Proteomics Analysis of SsNsd1-Mediated Compound Appressoria Formation in Sclerotinia sclerotiorum

 ,
,  ,
,
Abstract

1. Introduction
2. Results
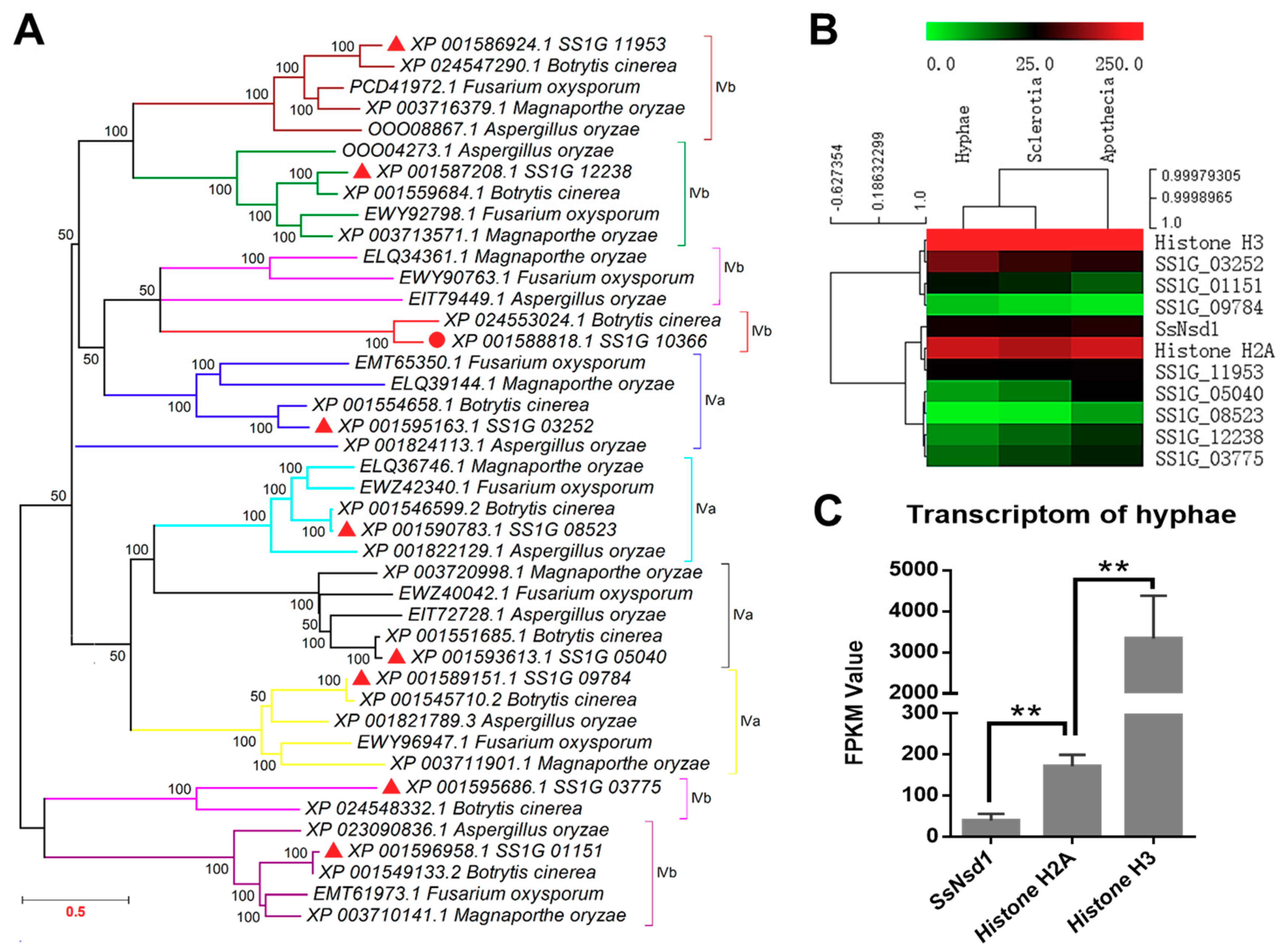
2.1. Phylogenetic Analysis of SsNsd1 and Other GATA-Type Proteins
2.2. Transcript Accumulation of SsNsd1 and Other GATA-Type Proteins
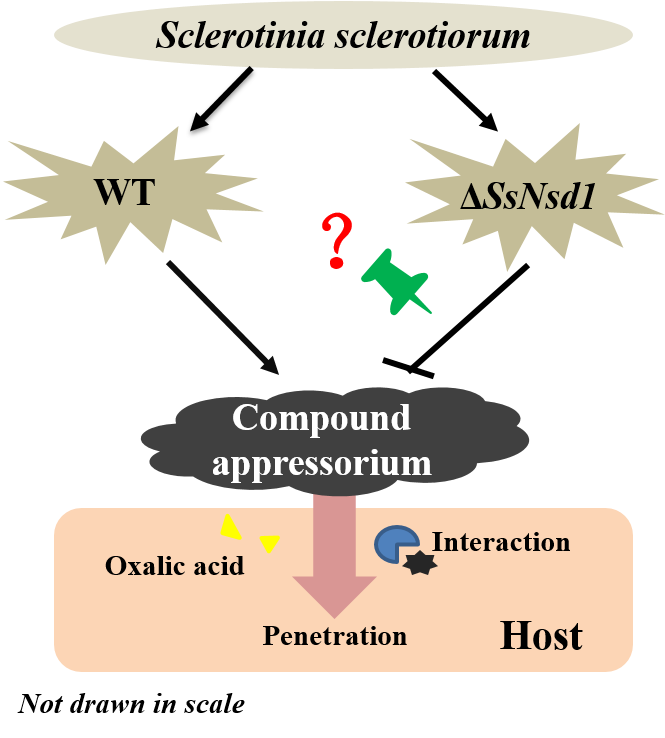
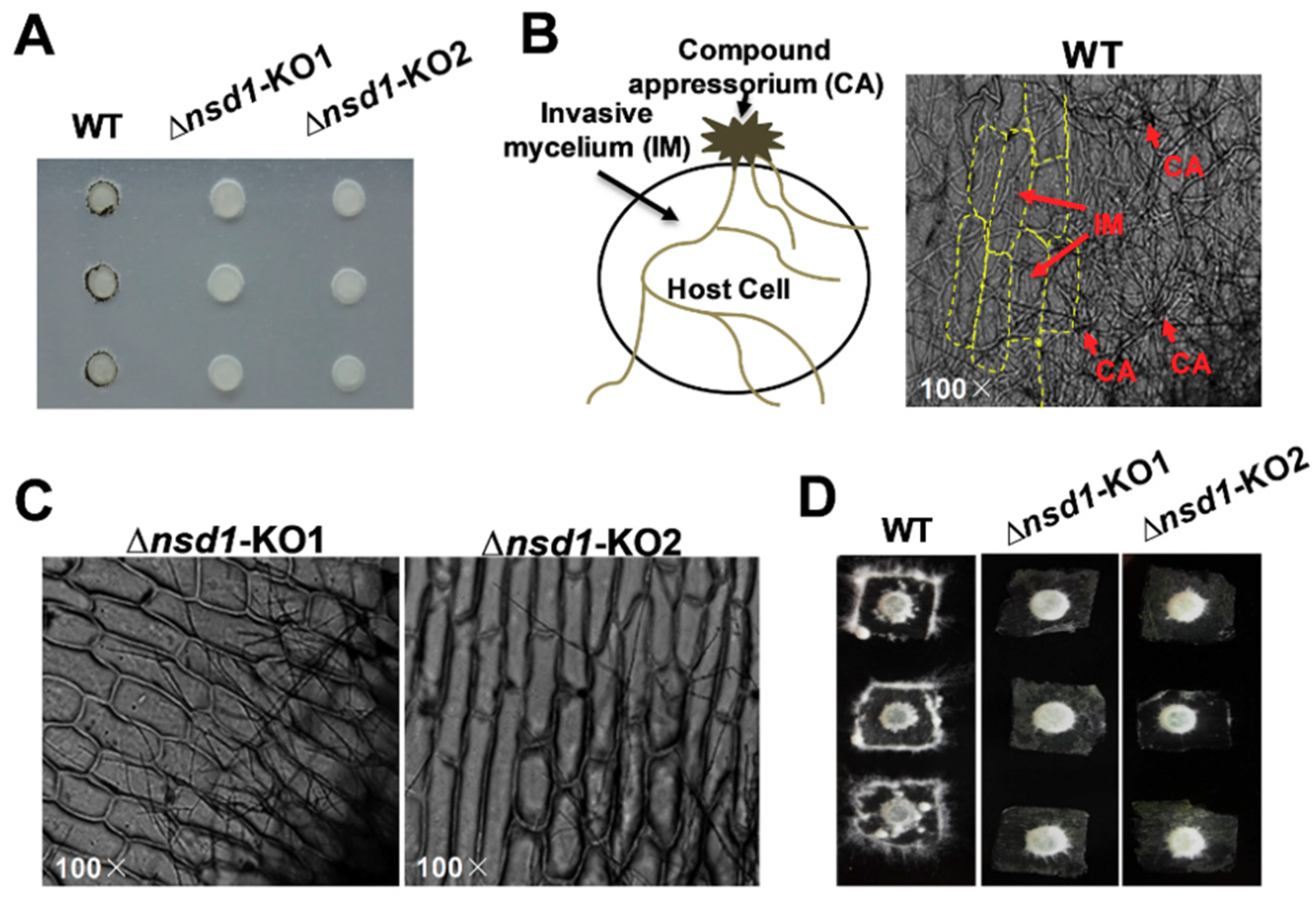
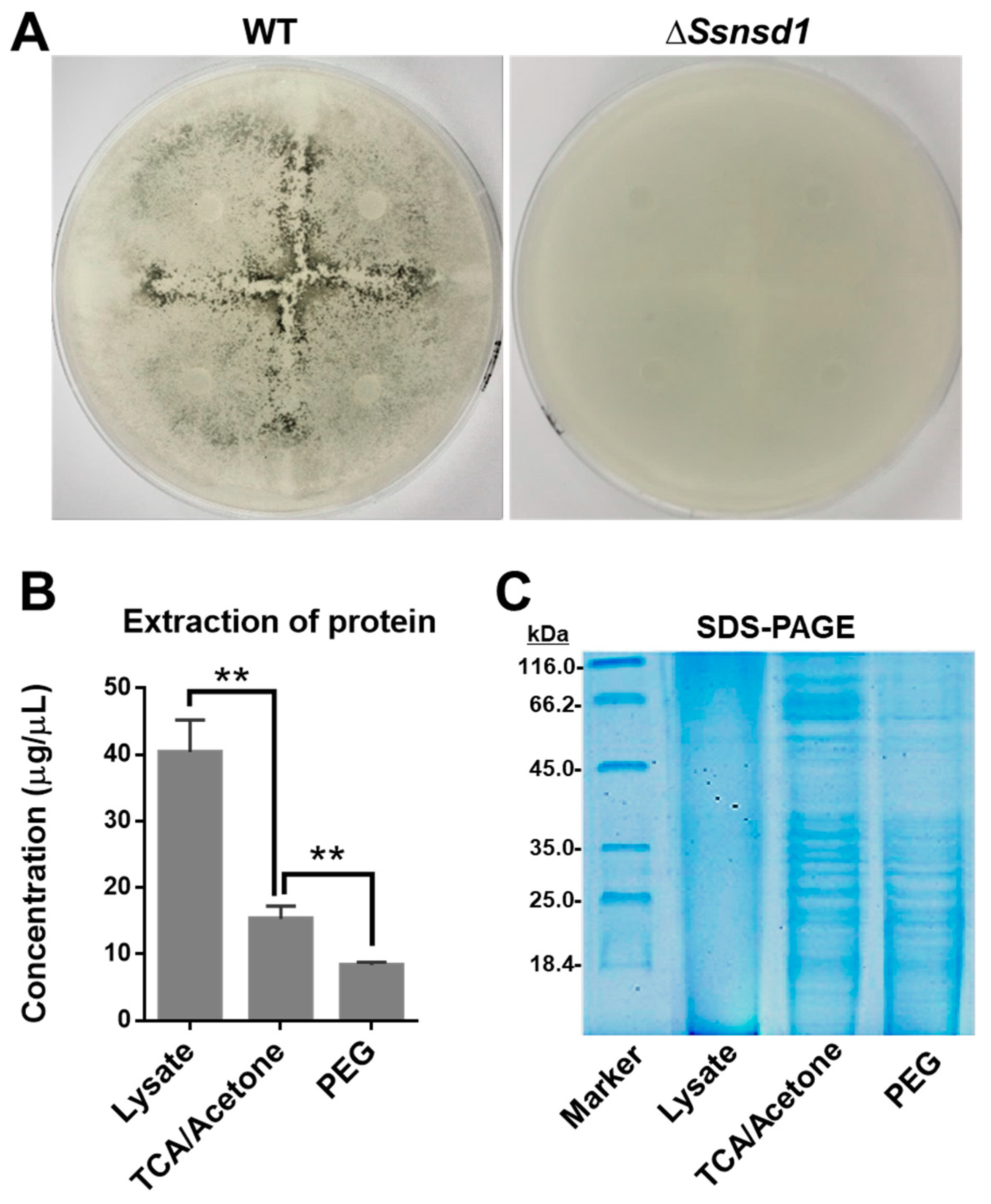
2.3. ΔSsNsd1 Mutant Suppressed Compound Appressorium Formation
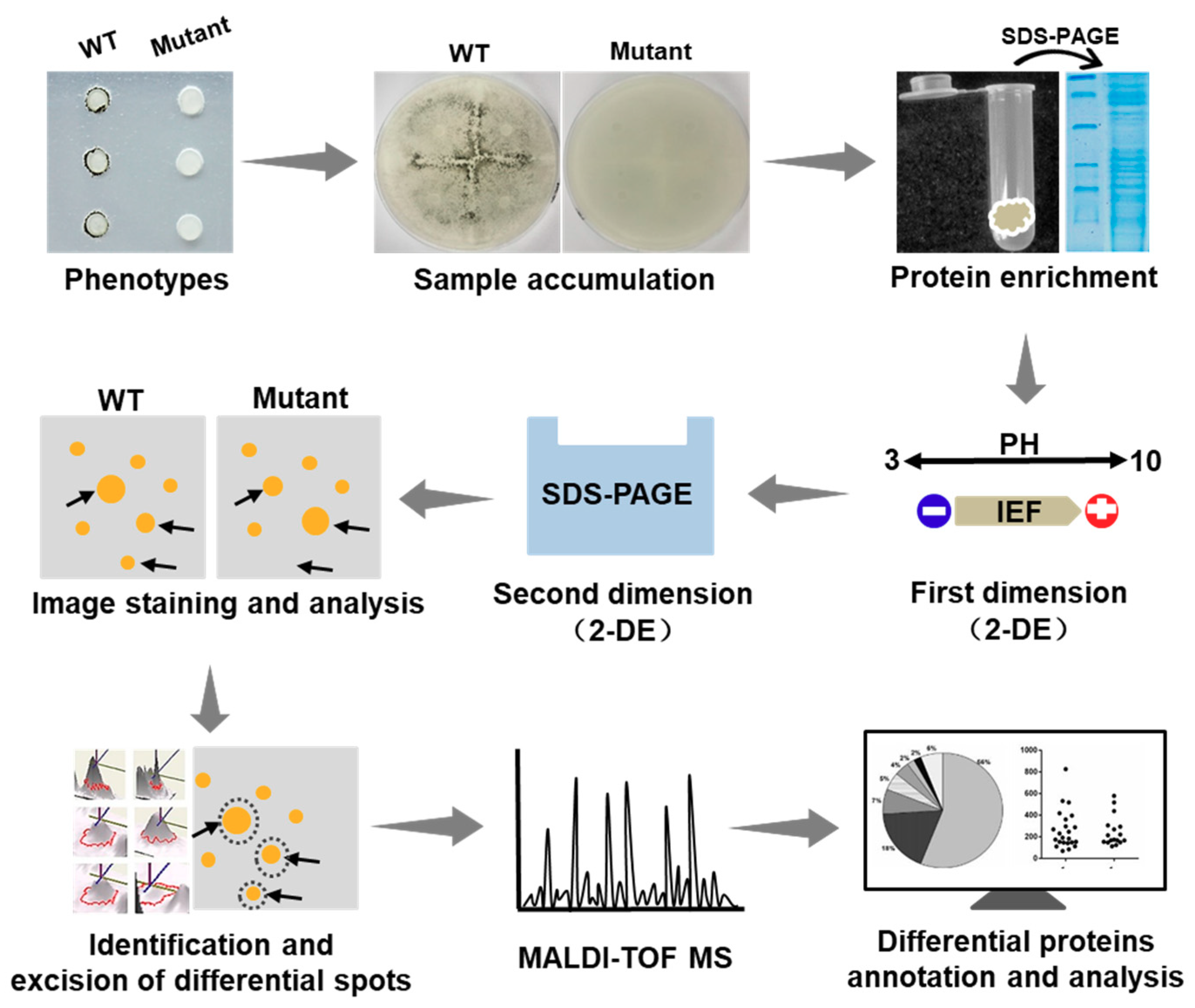
2.4. Diagram of the Identification of Differential Proteins
2.5. Protein Extraction from Enriched Compound Appressoria
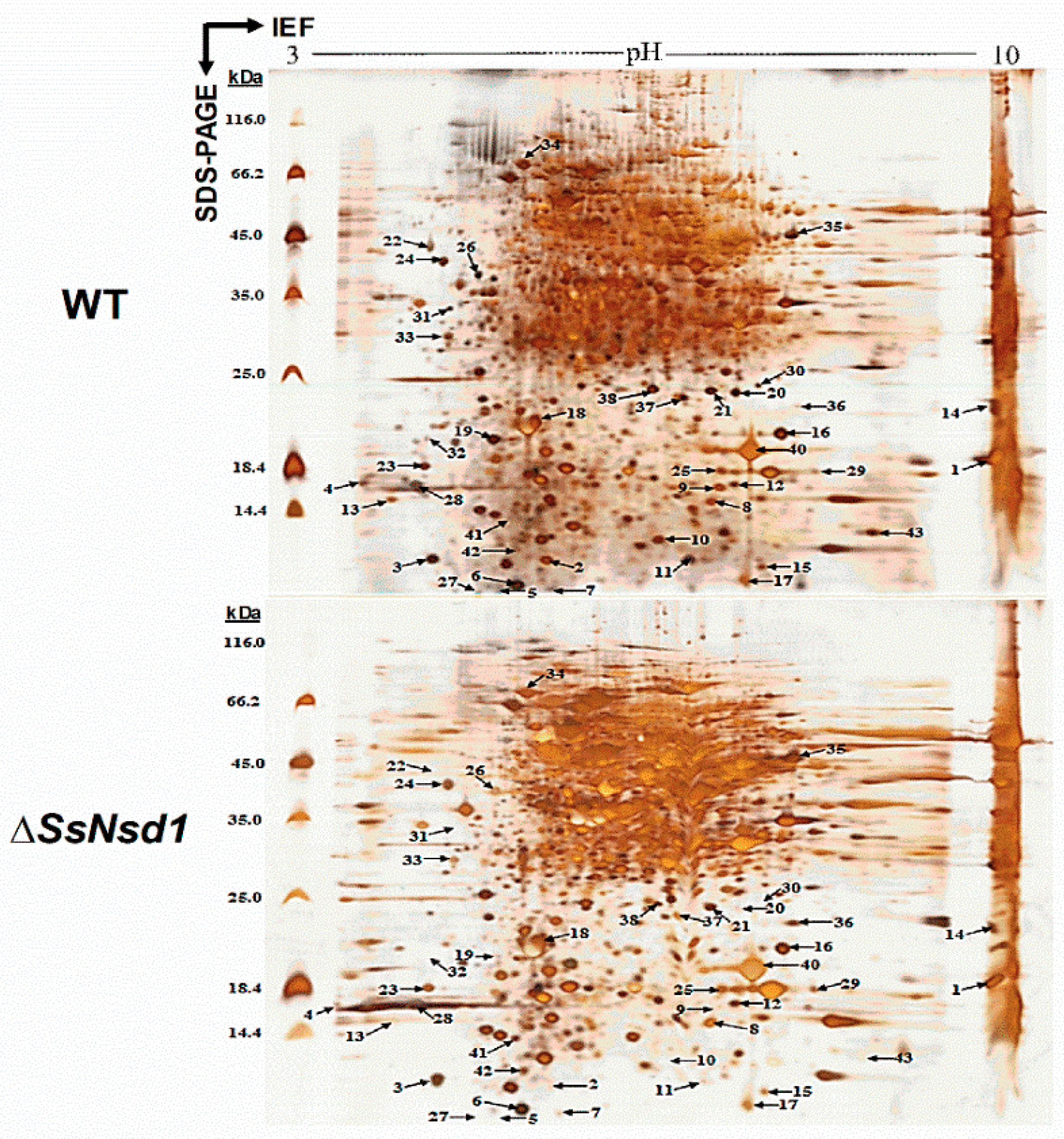
2.6. Identification of the Differential Protein Spots by 2-DE Analysis
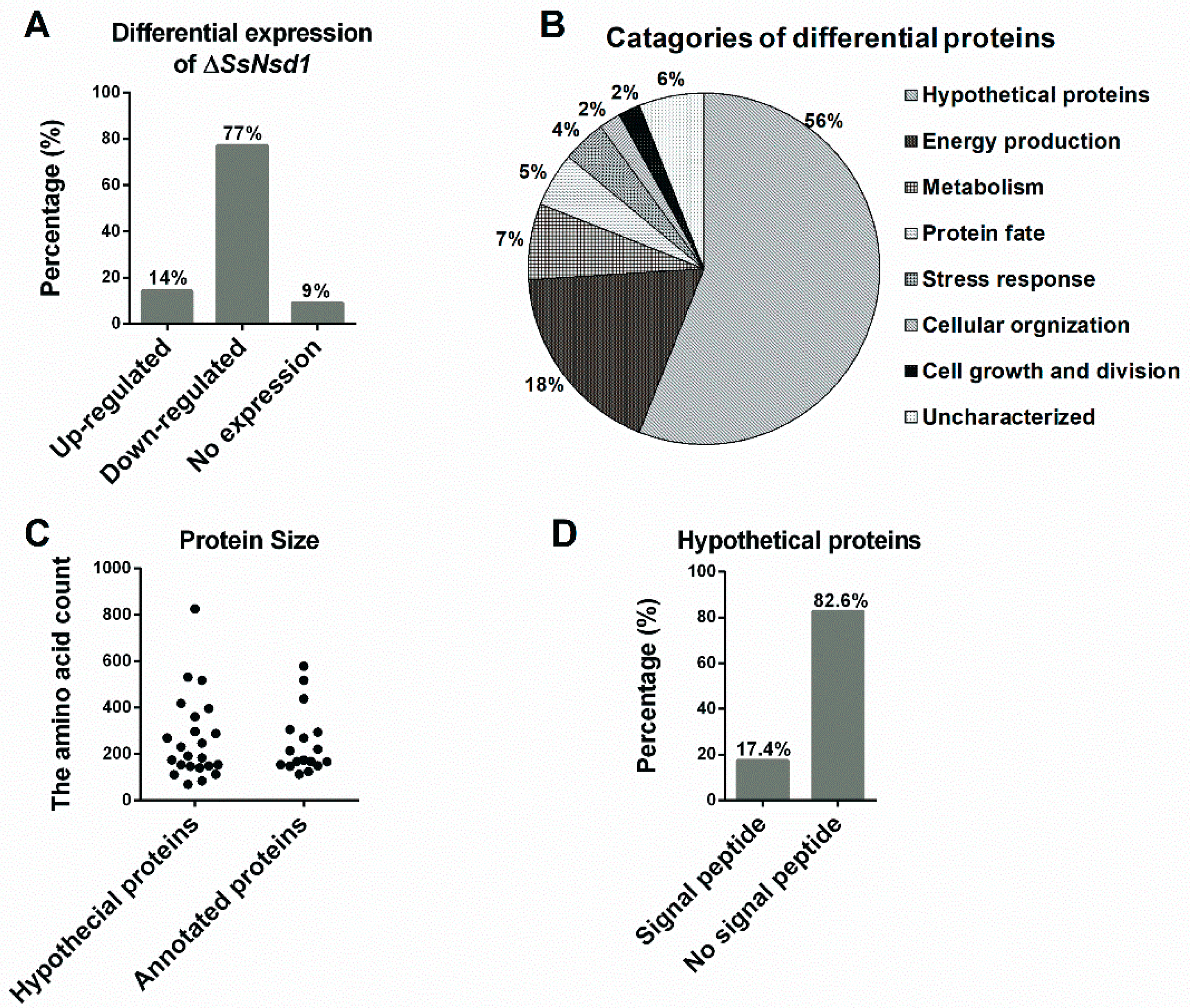
2.7. Prediction and Characteristics of the Identified Proteins
2.8. Functional Analysis of Annotated Proteins
3. Discussion
4. Materials and Methods
4.1. Fungal Strains and Culture Conditions
4.2. Phylogenetic Analysis of SsNsd1 and Other GATA-Type Proteins
4.3. Digital Gene Expression of SsNsd1 and Other GATA-Type Proteins
4.4. Compound Appressorium Assays
4.5. Two-Dimensional Gel Electrophoresis (2-DE) Strategy in This Study
4.6. Protein Extraction and Optimization
4.7. The 2-DE Assay and Image Analysis
4.8. MALDI-TOF Analysis and Prediction of Differential Proteins
4.9. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boland, G.J.; Hall, R. Index of plant hosts of Sclerotinia sclerotiorum. Can. J. Plant Pathol. 1994, 16, 93–108. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Zhang, Y.; Yu, P.-L.; Pan, H.; Rollins, J.A. Introduction of large sequence inserts by CRISPR-Cas9 to create pathogenicity mutants in the multinucleate filamentous pathogen Sclerotinia sclerotiorum. mBio 2018, 9, e00567-18. [Google Scholar] [CrossRef] [PubMed]
- Bardin, S.D.; Huang, H.C. Research on biology and control of sclerotinia diseases in Canada. Can. J. Plant Pathol. 2001, 23, 88–98. [Google Scholar] [CrossRef]
- Li, J.; Mu, W.; Veluchamy, S.; Liu, Y.; Zhang, Y.; Pan, H.; Rollins, J.A. The GATA-type IVb zinc-finger transcription factor SsNsd1 regulates asexual–sexual development and appressoria formation in Sclerotinia sclerotiorum. Mol. Plant Pathol. 2018, 19, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.D.; Thomma, B.P.H.J.; Nelson, B.D. Sclerotinia sclerotiorum (Lib.) de Bary: Biology and molecular traits of a cosmopolitan pathogen. Mol. Plant Pathol. 2006, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.L.; Stotz, H.U. Oxalate production by Sclerotinia sclerotiorum deregulates guard cells during infection. Plant Physiol. 2004, 136, 3703–3711. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Hua, L.; Sivasithamparam, K.; Kuo, J.; Barbetti, M.J. The infection processes of Sclerotinia sclerotiorum in cotyledon tissue of a resistant and a susceptible genotype of Brassica napus. Ann. Bot. 2010, 106, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, R.D.; Dow, R.L. Histopathology of Sclerotinia sclerotiorum infection of bean. Phytopathology 1973, 63, 708. [Google Scholar] [CrossRef]
- Xiao, X.; Xie, J.; Cheng, J.; Li, G.; Yi, X.; Jiang, D.; Fu, Y. Novel secretory protein Ss-Caf1 of the plant-pathogenic fungus Sclerotinia sclerotiorum is required for host penetration and normal sclerotial development. Mol. Plant Microbe Interact. 2014, 27, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Liberti, D.; Li, M.; Kim, Y.-T.; Hutchens, A.; Wilson, R.; Rollins, J.A. Oxaloacetate acetylhydrolase gene mutants of Sclerotinia sclerotiorumdo not accumulate oxalic acid, but do produce limited lesions on host plants. Mol. Plant Pathol. 2015, 16, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Moomaw, E.W.; Rollins, J.A. Fungal oxalate decarboxylase activity contributes to Sclerotinia sclerotiorum early infection by affecting both compound appressoria development and function. Mol. Plant Pathol. 2015, 16, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Zuppini, A.; Navazio, L.; Sella, L.; Castiglioni, J.; Favaron, F.; Mariani, P. An endopolygalacturonase from Sclerotinia sclerotiorum induces calcium-mediated signaling and programmed cell death in soybean cells. Mol. Plant Microbe Interact. 2005, 18, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Yajima, W.; Liang, Y.; Kav, N.N.V. Gene disruption of an arabinofuranosidase/beta-xylosidase precursor decreases Sclerotinia sclerotiorum virulence on canola tissue. Mol. Plant Microbe Interact. 2009, 22, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wei, W.; Fu, Y.; Cheng, J.; Xie, J.; Li, G.; Yi, X.; Kang, Z.; Dickman, M.B.; Jiang, D. A secretory protein of necrotrophic fungus Sclerotinia sclerotiorum that suppresses host resistance. PLoS ONE 2013, 8, e53901. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, J.; Zhu, W.; Yang, Y.; Mei, J.; Bi, C.; Qian, W.; Qing, L.; Tan, W. Ss-Rhs1, a secretory Rhs repeat-containing protein, is required for the virulence of Sclerotinia sclerotiorum. Mol. Plant Pathol. 2017, 18, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liang, X.; Rollins, J.A. Sclerotinia sclerotiorum γ-Glutamyl transpeptidase (Ss-Ggt1) is required for regulating glutathione accumulation and development of sclerotia and compound appressoria. Mol. Plant Microbe Interact. 2012, 25, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Jurick, W.M.; Rollins, J.A. Deletion of the adenylate cyclase (sac1) gene affects multiple developmental pathways and pathogenicity in Sclerotinia sclerotiorum. Fungal Genet. Biol. 2007, 44, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Erental, A.; Harel, A.; Yarden, O. Type 2A phosphoprotein phosphatase is required for asexual development and pathogenesis of Sclerotinia sclerotiorum. Mol. Plant Microbe Interact. 2007, 20, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Simon, A.; Cohrs, K.C.; Viaud, M.; Tudzynski, P. The transcription factor BcLTF1 regulates virulence and light responses in the necrotrophic plant pathogen Botrytis cinerea. PLoS Genet. 2014, 10, e1004040. [Google Scholar] [CrossRef] [PubMed]
- Berth, M.; Moser, F.M.; Kolbe, M.; Bernhardt, J. The state of the art in the analysis of two-dimensional gel electrophoresis images. Appl. Microbiol. Biotechnol. 2007, 76, 1223–1243. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837. [Google Scholar] [CrossRef] [PubMed]
- Michael, F.; Peter, B.; Hanno, L.; Laura, S. The rat liver mitochondrial proteins. Electrophoresis 2002, 23, 311–328. [Google Scholar]
- Mayer, K.; Albrecht, S.; Schaller, A. Targeted Analysis of Protein Phosphorylation by 2D Electrophoresis. In Plant Phosphoproteomics: Methods and Protocols; Schulze, W.X., Ed.; Springer: New York, NY, USA, 2015; pp. 167–176. [Google Scholar]
- Makrantoni, V.; Antrobus, R.; Botting, C.H.; Coote, P.J. Rapid enrichment and analysis of yeast phosphoproteins using affinity chromatography, 2D-PAGE and peptide mass fingerprinting. Yeast 2005, 22, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Voedisch, M.; Scherlach, K.; Winkler, R.; Hertweck, C.; Horn, U.; Brakhage, A.A.; Kniemeyer, O. Characterisation of the hypoxic response of the human-pathogenic fungus Aspergillus fumigatus by 2D-gel electrophoresis. Infection 2009, 37, 19. [Google Scholar]
- Zakharova, K.; Sterflinger, K.; Razzazi-Fazeli, E.; Noebauer, K.; Marzban, G. Global proteomics of the extremophile black fungus Cryomyces antarcticus using 2D-electrophoresis. Nat. Sci. 2014, 6, 978–995. [Google Scholar]
- Dharmasiri, S.; Harrington, H.M.; Dharmasiri, N. Heat shock modulates phosphorylation status and activity of nucleoside diphosphate kinase in cultured sugarcane cells. Plant Cell Rep. 2010, 29, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Han, Y.-J.; Kim, J.-I.; Song, P.-S. Arabidopsis nucleoside diphosphate kinase-2 as a plant GTPase activating protein. BMB Rep. 2008, 41, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Hetmann, A.; Kowalczyk, S. Nucleoside diphosphate kinase isoforms regulated by phytochrome A isolated from oat coleoptiles. Acta Biochim. Pol. 2009, 56, 143–153. [Google Scholar] [PubMed]
- Wang, X.-L.; Cai, H.-P.; Ge, J.-H.; Su, X.-F. Detection of eukaryotic translation initiation factor 4E and its clinical significance in hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 2540–2544. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-D.; Shi, X.-B.; Sun, L.-B.; Zhou, Q.-Y.; Zheng, D.-W.; Shi, H.-S.; Che, Y.-L.; Wang, Z.-S.; Shao, G.-F. Down-regulation of eIF5A-2 prevents epithelial-mesenchymal transition in non-small-cell lung cancer cells. J. Zhejiang Univ. Sci. B 2013, 14, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Achilonu, I.; Elebo, N.; Hlabano, B.; Owen, G.R.; Papathanasopoulos, M.; Dirr, H.W. An update on the biophysical character of the human eukaryotic elongation factor 1 beta: Perspectives from interaction with elongation factor 1 gamma. J. Mol. Recognit. 2018, 31, e2708. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Sun, X.-X.; Lu, H. Ribosomal protein L11 associates with c-Myc at 5 S rRNA and tRNA genes and regulates their expression. J. Biol. Chem. 2010, 285, 12587–12594. [Google Scholar] [CrossRef] [PubMed]
- Cushman, I.; Bowman, B.R.; Sowa, M.E.; Lichtarge, O.; Quiocho, F.A.; Moore, M.S. Computational and biochemical identification of a nuclear pore complex binding site on the nuclear transport carrier NTF2. J. Mol. Biol. 2004, 344, 303–310. [Google Scholar] [CrossRef] [PubMed]
- De Boor, S.; Knyphausen, P.; Kuhlmann, N.; Wroblowski, S.; Brenig, J.; Scislowski, L.; Baldus, L.; Nolte, H.; Krüger, M.; Lammers, M. Small GTP-binding protein Ran is regulated by posttranslational lysine acetylation. Proc. Natl. Acad. Sci. USA 2015, 112, E3679–E3688. [Google Scholar] [CrossRef] [PubMed]
- Uben. Ubiquitin-conjugating Enzyme, E2. In Encyclopedic Reference of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; p. 942. [Google Scholar]
- Takayama, Y.; Mamnun, Y.M.; Trickey, M.; Dhut, S.; Masuda, F.; Yamano, H.; Toda, T.; Saitoh, S. Hsk1- and SCFPof3-dependent proteolysis of S. pombe Ams2 ensures histone homeostasis and centromere function. Dev. Cell 2010, 18, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, H.A.; Hamelberg, D. Conformational selection in the recognition of phosphorylated substrates by the catalytic domain of human Pin1. Biochemistry 2011, 50, 9605–9615. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.-C.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011, 36, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, A.; Remington, S.J. Citrate synthase: Structure, control, and mechanism. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Quispe, C.F. GATA-Family Transcription Factors in Magnaporthe oryzae. Master’s Thesis, University of Nebraska-Lincoln, Lincoln, NE, USA, 2011. [Google Scholar]
- Liu, L.; Wang, Q.; Sun, Y.; Zhang, Y.; Zhang, X.; Liu, J.; Yu, G.; Pan, H. Sssfh1, a gene encoding a putative component of the RSC chromatin remodeling complex, is involved in hyphal growth, reactive oxygen species accumulation, and pathogenicity in Sclerotinia sclerotiorum. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Tsitsigiannis, D.I.; Kowieski, T.M.; Zarnowski, R.; Keller, N.P. Endogenous lipogenic regulators of spore balance in Aspergillus nidulans. Eukaryot. Cell 2004, 3, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Han, K.-H.; Han, K.-Y.; Yu, J.-H.; Chae, K.-S.; Jahng, K.-Y.; Han, D.-M. The nsdD gene encodes a putative GATA-type transcription factor necessary for sexual development of Aspergillus nidulans. Mol. Microbiol. 2001, 41, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-K.; Kwon, N.-J.; Lee, I.-S.; Jung, S.; Kim, S.-C.; Yu, J.-H. Negative regulation and developmental competence in Aspergillus. Sci. Rep. 2016, 6, 28874. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.; Görg, A. High-resolution two-dimensional electrophoresis. Methods Mol. Biol. 2009, 564, 13–32. [Google Scholar] [PubMed]
- Weiss, W.; Görg, A. Two-dimensional electrophoresis for plant proteomics. Methods Mol. Biol. 2007, 355, 121–143. [Google Scholar] [PubMed]
- Görg, A.; Weiss, W.; Dunn, M.J. Current two-dimensional electrophoresis technology for proteomics. Proteomics 2004, 4, 3665–3685. [Google Scholar] [CrossRef] [PubMed]
- Görg, A.; Weiss, W. Two-Dimensional Electrophoresis with Immobilized pH Gradients. In Proteome Research: Two-Dimensional Gel Electrophoresis and Identification Methods; Rabilloud, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 57–106. [Google Scholar]
- Yu, Z.; Dan, H.; Wang, S. Comparison of methods for extraction of protein secreted from Aspergillus fumigatus. Chin. J. Biol. 2010, 23, 313–316. [Google Scholar]
- Smith, R. Two-Dimensional Electrophoresis: An Overview. In Two-Dimensional Electrophoresis Protocols; Tyther, R., Sheehan, D., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 2–17. [Google Scholar]
- Pedreschi, R.; Hertog, M.L.; Carpentier, S.C.; Lammertyn, J.; Robben, J.; Noben, J.P.; Panis, B.; Swennen, R.; Nicolaï, B.M. Treatment of missing values for multivariate statistical analysis of gel-based proteomics data. Proteomics 2008, 8, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; van der Geer, P.; Hunter, T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991, 201, 110–149. [Google Scholar] [PubMed]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255. [Google Scholar] [CrossRef] [PubMed]
- Schulze, W.X. Proteomics approaches to understand protein phosphorylation in pathway modulation. Curr. Opin. Plant Biol. 2010, 13, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, J.; Hiromasa, Y.; Pan, H.; Bai, G. Differentially expressed proteins associated with fusarium head blight resistance in wheat. PLoS ONE 2013, 8, e82079. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, M.; Tatsumi, Y.; Tamesui, N.; Kumada, Y.; Horiuchi, J.I.; Okumura, K. Dynamic analysis of the silver staining gel image of 2-DE for protein spots segmentation. J. Electrophor. 2010, 54, 1–7. [Google Scholar] [CrossRef]
- Li, J.; Yu, G.; Sun, X.; Zhang, X.; Liu, J.; Pan, H. AcEBP1, an ErbB3-Binding Protein (EBP1) from halophyte Atriplex canescens, negatively regulates cell growth and stress responses in Arabidopsis. Plant Sci. 2016, 248, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Teakle, G.R.; Gilmartin, P.M. Two forms of type IV zinc-finger motif and their kingdom-specific distribution between the flora, fauna and fungi. Trends Biochem. Sci. 1998, 23, 100–102. [Google Scholar] [CrossRef]
- Reyes, J.C.; Muro-Pastor, M.I.; Florencio, F.J. The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol. 2004, 134, 1718–1732. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.-B.; Zhang, X.-J.; Liu, C.-Z.; Wei, J.-K.; Li, F.-H.; Xiang, J.-H. Horizontally transferred genes in the genome of pacific white shrimp, Litopenaeus vannamei. BMC Evol. Biol. 2013, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, X.; Yu, G.; Jia, C.; Liu, J.; Pan, H. Generation and analysis of expressed sequence tags (ESTs) from halophyte Atriplex canescens to explore salt-responsive related genes. Int. J. Mol. Sci. 2014, 15, 11172–11189. [Google Scholar] [CrossRef] [PubMed]
- Papura, D.; Jacquot, E.; Dedryver, C.A.; Luche, S.; Riault, G.; Bossis, M.; Rabilloud, T. Two-dimensional electrophoresis of proteins discriminates aphid clones of Sitobion avenae differing in BYDV-PAV transmission. Arch. Virol. 2002, 147, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- Méchin, V.; Damerval, C.; Zivy, M. Total protein extraction with TCA-acetone. Methods Mol. Biol. 2007, 355, 1–8. [Google Scholar] [PubMed]
- Sim, S.-L.; He, T.; Tscheliessnig, A.; Mueller, M.; Tan, R.B.H.; Jungbauer, A. Protein precipitation by polyethylene glycol: A generalized model based on hydrodynamic radius. J. Biotechnol. 2012, 157, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot a | Protein Name b | Locus c | Accession No. d | Score e | PM f | Cov g |
|---|---|---|---|---|---|---|
| 01↓ | nucleoside diphosphate kinase | gi/156063126 | XP_001597486.1 | 360 | 6 | 44 |
| 03↓ | nuclear transport factor 2 | gi/156052963 | XP_001592408.1 | 364 | 4 | 34 |
| 04↓ | 60 s ribosomal protein L23 | gi/154703604 | EDO03343.1 | 85 | 6 | 47 |
| 08↓ | ubiquitin-conjugating enzyme E2 | gi/156042748 | XP_001587931.1 | 141 | 5 | 48 |
| 09↓ | nucleoside diphosphate kinase | gi/156063126 | XP_001597485.1 | 130 | 8 | 74 |
| 12↓ | peptidyl-prolyl cis-trans isomerase | gi/156054872 | XP_001593362.1 | 532 | 9 | 53 |
| 16↓ | peptidyl-prolyl cis-trans isomerase B | gi/156042834 | XP_001587974.1 | 583 | 10 | 37 |
| 17↓ | nucleoside diphosphate kinase | gi/156063126 | XP_001597485.1 | 281 | 5 | 39 |
| 18↓ | eukaryotic translation initiation factor 5A-1 | gi/156063512 | XP_001597678.1 | 97 | 2 | 44 |
| 25↓ | peptidyl-prolyl cis-trans isomerase | gi/156054872 | XP_001593362.1 | 620 | 12 | 57 |
| 26↓ | elongation factor 1-beta | gi/156053087 | XP_001592470.1 | 107 | 2 | 31 |
| 28↓ | 60 s ribosomal protein L23 | gi/154703604 | EDO03343.1 | 59 | 2 | 62 |
| 29↑ | peptidyl-prolyl cis-trans isomerase | gi/156054872 | XP_001593362.1 | 406 | 8 | 46 |
| 30↓ | GTP-binding nuclear protein GSP1/Ran | gi/156057585 | XP_001594716.1 | 215 | 7 | 47 |
| 32- | SCF complex subunit Skp1 | gi/156065065 | XP_001598454.1 | 603 | 13 | 69 |
| 35↓ | citrate synthase, mitochondrial precursor | gi/156063018 | XP_001597431.1 | 104 | 9 | 42 |
| 39↓ | peptidyl-prolyl cis-trans isomerase | gi/156054872 | XP_001593362.1 | 498 | 11 | 51 |
| Spot a | Protein Name b | Locus c | Accession No. d | Score e | PM f | SignalP g |
|---|---|---|---|---|---|---|
| 02↓ | hypothetical proteins | SS1G_05791 | gi|156053886 | 412 | 6 | No |
| 05- | hypothetical proteins | SS1G_08534 | gi|156049655 | 147 | 5 | No |
| 06↓ | hypothetical proteins | SS1G_02967 | gi|156061643 | 1090 | 16 | Yes |
| 10↓ | hypothetical proteins | SS1G_13380 | gi|156033320 | 329 | 7 | No |
| 11↓ | hypothetical proteins | SS1G_10490 | gi|156044772 | 136 | 5 | No |
| 14↑ | hypothetical proteins | SS1G_05792 | gi|156053888 | 404 | 6 | No |
| 15↓ | hypothetical proteins | SS1G_03527 | gi|156059030 | 58 | 3 | No |
| 19↓ | hypothetical proteins | SS1G_03843 | gi|156059662 | 545 | 10 | No |
| 20↓ | hypothetical proteins | SS1G_06246 | gi|156054796 | 143 | 4 | No |
| 21↓ | hypothetical proteins | SS1G_06246 | gi|156054796 | 658 | 10 | No |
| 22↓ | hypothetical proteins | SS1G_11818 | gi|156039363 | 1040 | 14 | Yes |
| 24↑ | hypothetical proteins | SS1G_04923 | gi|156055144 | 199 | 5 | Yes |
| 27- | hypothetical proteins | SS1G_09167 | gi|156045782 | 46 | 7 | No |
| 31↓ | hypothetical proteins | SS1G_02387 | gi|156060497 | 237 | 5 | No |
| 33↓ | hypothetical proteins | SS1G_06394 | gi|156052457 | 389 | 4 | No |
| 34↓ | hypothetical proteins | SS1G_02266 | gi|156060255 | 1510 | 32 | Yes |
| 36↑ | hypothetical proteins | SS1G_03994 | gi|156056527 | 61 | 5 | No |
| 37↓ | hypothetical proteins | SS1G_12818 | gi|156036260 | 383 | 8 | No |
| 38↓ | hypothetical proteins | SS1G_14285 | gi|156031009 | 491 | 8 | No |
| 40↓ | hypothetical proteins | SS1G_03527 | gi|156059030 | 421 | 11 | No |
| 41↑ | hypothetical proteins | SS1G_08260 | gi|156049107 | 57 | 2 | No |
| 42↑ | hypothetical proteins | SS1G_06959 | gi|156051104 | 155 | 4 | No |
| 43- | hypothetical proteins | SS1G_06561 | gi|156052787 | 186 | 3 | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, X.; Li, L.; Liu, J.; Zhang, Y.; Pan, H. Proteomics Analysis of SsNsd1-Mediated Compound Appressoria Formation in Sclerotinia sclerotiorum. Int. J. Mol. Sci. 2018, 19, 2946. https://doi.org/10.3390/ijms19102946
Li J, Zhang X, Li L, Liu J, Zhang Y, Pan H. Proteomics Analysis of SsNsd1-Mediated Compound Appressoria Formation in Sclerotinia sclerotiorum. International Journal of Molecular Sciences. 2018; 19(10):2946. https://doi.org/10.3390/ijms19102946
Chicago/Turabian StyleLi, Jingtao, Xianghui Zhang, Le Li, Jinliang Liu, Yanhua Zhang, and Hongyu Pan. 2018. "Proteomics Analysis of SsNsd1-Mediated Compound Appressoria Formation in Sclerotinia sclerotiorum" International Journal of Molecular Sciences 19, no. 10: 2946. https://doi.org/10.3390/ijms19102946
APA StyleLi, J., Zhang, X., Li, L., Liu, J., Zhang, Y., & Pan, H. (2018). Proteomics Analysis of SsNsd1-Mediated Compound Appressoria Formation in Sclerotinia sclerotiorum. International Journal of Molecular Sciences, 19(10), 2946. https://doi.org/10.3390/ijms19102946