Abstract
Myeloid hematological malignancies are clonal bone marrow neoplasms, comprising of acute myeloid leukemia (AML), the myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML), the myeloproliferative neoplasms (MPN) and systemic mastocytosis (SM). The field of epigenetic regulation of normal and malignant hematopoiesis is rapidly growing. In recent years, heterozygous somatic mutations in genes encoding epigenetic regulators have been found in all subtypes of myeloid malignancies, supporting the rationale for treatment with epigenetic modifiers. Histone deacetylase inhibitors (HDACi) are epigenetic modifiers that, in vitro, have been shown to induce growth arrest, apoptotic or autophagic cell death, and terminal differentiation of myeloid tumor cells. These effects were observed both at the bulk tumor level and in the most immature CD34+38− cell compartments containing the leukemic stem cells. Thus, there is a strong rationale supporting HDACi therapy in myeloid malignancies. However, despite initial promising results in phase I trials, HDACi in monotherapy as well as in combination with other drugs, have failed to improve responses or survival. This review provides an overview of the rationale for HDACi in myeloid malignancies, clinical results and speculations on why clinical trials have thus far not met the expectations, and how this may be improved in the future.
1. Introduction to Myeloid Hematological Diseases and Their Treatment
The myelodysplastic syndromes (MDS) are a heterogenous group of clonal myeloid hematological diseases, with a median age of onset of 70 years and a survival of 0.5–8 years, and a 30% risk of transformation to acute myeloid leukemia (AML) [1]. MDS is curable only by allogeneic stem cell transplant, for which only a minority of patients are eligible. The only approved therapy for higher risk MDS is treatment with hypomethylating agents, where two drugs are available, azacitidine and decitabine. Approximately 50–60% of patients respond to therapy [2]. Azacitidine and decitabine are considered to be DNA demethylating agents, as preclinical studies in cell lines have shown a quick and profound global DNA demethylation, as well as site specific promoter demethylation of e.g., P15/INK4B [3,4,5]. However, in vivo and ex vivo studies of MDS patient CD34+ progenitor cells do not show a clear DNA demethylation in response to azacitidine, and thus the in vivo mechanism of action of the drugs remains unclear [6,7].
The Philadelphia chromosome negative myeloproliferative neoplasms (MPN) comprises of polycythemia vera (PV), essential thrombocytemia (ET), and myelofibrosis (MF) [1]. The rare diseases chronic neutrophil leukemia and chronic eosinophil leukemia will not be discussed further in this review, nor will the Philadelphia chromosome positive chronic myeloid leukemia (CML). The, Janus kinase 2 (JAK2) V617F mutation occurs in around 95% of PV, and 50% of ET and MF, causing auto-phosphorylation of cytokine receptors and increased JAK-STAT pathway activation [8]. Although the MPNs share a common driver mutation, there are major clinical differences as ET has a largely normal life expectancy, PV has a long life expectancy, whereas MF has a significantly shortened life expectancy [9]. In higher risk disease, oral cytoreductive treatment hydroxycarbamide is used, and for MF, and now also for PV, the JAK2 inhibitor Ruxolitinib may be applied. Allogeneic stem cell transplant may be an option for younger, high risk MF patients [10].
Chronic myelomonocytic leukemia (CMML) is the largest group of MDS/MPN overlap syndromes [1], where proliferative CMML disease is treated like MPN and dysplastic CMML is treated like MDS [11], however responses to azacitidine or decitabine are never long lasting, and there is an imminent need for new treatment options in CMML.
Systemic mastocytosis (SM) is a rare myeloid malignancy [1]. Over 90% of patients carry the D816V activating point mutation in the KIT gene. There are two clinical phenotypes, indolent SM with a normal life expectancy and aggressive SM with a poor prognosis [12]. In the aggressive SM group, several therapies have been tested, most recently the pan tyrosine kinase inhibitor Midostaurin [12,13], however to date, no therapy except allogeneic stem cell transplantation has been shown to improve survival, and as the median age at disease onset is around age 70, only a handful of patients are eligible for transplant [14].
Overall, 30% of MDS and CMML patients progress to acute myeloid leukemia (AML). AML is defined by over 20% myeloblasts in the bone marrow, and may be primary (around 75% of all AML cases), secondary (to e.g., MDS, MPN, around 15% of cases) or treatment related e.g., after chemotherapy of hematological or other tumors (10% of cases). Subtypes of AML are well defined according to cell of origin, specific cytogenetic or other aberrations outlined in the WHO diagnostic criteria [1]. In general, patients that are not elderly and unfit, are given intensive chemotherapy (including cytarabine and anthracyclines) to achieve complete remission, and thereafter chemotherapy consolidation or consolidation with allogeneic stem cell transplant in case of high risk disease [15]. However, a large portion of patients are elderly and fragile, and for these patients lower intensity treatment, often similar to regimens given to MDS-patients, can be considered if the AML is not highly proliferative [16]. The 5-year overall survival in adult AML is around 20%.
2. Epigenetic Regulation of Normal and Malignant Hematopoiesis
Our understanding of how epigenetic regulation of hematopoiesis is orchestrated is rapidly growing. Epigenetics include DNA methylation (Figure 1) as well as covalent, reversible histone modifications (Figure 2) [17,18]. DNA methylation is associated with transcriptional repression via formation of heterochromatin. This is achieved by methylation of the 5-cytosine by DNA methyl transferase (DNMT) enzymes, where maintenance methylation is exerted by DNMT1, and de novo methylation is exerted by DNMT3A and 3B (Figure 1). The gene encoding the DNMT3A enzyme is commonly mutated in AML, leading to loss of function (Table 1). DNA demethylation is a multistep process exerted by the TET enzymes that oxidize 5-methyl cytosine to cytosine (Figure 1). This oxidation process requires α-keto-glutarate, which is produced from isocitrate by isocitrate dehydrogenases 1 and 2 (IDH1 and 2) (Figure 1). Mutations in IDH1 and 2 are common in AML, and TET2 mutations are common in all myeloid malignancies (Table 1). Table 1 summarizes the currently known mutations in epigenetic regulators found in myeloid malignancies, and is compiled from the pivotal studies of Papaemmanuil [19] and Haferlach [20] for MDS, and Ley for AML [21], as well as reviews for CMML [22], MPN [23], SM [24], and references [17,18,25,26] for comparison of mutation frequencies reported.
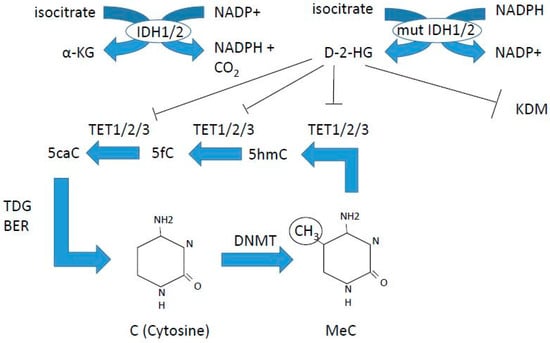
Figure 1.
DNA methylation and demethylation. DNMT3A is commonly mutated in acute myeloid leukemia (AML), IDH1, 2 mutations are found in AML, and TET2 is frequently mutated in all myeloid malignancies. Azacitidine and decitabine are DNA demethylating agents, inhibiting DNA methyl transferases (DNMTs).
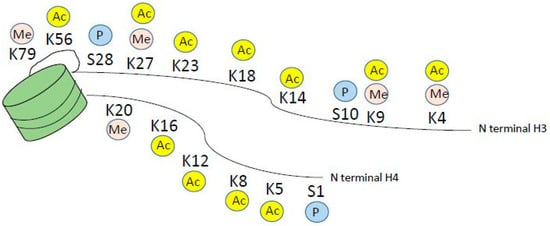
Figure 2.
Histone modifications on the N terminal tail of histone H3 and H4. For simplicity, only methylation, acetylation and phosphorylation are depicted, however modifications also include arginine methylation and ubiquitination marks.

Table 1.
In all types of myeloid malignancies, genetic alterations in epigenetic modifiers are found, however the mutation frequency varies between diseases. For references please see text.
Histone modifications occur on the n-terminal protruding tail of predominantly histone H3 and H4, and consist of lysine residues being acetylated or methylated, and serine residues can be phosphorylated (Figure 2). In addition, arginine may be methylated however this will not be further discussed as it is outside the scope of this paper. Histone acetylation occurs via a family of lysine acetyl transferases (KAT), formerly called histone acetyl transferases (HAT), and histone lysine methyl transferases (KMT) methylate histones. KAT and KMT enzymes along with protein arginine methyl transferases are called epigenetic writers, and HDAC, histone lysine demethylases (KDM) and phosphatases are called epigenetic erasers (Figure 3), reviewed in reference [27]. In addition, there are readers that read the epigenetic code. These are bromodomain containing proteins like the BET family, chromodomain, PHD finger and WD 40 repeats (Figure 3). Inhibitors to both writers, e.g., DOT1L [28,29] and readers e.g., BET inhibitor JQ1 [30] are in development and early clinical trials for myeloid malignancies
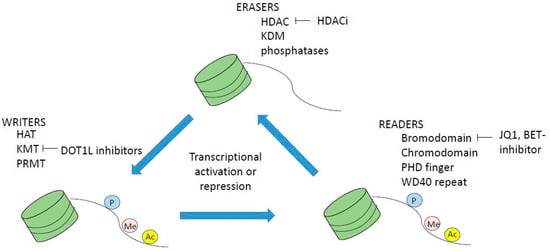
Figure 3.
Epigenetic writers are histone acetyl transferases HAT (KAT), histone lysine methyl transferase (KMT) and PRMTs (protein arginine methyl transferases), readers are bromodomain proteins like BET family proteins, and erasers are histone deacetylase inhibitors (HDACi), KDM (lysine/histone demethylases) and phosphatases. Inhibitors or writers, readers and erasers are being developed and are in clinical trials for myeloid malignancies, for example HDACi, bromodomain BET inhibitor Q1 and DOT1L inhibitors, of which the latter are in clinical phase I trials.
3. Dysregulation of Histone Acetylation and Methylation in Myeloid Malignancies
KATs modulate the process of hematopoiesis both via altering the epigenetic status of chromatin via histone lysine acetylation [18], and via regulation of non-histone protein acetylation [31]. Mutations in KATs, e.g., CBP and p300 have been described in myeloid malignancies, although they are rare events (Table 1, Figure 4). KAT6A (MOZ/myst3, part of the MYST family of KATs) is important in regulating hematopoietic stem cells, and is a target of translocations causing AML. Recently, Baell et al. elegantly showed that KAT6A/B (MOZ/myst 3 and myst 4) inhibitors arrest tumor growth and induce senescence in AML cells, in vitro and in vivo [32]. UTX (KDM6A) acetylates H3K27ac thus mediating active chromatin, and UTX mutations are found in AML (Table 1) [33].
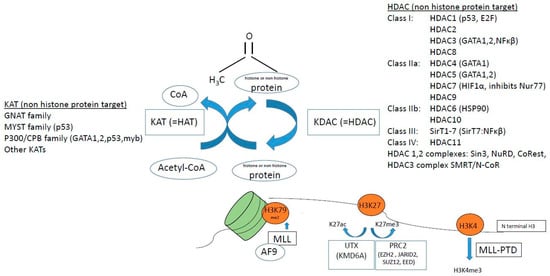
Figure 4.
Factors associated with histone or non-histone protein lysine acetylation/methylation, affected in hematological malignancies. In brackets are listed non-histone targets of HAT (KAT) and histone deacetylases (HDACs). See text for details on how these epigenetic factors are associated with myeloid malignancies.
In AML subtype MLL-PTD (MLL mixed lineage leukemia), a mutation causes a partial tandem duplication (PTD) that confers excessive tri-methylation of H3K4 (Figure 4). MLL, which is a KMT, may also have over 50 different translocation partners, one of which is MLL-AF9 that recruits DOT1L to methylate H3K79me2 (Figure 4). The myb oncogene requires myb–p300 interaction for leukemic transformation of AML oncogenes AML-ETO and MLL-AF9 [34], thus there are many implications of histone methylation in leukemia.
Mutations in the TP53 gene encoding the P53 tumor suppressor occurs in approximately 10% of AML and MDS patients, and is associated with a dismal prognosis. The TP53 gene requires coactivator CBP/p300 acetylation for full transcriptional activation, where the KAT p300 acetylates p53. P53 is normally de-acetylated by HDAC1. In addition, in AML with inv(16) or t(16;16), p53 activity is inhibited via interactions between the inv(16) fusion protein CBFβ-SMMHC with HDAC8, where HDAC8 aberrantly deacetylates p53, which promotes leukemia. Inhibition of HDAC8 restores p53 and induces apoptosis selectively in leukemic inv(16)+ CD34+ cells but spares normal CD34+cells [35].
The HDAC3 containing NCoR complex can be recruited by the oncogenic fusion proteins AML-ETO and PML-RARα, and HDAC1 knockdown increases survival in PML-RARα mediated APL. To add to the complexity of HDAC function, a specific HDAC may have different roles over time in leukemia development. The most well studied example is acute promyelocytic leukemia (APL), where a translocation t(15;17) generates the PML-RARα fusion protein. Normally, RAR (retinoic acid receptor) is a transcription factor and when retinoic acid is absent, RAR associates with HDAC1/2 containing complex SMRT/N-CoR and represses transcription. Retinoic acid causes release of the corepressor complex and leads to transcription. In APL cells, retinoic acid does not release the corepressor complex, resulting in a differentiation block. However, pharmacological doses of retinoic acid, as used in the clinic, degrade the fusion protein and the cells differentiate. Thus, HDAC play an important role in APL, however their role varies over time as HDAC1/2 knockdown in early leukemogenesis expands the leukemia, whereas in leukemic phase, knockdown of the same HDAC1/2 cause differentiation and apoptosis of APL cells, and increased survival of APL mice [36]. The polycomb repressive complex 2 (PRC2) silences H3K27 to H3K27me3, and several genes encoding components of the PRC2; EZH2, SUZ12, JARID2 and also mutations in ASXL1 coding for the PRC2 associated protein ASXL1, have been found to be mutated in myeloid malignancies (Table 1, Figure 4) [17,18,19,20,21,22,23,25,26].
HDAC enzymes (KDACs) are commonly mutated in solid tumors, with 30% of endometrial tumors having HDAC mutations, however only 2% of patients with AML have HDAC mutations [36]. A thorough investigation of HDAC gene expression in MDS and AML showed that in myeloid malignancies, HDAC expression is heterogenous, with no clear pattern of over- or under-expression of any HDAC [37]. However, in the CD34+ progenitor compartment of patients with MF, there is an increase in HDAC levels [38]. Interestingly, HDACs actually preceded the histone proteins phylogenetically, clearly indicating that HDACs primarily target non-histone protein substrates [39], and many of these non-histone proteins are the products of tumor suppressor genes, oncogenes or transcription factors important for hematopoiesis (Figure 4) [31], and as they are deacetylated by HDACs, they are targets of HDACi treatment.
4. Preclinical Experience of HDACi in Myeloid Malignancies
In general, HDACi treatment alters the expression of 5–10% of transcribed genes, depending on cell type. The specific antitumor activity of HDACi varies between tumor types, and the antitumor activity on a specific tumor type may vary between different HDACi. The general mechanisms for HDACi induced cell death include apoptosis or autophagy, increasing ROS production and decreasing scavengers, increasing DNA damage and decreasing DNA repair, decreasing oncoprotein expression and stability and stimulating immunogenic cell death [27,40,41]. In myeloid malignancies, HDACi treatment induces cell death, growth arrest or differentiation, activating chromosome degradation, altering angiogenesis, inactivating chaperone complexes, and inducing expression of cell cycle inhibitors e.g., p21, and of pro-apoptotic genes [42,43,44,45,46,47]. The DNA damage induced by HDACi can be repaired by normal cells but not by transformed cells [48].
Induction of apoptosis is the major route for HDACi induced cell death, and it may be via either the intrinsic (mitochondria) or extrinsic (death receptor) pathway. The specific changes in gene expression leading to apoptosis in myeloid malignancies varies between the different HDACi, but frequently, extrinsic pathway via TRAIL is used. In AML, HDACi MS275 treatment induces the expression of TRAIL by activating the TNFS10 gene that encodes TRAIL, triggering death signal via the extrinsic pathway, and additionally RNA interference against TRAIL blocked downstream caspase activation and inhibited MS275 mediated apoptosis, suggesting that at least in AML cells, MS275 mechanism of action is via TRAIL [44]. In K562 leukemic cells, VPA reduces the expression of c-FLP and Bcl.2/Bcl-xL anti-apoptotic factors, as well as sensitized cells to TRAIL/Apo2L mediated apoptosis, thus acting on both the intrinsic and extrinsic apoptotic pathways [49]. In APL and AML1-ETO mouse models in vivo and human cell lines in vitro, HDACi valproic acid upregulates TRAIL, DR5, FasL and Fas in leukemic cells but not normal progenitors, thus for the sensitivity of HDACi to leukemia a transformed phenotype is required [42].
In a recent study of several AML cell lines as well as CD33+ progenitor cells from AML and MDS patients, vorinostat induced gene expressions of COX2, p15, cFOS, genes that are downregulated in MDS and AML, and suppressed overexpressed genes cyclin D1 and c-MYC [50]. This led to cell cycle arrest, terminal differentiation and or apoptosis, via mechanisms including modulation of SP1 [50]. Recently, HDACi entinostat has been shown to restore the decreased orphan nuclear receptor Nur77 expression in AML cell lines and in AML patient leukemia cells, especially in the leukemic stem/very early CD34+/38− progenitors, and induce apoptosis, presenting a novel mechanism of action of HDACi and suggesting that Nur77 may be a biomarker for HDACi apoptotic effect [51]. Thus, HDACi have multiple and broad effects, and likely the mechanism of HDACi induced tumor cell death may be depending on the molecular defects of the target cell, as well as of the specific HDACi used [43].
In AML/ETO, single agent valproic acid inhibits not only the mature leukemic cells but also immature progenitors by targeting the AML1/ETO-HDAC complex SMRT/N-CoR, inducing differentiation [52]. In MPN, preclinical data strongly supports the effect of HDACi inhibiting proliferation and inducing apoptosis in JAK2 mutated cells, normalizing splenomegaly and blood counts in JAK2 mutant knock-in mice [53] and promoting proteasome mediated JAK2 degradation by disrupting HSP90 chaperone function. Treatment of JAK2 mutated CD34+ progenitor cells with panobinostat induces apoptosis and inhibits JAK2 expression and activity, subsequently reducing pSTAT3, pSTAT5, pAKT and pGATA1, and partially inhibiting the binding between HSP90 and JAK2, suggesting that acetylation of HSP90 could mediate JAK2 degradation [54]. The same study showed a synergistic effect of addition of JAK2 inhibitor to panobinostat.
HDACi therapy in SM was first assessed in a canine model of SM [55]. Our group has shown that several first and second generation HDACi dose dependently inhibit growth and induce apoptosis in KIT D816V mutated SM cell lines, and that vorinostat selectively kills KIT D816V mutated primary patient mast cells whereas normal mast cells are unaffected [56]. To support epigenetics in the pathogenesis of SM, a recent study shows a deficiency of lysine methylation in aggressive SM [57]. However, to date there have been no clinical trials of HDACi in SM.
5. Preclinical Rationale for Combination Therapy Including HDACi
There is a large body of preclinical evidence showing synergistic effects of various first and second generation HDACi in combination with azacitidine or decitabine in MDS and AML cell lines or ex vivo cultured patient cells. These include enhanced growth arrest, inhibition of DNA synthesis and loss of clonogenic potential, and synergistic effects in re-expressing silenced genes [58,59,60]. When combining panobinostat and decitabine in vitro, there were synergistic effects in attenuating DNMT1 and EZH2, de-repression of JunB and enhanced leukemic cell death [61]. Primary AML patient CD34+ cells were more sensitive than normal CD34+ cells to the treatment, indicating a specific anti-leukemic effect and that normal progenitors are spared. In a molecular study of clinical samples from the clinical trial of Tan et al., using azacitidine and panobinostat for MDS and AML [62], Liu et al. analyzed mRNA of Nur77, p15 and p21 in the clinical patient samples, and found that restored levels of Nur77 and p21 correlated with clinical responses to the combination therapy [59], in concordance with other studies suggesting Nur77 as a biomarker of HDACi mediated apoptosis also in the leukemic stem cell compartment [51]. In MPN, HDACi have been shown to synergize with JAK2 inhibitors in inducing apoptosis in JAK2 mutated cells [54].
6. Results from Clinical Studies of HDACi Monotherapy and Combination Therapy for Myeloid Malignancies
Single agent first and second generation HDACi have been tested in several small phase I and II studies for MDS and AML, showing low overall response rates and 0–10% partial or complete remissions, reviewed by Morabito et al. and by Stahl et al. [63,64], with the conclusion that combination treatment is needed to achieve a clinical effect. However, despite preclinical support for synergistic effects of combination therapy since the pivotal study of Cameron et al. in 1999, showing synergy of demethylaton and HDAC inhibition in re-expressing silenced genes in cancer [58], and several studies since references [59,60,61,62], and early phase I studies showed promising results, both for vorinostat in combination with decitabine [65] and panobinostat in combination with azacitidine [62,66], thus far, the randomized phase II clinical trials of various doses and various HDACi drugs in combination with azacitidine or decitabine for MDS, CMML and AML have not been able to show an improved clinical outcome. The recent phase II studies in MDS, CMML and AML are summarized in Table 2. In addition, a meta-analysis of these trials has been recently published [67]. Of note, there are currently 156 clinical trials of HDACi mono- or combination therapy registered at clinicaltrials.gov, using a plethora of HDACi agents (Table 3).
Table 2.
Phase I/II trials with combination treatment of HDACi and hypomethylating agents, in AML and MDS, sometimes including CMML. OS = overall survival, ORR = overall response rate, CR = complete remission. In the studies by Uy et al., and Tan et al., there was no control arm thus a comparison of efficacy to monotherapy could not be made. Out of five evaluable studies, none showed an advantage of combination therapy. 1 Azacitidine 75 mg/m2 Day 1–5/28, 2 panobinostat 3 days/w 7 doses/28 days, phase II 30 mg oral daily Day 1–7/28, 3 decitabine 20 mg/m2 iv Day 1–5, 4 valproic acid 50 mg/kg oral Day 1–7/28, 5 azacitidine 75 mg/m2 Day 1–7/28, 6 vorinostat 300 mg twice daily Day 3–9/28, 7 panobinostat 20–40 mg Day 3, 5, 8, 10, 12, 15, in phase IIb 40 mg, 8 pracinostat 60 mg or placebo oral every 2 days Day 1–21/28, 9 azacitidine 50 mg/m2 10 days, 10 entinostt 4 mg/m2 Day 3, 10/28, 11 panobinostat three times/week during two weeks/4, phase I dose escalation to 50 mg, phase II 40 mg.
Table 3.
HCACi that are listed at clinicaltrials.gov, with at least one listed phase I clinical trial.
A review of valproic acid effects on AML cells conclude that single agent valproic acid may stabilize disease in the many old and fragile AML patients that are unfit for more intensive therapy, however as of yet, no randomized studies have been conducted [75]. However, there is an ongoing prospective randomized multicenter phase II trial of low dose decitabine alone or in combination with valproic acid and all-trans-retinoic acid in patients with AML, ineligible for induction chemotherapy, is also ongoing and an interim report has been published [76]. Thus, there may be therapeutic options of epigenetic drugs also for the elderly, fragile patients that are not eligible for more intense therapy.
A number of HDACi have been investigated in clinical trials in MPN, recently reviewed by Bose and Verstovsek [77]. Overall, HDACi monotherapy is effective in MPN however not well tolerated in ET and PV patients, where published studies show significant drop out due to toxicity, even if HDACi monotherapy clearly is active and also decreases JAK2 mutation burden in PV and ET [78,79,80]. Some studies have reported toxicity and high dropout rate also in MF [81], however a recent follow up study on panobinostat monotherapy in primary MF and post PV/ET MF showed a response rate of 36% according to IWG-MRT criteria, with a median spleen volume reduction of 34% in eight evaluable patients, of which one obtained a complete molecular response and six patients remained on therapy for a median of 18 months [82]. Bose and Verstovsek conclude that that the combination of HDACi with JAK2 inhibitor in MF is the most promising approach, however toxicity and long-term tolerability may be future concerns [77]. Currently, three clinical phase I/II trials using HDACi in combination with JAK2 inhibitor ruxolitinib are ongoing, NCT01693601 (the Prime study, panobinostat and ruxolitinib, likely to end Feb 2019), NCT01433445 with panobinostat and ruxolitinib, currently in expansion phase, and NCT02267278 with pracinostat and ruxolitinib). Overall, preclinical data for combination therapy in MF is solid and there are great expectations on the ongoing combination trials of HDACi and ruxolitinib in MF. For SM, there have been no clinical trials including HDACi therapy until now.
7. Why Have the Clinical Studies Failed?
Four recent reviews on the combination trials of azacitidine or decitabine with HDACi conclude that there may be still a future for the drug combination, despite the lack of beneficial results in phase II trials [64,83,84,85]. As preclinical data on how to best combine the drugs is lacking, it may well be that the clinical trials have administered the drugs with suboptimal timing. Simultaneous administration with varying dose intervals has been used, perhaps inducing pharmacological antagonism as azacitidine requires cell division and DNA replication to exert its effects, and HDACi inhibit cell division and proliferation, thus potentially antagonizing the effect of azacitidine. Another issue that must be met is how to choose the optimal HDACi for the specific target patient population, regarding selectivity of inhibition of target proteins, and regarding the pattern of somatic mutations and chromosomal abnormalities of each patient. Here, novel more selective HDACi are being developed, with focused targets. In addition, the mechanism of action of HDACi in MDS and AML is unclear. In fact, despite azacitidine and decitabine being widely used for over 10 years, the mechanism of action of these drugs in vivo is still unknown, and we, as well as others, have failed to demonstrate demethylation of MDS progenitor cells upon azacitidine treatment [6,7]. In addition, there are to date no established biomarkers to assess azacitidine or decitabine effects, nor are there any established biomarkers for monitoring HDACi effects. Thus, we have no readout for either of the drugs and thus no means of elucidating which drug is failing, when we combine them in the clinical setting. Currently our only readout is remission and survival, and possibly decrease of a mutated clone size, however we cannot measure if the drug effects are counteracting each other as we have no biomarkers of treatment effect. Thus, before attempting new clinical trials, we need to solve the issue of how to combine the drugs to optimize synergy and decrease the risk of antagonism or inhibition, and in addition we imperatively need to establish reliable biomarkers of drug effects.
8. Summary
Despite a theoretical rationale and profound preclinical proof of HDACi efficacy in myeloid malignancies, all phase II randomized clinical trials have failed, except for the combination of HDACi with JAK2 inhibitor ruxolitinib in MF. However, optimizing combination treatment strategy, e.g., sequential treatment and not simultaneous, will be a key issue to avoid pharmacological antagonism, and requires further basic in vitro studies of optimizing drug scheduling and doses, as well as biomarkers to follow in vivo drug effects. In addition, novel, more selective HDACi should be preferred, avoiding off target effects. In conclusion, there may still be a role for HDACi in myeloid malignancies, beyond the promising combination therapy of HDACi and ruxolitinib for MF.
I apologize to all authors that have made significant contributions to the field but was not cited in the current review, due to practical space limitations.
Conflicts of Interest
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Madanat, Y.; Sekeres, M.A. Optimizing the use of hypomethylating agents in myelodysplastic syndromes: Selecting the candidate, predicting the response, and enhancing the activity. Semin. Hematol. 2017, 54, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, S.; Heil, O.; Lyko, F.; Brueckner, B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS ONE 2011, 6, e17388. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, M.; Nguyen, T.T.; Nguyen, C.; Guldberg, P.; Kohler, G.; Wijermans, P.; Jones, P.A.; Lubbert, M. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood 2002, 100, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Schmidt-Mende, J.; Karimi, M.; Gogvadze, V.; Hassan, M.; Ekstrom, T.J.; Zhivotovsky, B.; Hellstrom-Lindberg, E. Hypomethylation and apoptosis in 5-azacytidine-treated myeloid cells. Exp. Hematol. 2008, 36, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Tobiasson, M.; Abdulkadir, H.; Lennartsson, A.; Katayama, S.; Marabita, F.; de Paepe, A.; Karimi, M.; Krjutskov, K.; Einarsdottir, E.; Grovdal, M.; et al. Comprehensive mapping of the effects of azacitidine on DNA methylation, repressive/permissive histone marks and gene expression in primary cells from patients with MDS and MDS-related disease. Oncotarget 2017, 8, 28812–28825. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.F.; Micklem, C.N.; Taguchi, M.; Itonaga, H.; Sawayama, Y.; Imanishi, D.; Nishikawa, S.; Miyazaki, Y.; Jakt, L.M. Longitudinal Analysis of DNA Methylation in CD34+ Hematopoietic Progenitors in Myelodysplastic Syndrome. Stem Cells Transl. Med. 2014, 3, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F. How I treat myelofibrosis. Blood 2014, 124, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Solary, E.; Itzykson, R. How I treat chronic myelomonocytic leukemia. Blood 2017, 130, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Chandesris, M.O.; Damaj, G.; Canioni, D.; Brouzes, C.; Lhermitte, L.; Hanssens, K.; Frenzel, L.; Cherquaoui, Z.; Durieu, I.; Durupt, S.; et al. Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2605–2607. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.A.; Shore, T.; et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Tenti, E.; Papayannidis, C.; Marconi, G.; Parisi, S.; Simonetti, G.; Paolini, S.; Sartor, C.; Ottaviani, E.; Testoni, N.; Martinelli, G. Efficacy of Azacitidine in the treatment of adult patients aged 65 years or older with AML. Expert Opin. Pharmacother. 2016, 17, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Kitamura, T. Epigenetics in normal and malignant hematopoiesis: An overview and update 2017. Cancer Sci. 2017, 108, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Man, N.; Tan, Y.; Nimer, S.D.; Wang, L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Front Oncol. 2015, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome, C. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2018 update on diagnosis, risk stratification and management. Am. J. Hematol. 2018, 93, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Schischlik, F.; Kralovics, R. Mutations in myeloproliferative neoplasms-their significance and clinical use. Expert Rev. Hematol. 2017, 10, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug. Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Marschalek, R. How to effectively treat acute leukemia patients bearing MLL-rearrangements? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Waters, N.J. Preclinical Pharmacokinetics and Pharmacodynamics of Pinometostat (EPZ-5676), a First-in-Class, Small Molecule S-Adenosyl Methionine Competitive Inhibitor of DOT1L. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Caputo, V.; Karadimitris, A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: Emerging preclinical and clinical evidence. Ther. Adv. Hematol. 2015, 6, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Leaver, D.J.; Hermans, S.J.; Kelly, G.L.; Brennan, M.S.; Downer, N.L.; Nguyen, N.; Wichmann, J.; McRae, H.M.; Yang, Y.; et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 2018, 560, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Puda, A.; Milosevic, J.D.; Berg, T.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, B.; Rumi, E.; Pietra, D.; Malcovati, L.; Elena, C.; et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am. J. Hematol. 2012, 87, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Singh, S.; Hua, W.K.; Cai, Q.; Chao, S.W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 Inhibition Specifically Targets Inv (16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of class I and II histone deacetylase gene expression in human leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Chen, C.; Dumlao, T.; Naik, S.; Chang, T.; Xiao, Y.Y.; Sominsky, I.; Burton, J. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk. Lymphoma 2008, 49, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Marks, P.A. Histone deacetylase inhibitors in the therapy of cancer: Much to learn. Epigenomics 2010, 2, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, H.; Towatari, M.; Hatano, S.; Kitamura, K.; Kiyoi, H.; Kinoshita, T.; Tanimoto, M.; Murate, T.; Kawashima, K.; Saito, H.; et al. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: A new approach to anti-leukemia therapy. Leukemia 1999, 13, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Saunthararajah, Y.; Redner, R.L.; Liu, J.M. Inhibitors of histone deacetylase relieve ETO-mediated repression and induce differentiation of AML1-ETO leukemia cells. Cancer Res. 1999, 59, 2766–2769. [Google Scholar] [PubMed]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Medici, M.C.; Russo, G.L. Valproic acid sensitizes K562 erythroleukemia cells to TRAIL/Apo2L-induced apoptosis. Anticancer Res. 2008, 28, 855–864. [Google Scholar] [PubMed]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS ONE 2013, 8, e53766. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ruvolo, V.R.; McQueen, T.; Chen, W.; Samudio, I.J.; Conneely, O.; Konopleva, M.; Andreeff, M. HDAC inhibition by SNDX-275 (Entinostat) restores expression of silenced leukemia-associated transcription factors Nur77 and Nor1 and of key pro-apoptotic proteins in AML. Leukemia 2013, 27, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Klisovic, R.B.; Vukosavljevic, T.; Yu, J.; Paschka, P.; Huynh, L.; Pang, J.; Neviani, P.; Liu, Z.; Blum, W.; et al. Targeting AML1/ETO-histone deacetylase repressor complex: A novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J. Pharmacol. Exp. Ther. 2007, 321, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Akada, H.; Akada, S.; Gajra, A.; Bair, A.; Graziano, S.; Hutchison, R.E.; Mohi, G. Efficacy of vorinostat in a murine model of polycythemia vera. Blood 2012, 119, 3779–3789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fiskus, W.; Chong, D.G.; Buckley, K.M.; Natarajan, K.; Rao, R.; Joshi, A.; Balusu, R.; Koul, S.; Chen, J.; et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009, 114, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Fenger, J.; Murahari, S.; Bear, M.D.; Kulp, S.K.; Wang, D.; Chen, C.S.; Kisseberth, W.C.; London, C.A. AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated Kit. Blood 2010, 115, 4217–4225. [Google Scholar] [CrossRef] [PubMed]
- Lyberg, K.; Ali, H.A.; Grootens, J.; Kjellander, M.; Tirfing, M.; Arock, M.; Hagglund, H.; Nilsson, G.; Ungerstedt, J. Histone deacetylase inhibitor SAHA mediates mast cell death and epigenetic silencing of constitutively active D816V KIT in systemic mastocytosis. Oncotarget 2017, 8, 9647–9659. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Mancini, M.; De Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2018, 32, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Urbanavicius, D.; Tan, P.; Spencer, A.; Dear, A.E. Mechanisms and potential molecular markers of early response to combination epigenetic therapy in patients with myeloid malignancies. Int. J. Oncol. 2014, 45, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Shaker, S.; Bernstein, M.; Momparler, L.F.; Momparler, R.L. Preclinical evaluation of antineoplastic activity of inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone deacetylation (trichostatin A, depsipeptide) in combination against myeloid leukemic cells. Leuk. Res. 2003, 27, 437–444. [Google Scholar] [CrossRef]
- Fiskus, W.; Buckley, K.; Rao, R.; Mandawat, A.; Yang, Y.; Joshi, R.; Wang, Y.; Balusu, R.; Chen, J.; Koul, S.; et al. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol. Ther. 2009, 8, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Wei, A.; Mithraprabhu, S.; Cummings, N.; Liu, H.B.; Perugini, M.; Reed, K.; Avery, S.; Patil, S.; Walker, P.; et al. Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Cancer J. 2014, 4, e170. [Google Scholar] [CrossRef] [PubMed]
- Morabito, F.; Voso, M.T.; Hohaus, S.; Gentile, M.; Vigna, E.; Recchia, A.G.; Iovino, L.; Benedetti, E.; Lo-Coco, F.; Galimberti, S. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin. Investig. Drugs 2016, 25, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Gore, S.D.; Vey, N.; Prebet, T. Lost in translation? Ten years of development of histone deacetylase inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Expert Opin. Investig. Drugs 2016, 25, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Gojo, I.; Goldberg, S.L.; Bredeson, C.; Kujawski, L.A.; Yang, A.; Marks, P.; Frankel, P.; Sun, X.; Tosolini, A.; et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br. J. Haematol. 2014, 167, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Munakata, W.; Ogura, M.; Uchida, T.; Taniwaki, M.; Kobayashi, T.; Shimada, F.; Yonemura, M.; Matsuoka, F.; Tajima, T.; et al. Phase I study of panobinostat and 5-azacitidine in Japanese patients with myelodysplastic syndrome or chronic myelomonocytic leukemia. Int. J. Hematol. 2018, 107, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Qi, J.; You, T.; Yang, L.; Wu, D.; Han, Y.; Zhu, L. Addition of histone deacetylase inhibitors does not improve prognosis in patients with myelodysplastic syndrome and acute myeloid leukemia compared with hypomethylating agents alone: A systematic review and meta-analysis of seven prospective cohort studies. Leuk. Res. 2018, 71, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Garcia-Manero, G.; Huang, X.; Cortes, J.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Brandt, M.; Pierce, S.; Kantarjian, H.M. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer 2015, 121, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Othus, M.; List, A.F.; Odenike, O.; Stone, R.M.; Gore, S.D.; Litzow, M.R.; Buckstein, R.; Fang, M.; Roulston, D.; et al. Randomized Phase II Study of Azacitidine Alone or in Combination with Lenalidomide or With Vorinostat in Higher-Risk Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: North American Intergroup Study SWOG S1117. J. Clin. Oncol. 2017, 35, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Sekeres, M.A.; Egyed, M.; Breccia, M.; Graux, C.; Cavenagh, J.D.; Salman, H.; Illes, A.; Fenaux, P.; DeAngelo, D.J.; et al. A phase 1b/2b multicenter study of oral panobinostat plus azacitidine in adults with MDS, CMML or AML with 30% blasts. Leukemia 2017, 31, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Montalban-Bravo, G.; Berdeja, J.G.; Abaza, Y.; Jabbour, E.; Essell, J.; Lyons, R.M.; Ravandi, F.; Maris, M.; Heller, B.; et al. Phase 2, randomized, double-blind study of pracinostat in combination with azacitidine in patients with untreated, higher-risk myelodysplastic syndromes. Cancer 2017, 123, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Sun, Z.; Figueroa, M.E.; Ketterling, R.; Melnick, A.; Greenberg, P.L.; Herman, J.; Juckett, M.; Smith, M.R.; Malick, L.; et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: Results of the US Leukemia Intergroup trial E1905. J. Clin. Oncol. 2014, 32, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Sun, Z.; Ketterling, R.P.; Zeidan, A.; Greenberg, P.; Herman, J.; Juckett, M.; Smith, M.R.; Malick, L.; Paietta, E.; et al. Eastern Cooperative Oncology G, North American Leukemia i: Azacitidine with or without Entinostat for the treatment of therapy-related myeloid neoplasm: Further results of the E1905 North American Leukemia Intergroup study. Br. J. Haematol. 2016, 172, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Duncavage, E.J.; Chang, G.S.; Jacoby, M.A.; Miller, C.A.; Shao, J.; Heath, S.; Elliott, K.; Reineck, T.; Fulton, R.S.; et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 2017, 31, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Gjertsen, B.T.; Bruserud, O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenetics 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Grishina, O.; Schmoor, C.; Dohner, K.; Hackanson, B.; Lubrich, B.; May, A.M.; Cieslik, C.; Muller, M.J.; Lubbert, M. DECIDER: Prospective randomized multicenter phase II trial of low-dose decitabine (DAC) administered alone or in combination with the histone deacetylase inhibitor valproic acid (VPA) and all-trans retinoic acid (ATRA) in patients >60 years with acute myeloid leukemia who are ineligible for induction chemotherapy. BMC Cancer 2015, 15, 430. [Google Scholar]
- Bose, P.; Verstovsek, S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin. Investig. Drugs 2016, 25, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; McMullin, M.F.; Ejerblad, E.; Zweegman, S.; Harrison, C.; Fernandes, S.; Bareford, D.; Knapper, S.; Samuelsson, J.; Lofvenberg, E.; et al. A phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br. J. Haematol. 2013, 162, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Vannucchi, A.M.; Martinelli, V.; Ruggeri, M.; Nobile, F.; Specchia, G.; Pogliani, E.M.; Olimpieri, O.M.; Fioritoni, G.; Musolino, C.; et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013, 161, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, A.; Dellacasa, C.M.; Finazzi, G.; Carobbio, A.; Ferrari, M.L.; Guglielmelli, P.; Gattoni, E.; Salmoiraghi, S.; Finazzi, M.C.; Di Tollo, S.; et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br. J. Haematol. 2010, 150, 446–455. [Google Scholar] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Estrov, Z.; Borthakur, G.; Cortes, J.; Verstovsek, S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 2012, 36, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Sandy, L.; Lu, M.; Yoon, J.; Petersen, B.; Zhang, D.; Ye, F.; Newsom, C.; Najfeld, V.; Hochman, T.; et al. A phase II study of panobinostat in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk. Res. 2017, 53, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Zeidan, A.; Gore, S.D.; Prebet, T. Hypomethylating agent combination strategies in myelodysplastic syndromes: Hopes and shortcomings. Leuk. Lymphoma 2017, 58, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. Epigenetic therapies in MDS and AML. Adv. Exp. Med. Biol. 2013, 754, 253–283. [Google Scholar] [PubMed]
- Stahl, M.; Zeidan, A.M. Hypomethylating agents in combination with histone deacetylase inhibitors in higher risk myelodysplastic syndromes: Is there a light at the end of the tunnel? Cancer 2017, 123, 911–914. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).