Design, Synthesis, and Cytotoxic Analysis of Novel Hederagenin–Pyrazine Derivatives Based on Partial Least Squares Discriminant Analysis


Abstract

1. Introduction
2. Result and Discussion
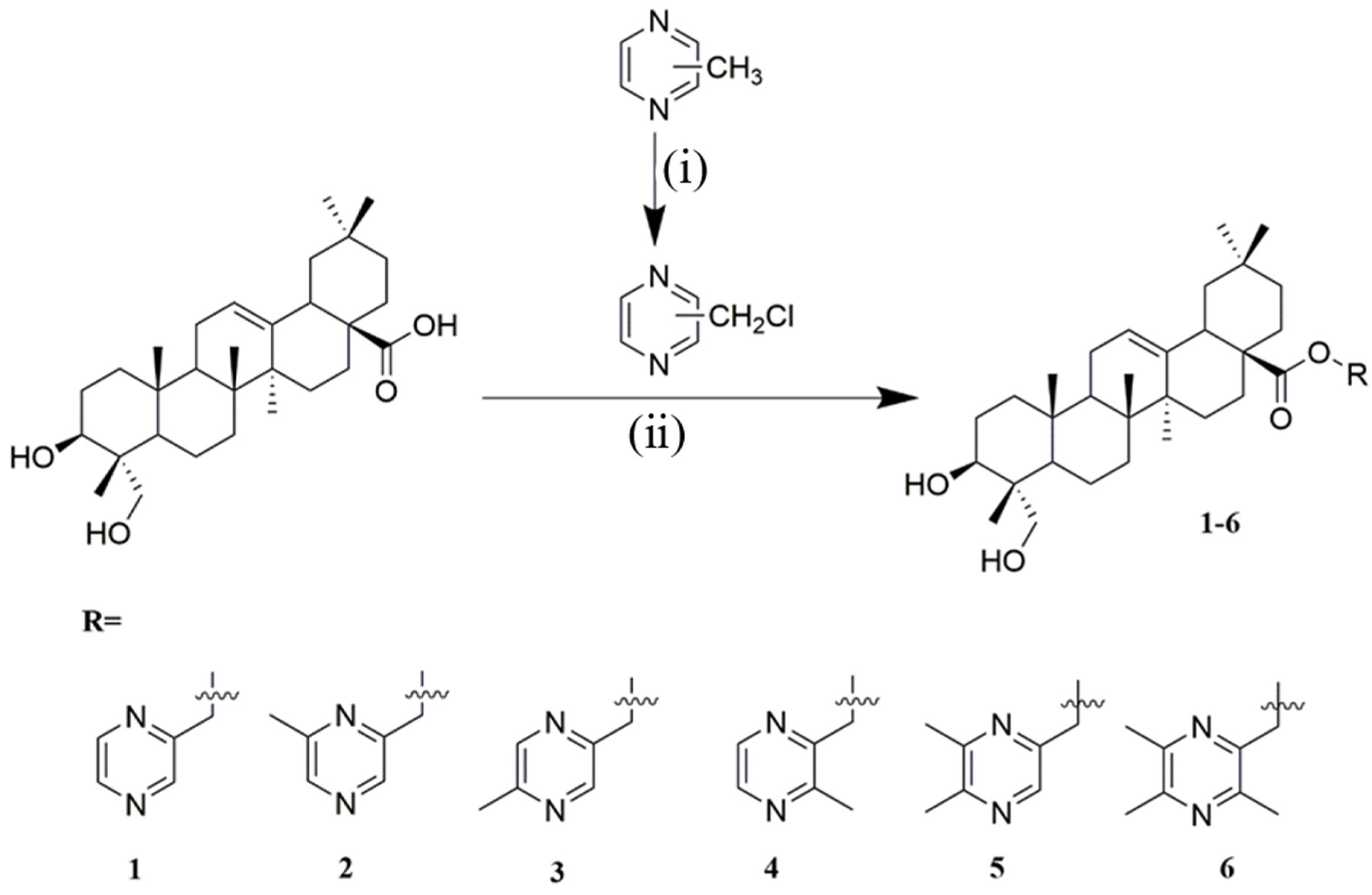
2.1. Chemistry
2.2. Biological Screening
2.2.1. Cytotoxicity Assay
2.2.2. Cluster Analysis
Principal Component Analysis (PCA)
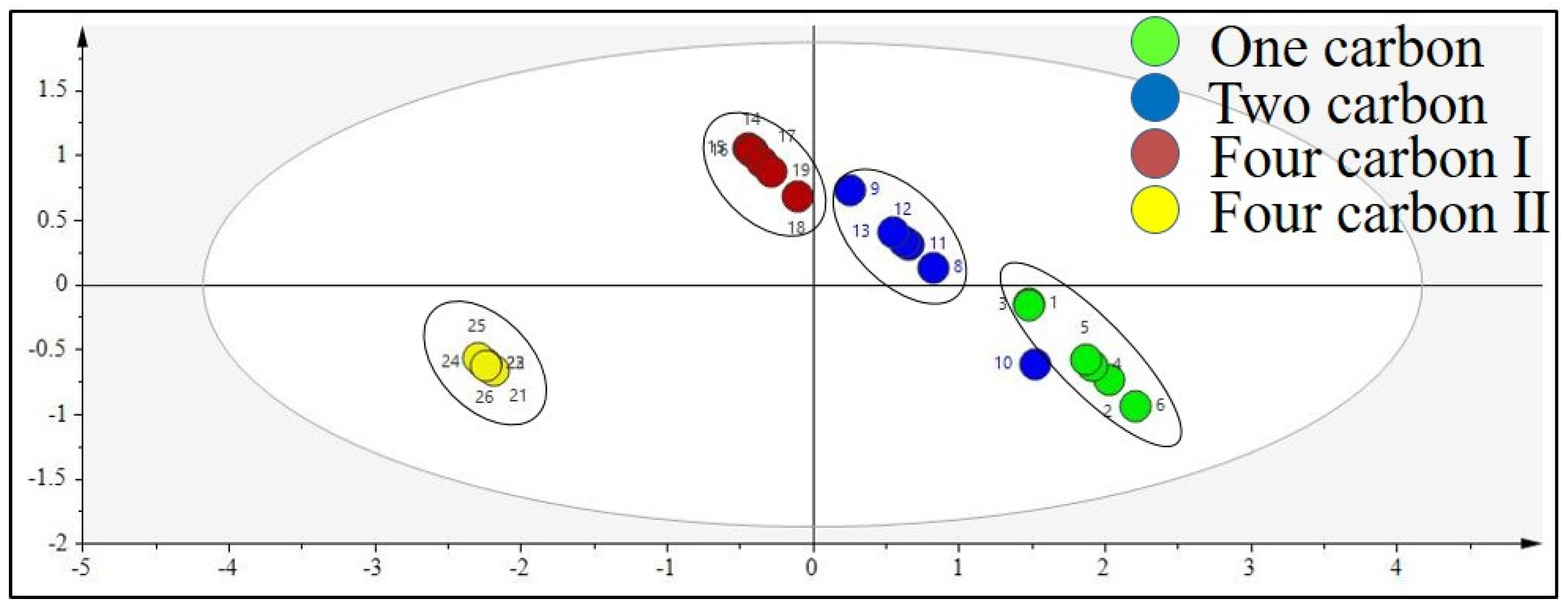
Partial Least Squares Discriminant Analysis (PLS-DA)
Structure–Activity Relationship Analysis
2.2.3. Analyses of Apoptosis
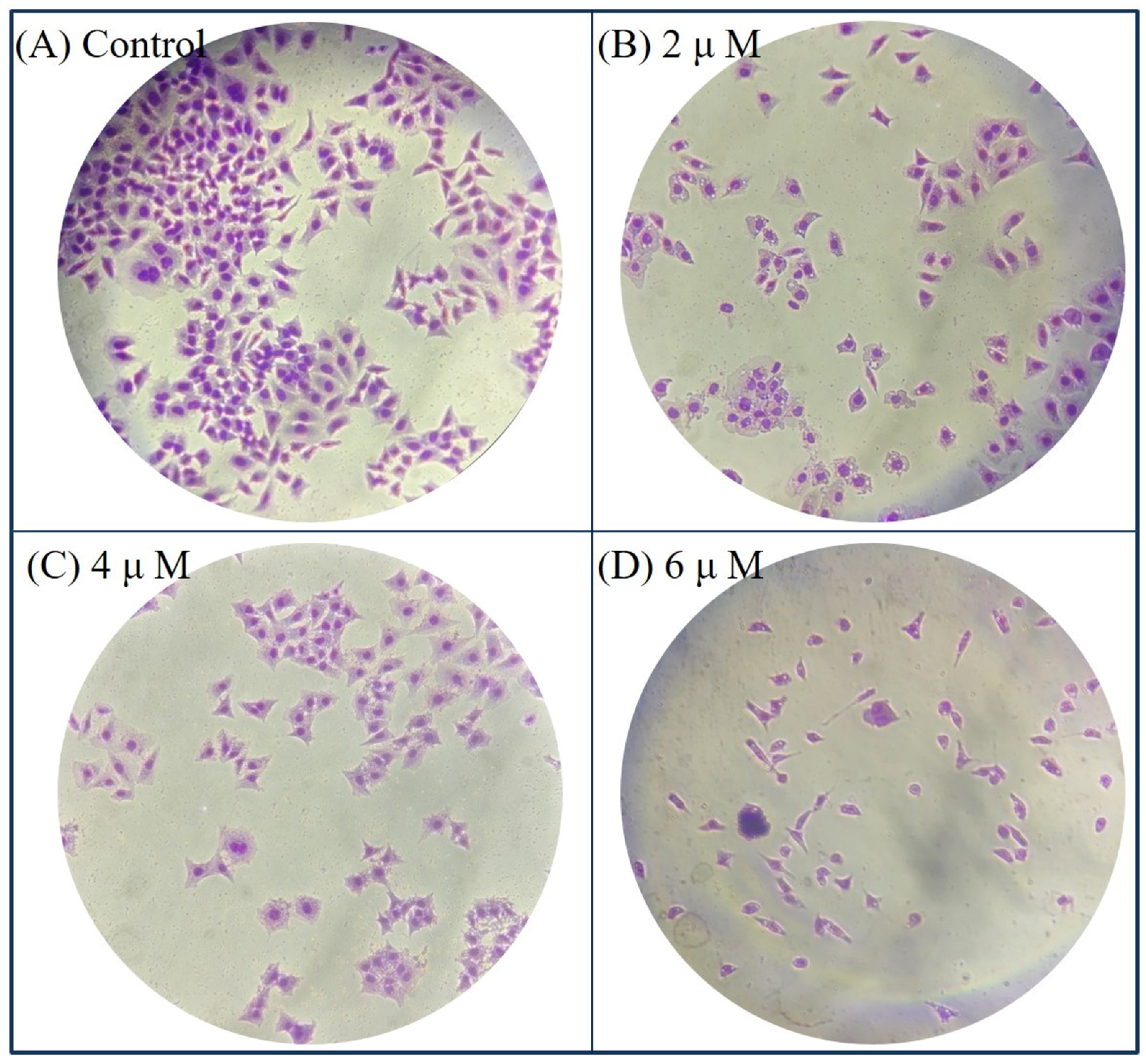
Morphological Detection of Apoptosis Using Giemsa Staining
4′,6-Diamidino-2-phenylindole (DAPI) Staining
Detection of Apoptosis Using Annexin V Fluorescein Isothiocyanate (FITC)/Propidium Iodide (PI) Staining
2.2.4. Detection of the Effect of Compound 9 on Cell-Cycle Progression
3. Discussion
4. Materials and Methods
4.1. General Aspects
4.1.1. Isolation of Hederagenin (He)
4.1.2. Preparation of Chloromethylpyrazine
4.1.3. Preparation of Pyrazinic Acid
4.1.4. Preparation of Compound 7
4.1.5. General Procedure for the Synthesis of Compounds 1–6
Synthesis of Compound 1
Synthesis of Compound 2
Synthesis of Compound 3
Synthesis of Compound 4
Synthesis of Compound 5
Synthesis of Compound 6
4.1.6. General Procedure for the Synthesis of Compounds 8–13
Synthesis of Compound 8
Synthesis of Compound 9
Synthesis of Compound 10
Synthesis of Compound 11
Synthesis of Compound 12
Synthesis of Compound 13
4.1.7. General Procedure for the Synthesis of Compounds 14–19
Synthesis of Compound 14
Synthesis of Compound 15
Synthesis of Compound 16
Synthesis of Compound 17
Synthesis of Compound 18
Synthesis of Compound 19
4.1.8. Procedure for the Synthesis of Compound 20
4.1.9. General Procedure for the Synthesis of Compounds 21–26
Synthesis of Compound 21
Synthesis of Compound 22
Synthesis of Compound 23
Synthesis of Compound 24
Synthesis of Compound 25
Synthesis of Compound 26
4.2. Biology Evaluation
4.2.1. Antitumor Activity
4.2.2. Giemsa Staining
4.2.3. DAPI Staining
4.2.4. Annexin V-FITC/PI Staining
4.2.5. Cell Cycle
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beghyn, T.; Deprezpoulain, R.; Willand, N.; Folleas, B.; Deprez, B. Natural compounds: leads or ideas? Bioinspired molecules for drug discovery. Chem. Biol. Drug Des. 2008, 72, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Misra, N.; Raj, K.; Srivastava, K.; Puri, S.K. Natural compounds in drug discovery: Pseudohybrid lupeol molecules as antimalarial. In Proceedings of the International Symposium on Current Trends and Drug Development and Research, Lucknow, India, 17–21 February 2007. [Google Scholar] [CrossRef]
- Petersen, F. Natural products based molecules for target and drug discovery in pharmaceutical research. Planta Med. Int. Open 2017, 4 (Suppl. 1). [Google Scholar]
- Manayi, A.; Nikan, M.; Haghighid, N.N.; Abdollahi, M. Advances in the anticancer value of the ursolic acid through nanodelivery. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Schwiebs, A.; Radeke, H. Immunopharmacological activity of betulin in inflammation-associated carcinogenesis. Anticancer Agents Med. Chem. 2018, 18, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; An, H.N.; Kumar, A.P.; Tan, B.K.H.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, B.M.F.; Salvador, J.A.R.; Marín, S.; Cascante, M. Synthesis and anticancer activity of novel fluorinated asiatic acid derivatives. Eur. J. Med. Chem. 2016, 114, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yadav, P.; Jaswal, V.; Kashyap, D.; Tuli, H.S.; Garg, V.K.; Das, S.K.; Srinivas, R. Celastrol as a pentacyclic triterpenoid with chemopreventive properties. Pharm. Pat. Anal. 2018, 7, 155–167. [Google Scholar]
- Fang, L.; Wang, M.; Gou, S.; Liu, X.; Zhang, H.; Cao, F. Combination of amino acid/dipeptide with nitric oxide donating oleanolic acid derivatives as PepT1 targeting antitumor prodrugs. J. Med. Chem. 2014, 57, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.W.; He, Y.; Wang, J.; Gao, W.; Liu, T.; Qin, M.; Wang, X.; Gao, C.; Wang, Y.; Liu, M.Y.; et al. Synthesis of heterocycle-modified betulinic acid derivatives as antitumor agents. Eur. J. Med. Chem. 2015, 95, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.W.; Li, Y.G.; Wang, P.; Zhou, K.; Hou, J.; Jin, Q.; Hao, D.C.; Ge, G.B.; Yang, L. Design, synthesis, and structure-activity relationship study of glycyrrhetinic acid derivatives as potent and selective inhibitors against human carboxylesterase 2. Eur. J. Med. Chem. 2016, 112, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.Y.; Son, K.H.; Do, J.C. Triterpenoids from the roots of Dipsacus asper. Chem. Pharm. Bull. (Tokyo) 1993, 16, 32–35. [Google Scholar]
- Sieben, A.; Prenner, L.; Sorkalla, T.; Wolf, A.; Jakobs, D.; Runkel, F.; Häberlein, H. α-Hederin, but Not Hederacoside C and Hederagenin from Hedera helix, Affects the Binding Behavior, Dynamics, and Regulation of β2-Adrenergic Receptors. Biochemistry 2009, 48, 3477–3482. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.T.; Sohn, I.C.; Park, H.J.; Kim, D.W.; Jung, G.O.; Park, K.Y. Essential moiety for antimutagenic and cytotoxic activity of hederagenin monodesmosides and bisdesmosides isolated from the stem bark of Kalopanax pictus. Planta Med. 2000, 66, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Baek, S.; Shin, D.; Lee, J.; Roh, J.L. Hederagenin Induces Apoptosis in Cisplatin-Resistant Head and Neck Cancer Cells by Inhibiting the Nrf2-ARE Antioxidant Pathway. Oxid. Med. Cell Longev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.G.; Zeng, W.; Wong, V.K.; Zhu, Y.Z.; Lo, A.C.; Liu, L.; Law, B.Y. Hederagenin and α-hederin promote degradation of proteins in neurodegenerative diseases and improve motor deficits in MPTP-mice. Pharmacol. Res. 2017, 115, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.H.; Guan, J.H.; Huang, Y.L.; Pan, Y.W.; Yang, W.; Lan, H.; Huang, S.; Hu, J.; Zhao, G.P. Experimental Study of Antiatherosclerosis Effects with Hederagenin in Rats. Evid. Based Complement. Altern. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.X.Z.; Zhou, J.Y.; Li, Y.; Zou, X.; Wu, J.; Gu, J.F.; Yuan, J.R.; Zhao, B.J.; Feng, L.; Jia, X.B. Hederagenin from the leaves of ivy (Hedera helix L.) induces apoptosis in human LoVo colon cells through the mitochondrial pathway. BMC Complement. Altern. Med. 2014, 14, 412. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Shi, L.; Wu, J.; Zhou, X.; Li, X.; Sun, X.; Zhu, L.; Xia, T.S.; Ding, Q. A hederagenin saponin isolated fromClematis ganpinianainduces apoptosis in breast cancer cells via the mitochondrial pathway. Oncol. Lett. 2018, 15, 1737–1743. [Google Scholar] [PubMed]
- Rodriguez-Hernandez, D.; Demuner, A.J.; Barbosa, L.C.; Csuk, R.; Heller, L. Hederagenin as a triterpene template for the development of new antitumor compounds. Eur. J. Med. Chem. 2015, 105, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, D.; Demuner, A.J.; Barbosa, L.C.A.; Heller, L.; Csuk, R. Novel hederagenin–triazolyl derivatives as potential anti-cancer agents. Eur. J. Med. Chem. 2016, 115, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Rasul, M.G.; Chakraborty, M.; Mandal, A.; Saha, A. Microwave assisted one-pot synthesis of pyrazine derivatives of pentacyclic triterpenoids and their biological activity. IJC-B 2011, 50B, 1519–1523. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Zhu, P.; Liu, J.; Xu, S.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Design, synthesis and antitumor activity of triterpenoid pyrazine derivatives from 23-hydroxybetulinic acid. Eur. J. Med. Chem. 2015, 97, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chu, F.; Zhang, Y.; Wang, X.; Li, Q.; Liu, W.; Xu, X.; Xing, Y.; Chen, J.; Wang, P. A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2015, 16, 21035–21055. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yan, W.Q.; Xu, X.; Wu, G.R.; Zhang, C.Z.; Han, Y.T.; Chu, F.H.; Zhao, R.; Wang, P.L.; Lei, H.M. Combination of amino acid/dipeptide with ligustrazine-betulinic acid as antitumor agents. Eur. J. Med. Chem. 2017, 130, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Song, S.; Liu, X.; Zhang, A.; Miao, Z.; Ding, C. Novel Dihydroisoxazoline-Alkyl Carbon Chain Hybrid Artemisinin Analogues (Artemalogs): Synthesis and Antitumor Activities. Rsc. Adv. 2016. [Google Scholar] [CrossRef]
- Silva, H.; Valério, B.C.; de França, C.C.; de Almeida, M.V.; César, E.T.; Silveira, J.N.; Garniersuillerot, A.; Fc, S.D.P.; Pereiramaia, E.C.; Fontes, A.P. Impact of the carbon chain length of novel platinum complexes derived from N-alkyl-propanediamines on their cytotoxic activity and cellular uptake. J. Inorg. Biochem. 2008, 102, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.M.; Yin, T.L.; Zheng, S.J.; Yang, J.; Yuan, B.F.; Feng, Y.Q. Metal oxide-based dispersive solid-phase extraction coupled with mass spectrometry analysis for determination of ribose conjugates in human follicular fluid. Talanta 2017, 167, 506. [Google Scholar] [CrossRef] [PubMed]
- Ivorra, E.; Girón, J.; Sánchez, A.J.; Verdú, S.; Barat, J.M.; Grau, R. Detection of expired vacuum-packed smoked salmon based on PLS-DA method using hyperspectral images. J. Food Eng. 2013, 117, 342–349. [Google Scholar] [CrossRef]
- Chwalek, M.; Lalun, N.; Bobichon, H.; Ple, K.; Voutquenne-Nazabadioko, L. Structure-activity relationships of some hederagenin diglycosides: Haemolysis, cytotoxicity and apoptosis induction. Biochim. Biophys. Acta 2006, 1760, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Yang, Y.T.; Wang, X.; Wang, K.Y.; Liu, J.Q.; Lei, L.; Luo, X.M.; Zhai, R.; Fu, F.H.; Wang, H.B. Design, synthesis and biological evaluation of novel α-hederagenin derivatives with anticancer activity. Eur. J. Med. Chem. 2017, 141, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.K.; Jain, D.C. 13C NMR spectroscopy of oleanane triterpenoids. Prog. Nucl. Magn. Reson. Spectrosc. 1992, 24, 1–90. [Google Scholar] [CrossRef]
- Sarikahya, N.B. Aristatosides A-C, hederagenin-type triterpene saponins from Cephalaria aristata. Phytochem. Lett. 2014, 8, 149–155. [Google Scholar] [CrossRef]
- Zhang, W.-T. Research on Preparation Process, Quality Standards of Dipsacus Sapogenin and Its Inhibitory Effect on Alpha-Glucosidase. Master’s Thesis, Jilin University, Jilin, China, June 2011. [Google Scholar]
- Wang, P.; Zhao, R.; Yan, W.; Zhang, X.; Zhang, H.; Xu, B.; Chu, F.; Han, Y.; et al. Neuroprotection by new ligustrazine-cinnamon acid derivatives on CoCl2 -induced apoptosis in differentiated PC12 cells. Bioorg. Chem. 2018, 77, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; She, G.; Yang, Y.; Li, Q.; Zhang, H.; Liu, J.; Cao, Y.; Xu, X.; Lei, H. Synthesis and biological evaluation of new ligustrazine derivatives as antitumor agents. Molecules 2012, 17, 4972–4985. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Half Maximal Inhibitory Concentration (IC50) Values (μM) | |||||
|---|---|---|---|---|---|
| Compound | A549 | MCF-7 | HepG2 | MDCK | H9c2 |
| 1 | 17.62 ± 0.37 | 16.05 ± 0.01 | 18.79 ± 0.04 | 18.06 ± 0.25 | 14.73 ± 0.51 |
| 2 | 20.55 ± 0.29 | 15.78 ± 0.22 | 27.23 ± 1.82 | 26.71 ± 1.64 | 25.09 ± 0.43 |
| 3 | 15.03 ± 0.35 | 16.83 ± 9.25 | 18.93 ± 0.35 | 18.42 ± 3.76 | >30 |
| 4 | 17.07 ± 1.06 | 17.31 ± 0.48 | 25.53 ± 1.40 | 26.12 ± 1.02 | 20.42 ± 2.24 |
| 5 | 24.37 ± 0.37 | >30 | 24.94 ± 0.06 | 23.53 ± 0.67 | 28.46 ± 0.29 |
| 6 | 25.52 ± 0.26 | >30 | >30 | 24.08 ± 0.76 | >30 |
| 7 | >30 | >30 | >30 | >30 | >30 |
| 8 | 23.92 ± 0.31 | 25.35 ± 0.09 | 17.41 ± 0.75 | 20.13 ± 0.30 | 21.66 ± 0.86 |
| 9 | 3.45 ± 0.59 | 8.73 ± 1.49 | 8.71 ± 0.38 | 14.11 ± 0.04 | 16.69 ± 0.12 |
| 10 | 20.93 ± 0.68 | 27.24 ± 2.09 | 28.17 ± 0.21 | 25.42 ± 0.28 | 19.60 ± 3.29 |
| 11 | 14.12 ± 0.01 | 27.30 ± 0.57 | 14.84 ± 0.03 | 18.64 ± 0.18 | 14.72 ± 0.16 |
| 12 | 10.05 ± 2.47 | 17.28 ± 0.58 | 14.42 ± 0.03 | 18.64 ± 0.71 | 13.16 ± 0.20 |
| 13 | 8.15 ± 0.17 | 18.03 ± 0.18 | 13.24 ± 0.29 | 23.22 ± 0.01 | 10.51 ± 0.61 |
| 14 | 17.60 ± 2.33 | 31.23 ± 0.15 | 7.05 ± 0.17 | 18.10 ± 0.12 | 15.90 ± 0.22 |
| 15 | 17.98 ± 0.04 | 19.77 ± 1.27 | 6.71 ± 0.20 | 17.12 ± 1.01 | 17.56 ± 1.85 |
| 16 | 24.78 ± 0.66 | 28.49 ± 0.28 | 6.99 ± 0.63 | 20.12 ± 0.06 | 14.99 ± 0.12 |
| 17 | 19.57 ± 0.22 | 22.88 ± 0.66 | 8.15 ± 0.22 | 20.93 ± 3.03 | 22.31 ± 0.30 |
| 18 | 16.17 ± 0.15 | 28.29 ± 0.41 | 11.91 ± 0.99 | 16.55 ± 0.22 | >30 |
| 19 | 12.23 ± 0.4 | 27.57 ± 0.52 | 9.06 ± 0.32 | 16.43 ± 0.04 | 16.50 ± 0.22 |
| 20 | >30 | >30 | >30 | >30 | >30 |
| 21 | 9.29 ± 0.99 | 24.98 ± 0.18 | 7.99 ± 0.12 | 14.85 ± 0.06 | 16.68 ± 0.10 |
| 22 | 10.40 ± 0.43 | 20.53 ± 3.63 | 7.15 ± 0.43 | 16.34 ± 1.57 | 16.26 ± 0.07 |
| 23 | 18.67 ± 0.32 | 21.26 ± 1.28 | 7.17 ± 1.09 | 19.49 ± 2.47 | 23.35 ± 0.55 |
| 24 | 21.97 ± 0.87 | 17.95 ± 2.80 | 6.89 ± 0.21 | 19.27 ± 1.47 | 27.87 ± 0.62 |
| 25 | >30 | 15.63 ± 1.41 | 6.42 ± 0.24 | >30 | >30 |
| 26 | >30 | 18.30 ± 1.16 | 7.15 ± 0.85 | >30 | >30 |
| He | >50 | >50 | >50 | >50 | >50 |
| DDP | 3.85 ± 0.63 | 5.17 ± 0.28 | 3.42 ± 0.68 | 9.97 ± 1.12 | 5.31 ± 0.26 |
| Cell Types | R2X | Q2(cum) |
|---|---|---|
| A549 | 0.761 | 0.159 |
| HepG2 | 0.872 | 0.697 |
| MCF-7 | 0.748 | 0.148 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, K.; Zhang, X.-H.; Han, Y.-T.; Wu, G.-R.; Cai, D.-S.; Xue, N.-N.; Guo, W.-B.; Yang, Y.-Q.; Chen, M.; Zhang, X.-Y.; et al. Design, Synthesis, and Cytotoxic Analysis of Novel Hederagenin–Pyrazine Derivatives Based on Partial Least Squares Discriminant Analysis. Int. J. Mol. Sci. 2018, 19, 2994. https://doi.org/10.3390/ijms19102994
Fang K, Zhang X-H, Han Y-T, Wu G-R, Cai D-S, Xue N-N, Guo W-B, Yang Y-Q, Chen M, Zhang X-Y, et al. Design, Synthesis, and Cytotoxic Analysis of Novel Hederagenin–Pyrazine Derivatives Based on Partial Least Squares Discriminant Analysis. International Journal of Molecular Sciences. 2018; 19(10):2994. https://doi.org/10.3390/ijms19102994
Chicago/Turabian StyleFang, Kang, Xiao-Hua Zhang, Yao-Tian Han, Gao-Rong Wu, De-Sheng Cai, Nan-Nan Xue, Wen-Bo Guo, Yu-Qin Yang, Meng Chen, Xin-Yu Zhang, and et al. 2018. "Design, Synthesis, and Cytotoxic Analysis of Novel Hederagenin–Pyrazine Derivatives Based on Partial Least Squares Discriminant Analysis" International Journal of Molecular Sciences 19, no. 10: 2994. https://doi.org/10.3390/ijms19102994
APA StyleFang, K., Zhang, X.-H., Han, Y.-T., Wu, G.-R., Cai, D.-S., Xue, N.-N., Guo, W.-B., Yang, Y.-Q., Chen, M., Zhang, X.-Y., Wang, H., Ma, T., Wang, P.-L., & Lei, H.-M. (2018). Design, Synthesis, and Cytotoxic Analysis of Novel Hederagenin–Pyrazine Derivatives Based on Partial Least Squares Discriminant Analysis. International Journal of Molecular Sciences, 19(10), 2994. https://doi.org/10.3390/ijms19102994