cell-cycle arrest through down-regulation of the scfskp2 ubiquitin ligase pathway in mantle-cell and other non-hodgkin b-cell lymphomas. Blood 2007, 110, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Camilloni, C.; Fariselli, P.; Vendruscolo, M. The s2d method: Simultaneous sequence-based prediction of the statistical populations of ordered and disordered regions in proteins. J. Mol. Biol. 2015, 427, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. The dynamine webserver: Predicting protein dynamics from sequence. Nucleic Acids Res. 2014, 42, W264–W270. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. Iupred2a: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Morfpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-pi contacts are an overlooked protein feature relevant to phase separation. eLife 2018, 7, e31486. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.-H.; Lee, C.; Kang, G.; Park, M.; Ham, S. Structural and thermodynamic investigations on the aggregation and folding of acylphosphatase by molecular dynamics simulations and solvation free energy analysis. J. Am. Chem. Soc. 2011, 133, 7075–7083. [Google Scholar] [CrossRef] [PubMed]
- Konrat, R. The protein meta-structure: A novel concept for chemical and molecular biology. Cell. Mol. Life Sci. 2009, 66, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztányi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2p2: Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Micetic, I.; Camilloni, C.; Davey, N.; Dosztanyi, Z.; Meszaros, B.; Monzon, A.M.; et al. Mobidb 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018, 46, D471–D476. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008. [Google Scholar]
- Dowle, M.; Srinivasan, M. Data.Table: Extension of ‘Data.Frame’. R Package Version 1.10.4-3. 2017. Available online: https://cran.r-project.org/package=data.table (accessed on 4 October 2018).
- Gross, J.; Ligges, U. Nortest: Tests for Normality. R Package Version 1.0-4. 2015. Available online: https://cran.r-project.org/package=nortest (accessed on 4 October 2018).
- Ren, K.; Russell, K. Formattable: Create ‘Formattable’ Data Structures. R Package Version 0.2.0.1. 2016. Available online: https://cran.r-project.org/package=formattable (accessed on 4 October 2018).
- Carlson, M. Org.Hs.Eg.Db: Genome Wide Annotation for Human. R Package Version 3.5.0. 2017. Available online: http://bioconductor.org/packages/org.Hs.eg.db/ (accessed on 4 October 2018).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Set Comparison | Espritz-D # | Espritz-N # | Espritz-X # | IUPred-L # | IUPred-S # | PrDOS # | PV2 # | VLXT # | VSL2b # |
|---|---|---|---|---|---|---|---|---|---|
| Adsu vs. Nadsu | |||||||||
| IDR number in adsu | 487 | 1638 | 1210 | 1462 | 1517 | 2353 | 2445 | 2361 | 1966 |
| IDR number in nadsu * | 382 | 1036 | 796 | 847 | 892 | 1520 | 1794 | 1492 | 1431 |
| Adjusted p-value | 2.38 × 10−8 | 1.32 × 10−27 | 2.81 × 10−23 | 4.34 × 10−27 | 1.32 × 10−27 | 4.24 × 10−34 | 2.85 × 10−19 | 2.75 × 10−25 | 8.29 × 10−19 |
| Adsu_Down vs. Adsu_Up | |||||||||
| IDR number in adsu_down | 276 | 1072 | 760 | 983 | 1025 | 1507 | 1508 | 1572 | 1216 |
| IDR number in adsu_up * | 171 | 458 | 363 | 387 | 397 | 758 | 683 | 639 | 606 |
| Adjusted p-value | 3.51 × 10−78 | <1.00 × 10−99 | <1.00 × 10−99 | <1.00 × 10−99 | <1.00 × 10−99 | <1.00 × 10−99 | <1.00 × 10−99 | <1.00 × 10−99 | <1.00 × 10−99 |
| Gene Set Comparison | Espritz-D | Espritz-N | Espritz-X | IUPred-L | IUPred-S | PrDOS | PV2 | VLXT | VSL2b |
|---|---|---|---|---|---|---|---|---|---|
| Adsu vs. Nadsu | |||||||||
| Median (mean) IDR length (adsu) | 64 (125) | 57 (97) | 66 (98) | 54 (87) | 52 (67) | 61 (98) | 61 (90) | 48 (62) | 74 (128) |
| Median (mean) IDR length (nadsu) | 61 (98) | 56 (91) | 62 (93) | 54 (86) | 51 (68) | 55 (86) | 56 (82) | 47 (61) | 66 (107) |
| Adjusted p-value * | 6.04 × 10−2 | 7.33 × 10−2 | 2.50× 10−2 | 6.02 × 10−1 | 5.10 × 10−1 | 3.75× 10−11 | 4.97× 10−9 | 2.42× 10−2 | 4.97× 10−9 |
| Adsu_Down vs. Adsu_Up | |||||||||
| Median (mean) IDR length (adsu_down) | 68.5 (154) | 60 (104) | 76 (108) | 56 (93) | 53 (70) | 64 (107) | 65 (96) | 49 (65) | 79.5 (142) |
| Median (mean) IDR length (adsu_up) | 59 (89) | 54 (86) | 58 (83) | 50 (75) | 50 (62) | 58 (85) | 57 (80) | 46 (57) | 67 (104) |
| Adjusted p-value * | 1.24× 10−3 | 5.26× 10−3 | 2.14× 10−7 | 5.08× 10−3 | 1.66× 10−2 | 3.38× 10−4 | 1.42× 10−5 | 1.73× 10−3 | 7.05× 10−5 |
| Gene Set Comparison | Number of Proteins | Number (%) of Completely Disordered Proteins | Number (%) of Proteins with IDR | Median Percent IDR Per Protein (All Proteins) | Median Percent IDR Per Protein (IDR-Containing Proteins) |
|---|---|---|---|---|---|
| adsu_down | 445 | 19 (4.3) | 367 (82.5) | 38.1 | 48.1 |
| adsu_up | 556 | 11 (2) | 370 (66.5) | 18.1 | 37.6 |
| nadsu | 17,459 | 476 (2.7) | 11,248 (64.4) | 17.8 | 37.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arvidsson, G.; Wright, A.P.H. A Protein Intrinsic Disorder Approach for Characterising Differentially Expressed Genes in Transcriptome Data: Analysis of Cell-Adhesion Regulated Gene Expression in Lymphoma Cells. Int. J. Mol. Sci. 2018, 19, 3101. https://doi.org/10.3390/ijms19103101
Arvidsson G, Wright APH. A Protein Intrinsic Disorder Approach for Characterising Differentially Expressed Genes in Transcriptome Data: Analysis of Cell-Adhesion Regulated Gene Expression in Lymphoma Cells. International Journal of Molecular Sciences. 2018; 19(10):3101. https://doi.org/10.3390/ijms19103101
Chicago/Turabian StyleArvidsson, Gustav, and Anthony P. H. Wright. 2018. "A Protein Intrinsic Disorder Approach for Characterising Differentially Expressed Genes in Transcriptome Data: Analysis of Cell-Adhesion Regulated Gene Expression in Lymphoma Cells" International Journal of Molecular Sciences 19, no. 10: 3101. https://doi.org/10.3390/ijms19103101
APA StyleArvidsson, G., & Wright, A. P. H. (2018). A Protein Intrinsic Disorder Approach for Characterising Differentially Expressed Genes in Transcriptome Data: Analysis of Cell-Adhesion Regulated Gene Expression in Lymphoma Cells. International Journal of Molecular Sciences, 19(10), 3101. https://doi.org/10.3390/ijms19103101