Pleiotropic Effects of Metformin on Cancer


Abstract

1. Introduction
2. MTF Bioavailability
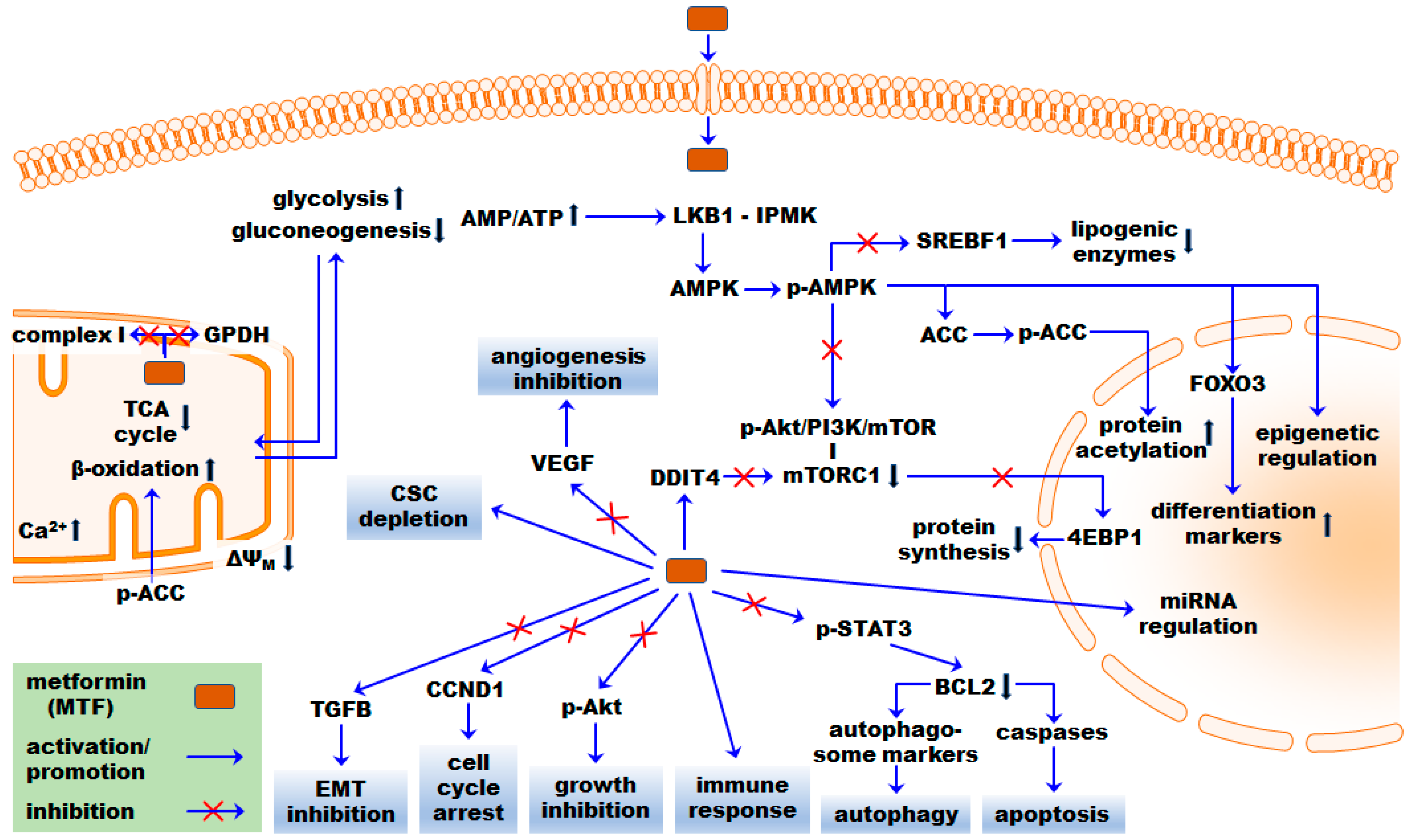
3. Pleiotropic MTF Effects on Cancer: Basic and Preclinical Cancer Research
3.1. Mitochondrial and Energetic Effects
3.2. Gastrointestinal Tract Effects
3.3. AMPK-Dependent Mechanisms
3.4. AMPK-Independent Mechanisms
3.5. Micro RNAs
3.6. Stress Effects
3.7. Antiproliferative Effects
3.8. Inhibition of Epithelial-to-Mesenchymal Transition
3.9. Antiangiogenic Effects
3.10. Autophagy
3.11. Apoptosis
3.12. Immune-Mediated Antitumor Response
3.13. Epigenetic Features
3.14. Hematological Malignancies
3.15. Cancer Stem Cells
3.16. MTF in Combination with Other Anticancer Drugs
3.17. Drug Delivery
3.18. MTF-Associated Resistance and Gene Polymorphisms
4. MTF Effects on Cancer: Population-Based Studies, Clinical Studies and Trials, and Recent Meta-Analyses
4.1. Population-Based Studies
4.2. Clinical Trials and Studies
4.3. Recent Meta-Analyses
5. Summary
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2DG | 2-deoxy-d-glucose |
| ACC | acetyl-CoA carboxylase |
| AMPK | AMP-activated protein kinase |
| CI | confidence interval |
| CSC | cancer stem cell |
| EMT | epithelial-to-mesenchymal transition |
| GPDH | glycerol-3-phosphate dehydrogenase |
| HOMA | homeostasis model assessment |
| HR | hazard ratio |
| IC50 | half maximal inhibitory concentration(s) |
| i.p. | intraperitoneal |
| MTF | metformin |
| OR | odds ratio |
| OXPHOS | oxidative phosphorylation |
| T2D | type 2 diabetes |
| TCA | tricarboxylic acid |
| TMZ | temozolomide |
References
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jimenez, C.; Gutierrez-Salmeron, M.; Chocarro-Calvo, A.; Garcia-Martinez, J.M.; Castano, A.; De la Vieja, A. From obesity to diabetes and cancer: Epidemiological links and role of therapies. Br. J. Cancer 2016, 114, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, A.; Drzewoski, J. Molecular action of metformin in hepatocytes: An updated insight. Curr. Diabetes Rev. 2015, 11, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Nurjhan, N.; Perriello, G.; Dailey, G.; Gerich, J.E. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1995, 333, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Jain, A.; Rohatgi, A. An observational study of vitamin b12 levels and peripheral neuropathy profile in patients of diabetes mellitus on metformin therapy. Diabetes Metab. Syndr. 2018, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, A.E.; Liebman, M.F.; Brien, W.; Parulekar, W.R.; Gelmon, K.A.; Shepherd, L.E.; Ligibel, J.A.; Hershman, D.L.; Rastogi, P.; Mayer, I.A.; et al. Effects of metformin versus placebo on vitamin B12 metabolism in non-diabetic breast cancer patients in CCTG MA.32. Breast Cancer Res. Treat. 2017, 164, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genomics 2012, 22, 820–827. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Landim, B.C.; de Jesus, M.M.; Bosque, B.P.; Zanon, R.G.; da Silva, C.V.; Goes, R.M.; Ribeiro, D.L. Stimulating effect of palmitate and insulin on cell migration and proliferation in PNT1A and PC3 prostate cells: Counteracting role of metformin. Prostate 2018, 78, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.B.; Matsuzaki, H.; Haorah, J.; Ulrich, A.; Standop, J.; Ding, X.Z.; Adrian, T.E.; Pour, P.M. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology 2001, 120, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Shikata, K.; Ninomiya, T.; Kiyohara, Y. Diabetes mellitus and cancer risk: Review of the epidemiological evidence. Cancer Sci. 2013, 104, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, LB.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Emami Riedmaier, A.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.W.; Bedwell, D.W.; Zamek-Gliszczynski, M.J. Ablation of both organic cation transporter (OCT)1 and OCT2 alters metformin pharmacokinetics but has no effect on tissue drug exposure and pharmacodynamics. Drug Metab. Dispos. 2012, 40, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Stage, T.B.; Brosen, K.; Christensen, M.M. A Comprehensive Review of Drug-Drug Interactions with Metformin. Clin. Pharmacokinet. 2015, 54, 811–824. [Google Scholar] [CrossRef] [PubMed]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathot, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet. Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.S.; Arnadottir, S.S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Lam, S.; Bassi, C.; Mouaaz, S.; Aman, A.; Kiyota, T.; Al-Awar, R.; Goodwin, P.J.; Stambolic, V. Metformin Pharmacokinetics in Mouse Tumors: Implications for Human Therapy. Cell Metab. 2016, 23, 567–568. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.B.; Horsman, M.R.; Jakobsen, S.; Jensen, J.B.; Garm, C.; Jessen, N.; Breining, P.; Frokiaer, J.; Busk, M. Results from (11)C-metformin-PET scans, tissue analysis and cellular drug-sensitivity assays questions the view that biguanides affects tumor respiration directly. Sci. Rep. 2017, 7, 9436. [Google Scholar] [CrossRef] [PubMed]
- Labuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopien, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Repiscak, P.; Erhardt, S.; Rena, G.; Paterson, M.J. Biomolecular mode of action of metformin in relation to its copper binding properties. Biochemistry 2014, 53, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Daley, B.; Gaskins, K.; Vasko, V.V.; Boufraqech, M.; Patel, D.; Sourbier, C.; Reece, J.; Cheng, S.Y.; Kebebew, E.; et al. Metformin Targets Mitochondrial Glycerophosphate Dehydrogenase to Control Rate of Oxidative Phosphorylation and Growth of Thyroid Cancer In Vitro and In Vivo. Clin Cancer Res. 2018, 24, 4030–4043. [Google Scholar] [CrossRef] [PubMed]
- Salani, B.; Marini, C.; Rio, A.D.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci. Rep. 2013, 3, 2070. [Google Scholar] [CrossRef] [PubMed]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; DeFronzo, R.A.; Rosenstock, J.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results From Short-term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care 2016, 39, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Song, R. Mechanism of Metformin: A Tale of Two Sites. Diabetes Care 2016, 39, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Horowitz, M.; Rayner, C.K. New insights into the anti-diabetic actions of metformin: From the liver to the gut. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Schommers, P.; Thurau, A.; Bultmann-Mellin, I.; Guschlbauer, M.; Klatt, A.R.; Rozman, J.; Klingenspor, M.; de Angelis, M.H.; Alber, J.; Gründemann, D.; et al. Metformin causes a futile intestinal–hepatic cycle which increases energy expenditure and slows down development of a type 2 diabetes-like state. Mol. Metab. 2017, 6, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Tokubuchi, I.; Tajiri, Y.; Iwata, S.; Hara, K.; Wada, N.; Hashinaga, T.; Nakayama, H.; Mifune, H.; Yamada, K. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE 2017, 12, e0171293. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Chen, Y.; Ahima, R.S.; Kim, S.F. Convergence of IPMK and LKB1-AMPK signaling pathways on metformin action. Mol. Endocrinol. 2014, 28, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.V.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Ikhlas, S.; Ahmad, M. Metformin: Insights into its anticancer potential with special reference to AMPK dependent and independent pathways. Life Sci. 2017, 185, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signaling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.J.; Kitagawa, H.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Repositioning metformin for cancer prevention and treatment. Trends. Endocrinol. Metab. 2013, 24, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.Q.; Wang, J.J.; Yan, J.S.; Huang, J.H.; Li, W.; Che, J.P.; Wang, G.C.; Liu, M.; Zheng, J.H. Metformin inhibits cell growth by upregulating microRNA-26a in renal cancer cells. Int. J. Clin. Exp. Med. 2014, 7, 3289–3296. [Google Scholar] [PubMed]
- Cabello, P.; Pineda, B.; Tormo, E.; Lluch, A.; Eroles, P. The Antitumor Effect of Metformin Is Mediated by miR-26a in Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1298. [Google Scholar] [CrossRef] [PubMed]
- Haga, N.; Saito, S.; Tsukumo, Y.; Sakurai, J.; Furuno, A.; Tsuruo, T.; Tomida, A. Mitochondria regulate the unfolded protein response leading to cancer cell survival under glucose deprivation conditions. Cancer Sci 2010, 101, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Loubiere, C.; Clavel, S.; Gilleron, J.; Harisseh, R.; Fauconnier, J.; Ben-Sahra, I.; Kaminski, L.; Laurent, K.; Herkenne, S.; Lacas-Gervais, S.; et al. The energy disruptor metformin targets mitochondrial integrity via modification of calcium flux in cancer cells. Sci Rep. 2017, 7, 5040. [Google Scholar] [CrossRef] [PubMed]
- Celestini, V.; Tezil, T.; Russo, L.; Fasano, C.; Sanese, P.; Forte, G.; Peserico, A.; Lepore Signorile, M.; Longo, G.; De Rasmo, D.; et al. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 2018, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, W.N.; Xue, Y.N.; Zhang, L.C.; Zhang, J.J.; Lu, S.Y.; Yan, X.Y.; Yu, H.M.; Su, J.; Sun, L.K. SIRT3 aggravates metformin-induced energy stress and apoptosis in ovarian cancer cells. Exp. Cell Res. 2018, 367, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.Q. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget 2018, 9, 2158–2174. [Google Scholar] [CrossRef] [PubMed]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, K.; Zheng, N.; Qiu, Y.; Xie, G.; Su, M.; Jia, W.; Li, H. Metformin suppressed the proliferation of LoVo cells and induced a time-dependent metabolic and transcriptional alteration. Sci. Rep. 2015, 5, 17423. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Rich, L.J.; Vincent-Chong, V.K.; Seshadri, M. Visualizing the effects of metformin on tumor growth, vascularity, and metabolism in head and neck cancer. J. Oral. Pathol. Med. 2018, 47, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, L.; Luo, N.; Zhao, Y.; Li, J.; Chen, Q.; Tian, Y. Metformin inhibits the proliferation and metastasis of osteosarcoma cells by suppressing the phosphorylation of Akt. Oncol. Lett. 2018, 15, 7948–7954. [Google Scholar] [CrossRef] [PubMed]
- Larsson, O.; Morita, M.; Topisirovic, I.; Alain, T.; Blouin, M.J.; Pollak, M.; Sonenberg, N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. USA 2012, 109, 8977–8982. [Google Scholar] [CrossRef] [PubMed]
- Li, N.S.; Zou, J.R.; Lin, H.; Ke, R.; He, X.L.; Xiao, L.; Huang, D.; Luo, L.; Lv, N.; Luo, Z. LKB1/AMPK inhibits TGF-β1 production and the TGF-β signaling pathway in breast cancer cells. Tumour Biol. 2016, 37, 8249–8258. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, N.; He, H.; Ying, Y.; Sunkara, S.; Luo, L.; Lv, N.; Huang, D.; Luo, Z. AMPK Inhibits the Stimulatory Effects of TGF-beta on Smad2/3 Activity, Cell Migration, and Epithelial-to-Mesenchymal Transition. Mol. Pharmacol. 2015, 88, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Cauchy, F.; Mebarki, M.; Albuquerque, M.; Laouirem, S.; Rautou, P.E.; Soubrane, O.; Raymond, E.; Bedossa, P.; Paradis, V. Anti-angiogenic effect of metformin in human liver carcinogenesis related to metabolic syndrome. Gut 2015, 64, 1498–1500. [Google Scholar] [CrossRef] [PubMed]
- Tadakawa, M.; Takeda, T.; Li, B.; Tsuiji, K.; Yaegashi, N. The anti-diabetic drug metformin inhibits vascular endothelial growth factor expression via the mammalian target of rapamycin complex 1/hypoxia-inducible factor-1alpha signaling pathway in ELT-3 cells. Mol. Cell. Endocrinol. 2015, 399, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Liang, S.; He, Z.; Zhu, X.; Chen, R.; Chen, J. Metformin, a first-line drug for type 2 diabetes mellitus, disrupts the MALAT1/miR-142-3p sponge to decrease invasion and migration in cervical cancer cells. Eur. J. Pharmacol. 2018, 830, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G.; Wang, Y.; Tang, S.; Sun, X.; Feng, X.; Li, Y.; Bao, G.; Li, P.; Mao, X.; et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget 2015, 6, 44579–44592. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, F.; Yamanaka, A.; Takebayashi, A.; Kita, N.; Takahashi, K.; Murakami, T. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ke, C.; Tang, Q.; Dong, H.; Zheng, X.; Lin, W.; Ke, J.; Huang, J.; Yeung, S.C.; Zhang, H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death. Dis. 2014, 5, e1088. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaidan, L.; El Ruz, R.A.; Malki, A.M. Screening Novel Molecular Targets of Metformin in Breast Cancer by Proteomic Approach. Front. Public Health 2017, 5, 277. [Google Scholar] [CrossRef] [PubMed]
- Sena, P.; Mancini, S.; Benincasa, M.; Mariani, F.; Palumbo, C.; Roncucci, L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. Int. J. Mol. Sci. 2018, 19, 1478. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Nair, V.; Karki, K. Metformin-induced anticancer activities: Recent insights. Biol. Chem. 2018, 399, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Sreevalsan, S.; Basha, R.; Abdelrahim, M.; Abudayyeh, A.; Rodrigues Hoffman, A.; Safe, S. Mechanism of metformin-dependent inhibition of mammalian target of rapamycin (mTOR) and Ras activity in pancreatic cancer: Role of specificity protein (Sp) transcription factors. J. Biol. Chem. 2014, 289, 27692–27701. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Tian, N.; Wang, J.; Yang, M.; Kong, L. Metabolic switching in the hypoglycemic and antitumor effects of metformin on high glucose induced HepG2 cells. J. Pharm. Biomed. Anal. 2018, 156, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Bikas, A.; Jensen, K.; Patel, A.; Costello, J., Jr.; McDaniel, D.; Klubo-Gwiezdzinska, J.; Larin, O.; Hoperia, V.; Burman, K.D.; Boyle, L.; et al. Glucose-deprivation increases thyroid cancer cells sensitivity to metformin. Endocr. Relat. Cancer 2015, 22, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Oliveras-Ferraros, C.; Cufi, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B.; Vazquez-Martin, A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Papanagnou, P.; Stivarou, T.; Tsironi, M. Unexploited Antineoplastic Effects of Commercially Available Anti-Diabetic Drugs. Pharmaceuticals (Basel) 2016, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.M.; Mukherjee, A.; Eckert, M.A.; Johnson, A.; Mills, K.A.; Pan, S.; Shridhar, V.; Lengyel, E.; Romero, I.L. Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 2015, 6, 23548–23560. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; de Castro, Í.A.; Braga, T.T.; Almeida, D.C.; Batista de Lima, A.G.U.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Sun, X.; Ma, Q.; Fu, G.F.; Cong, L.L.; Zhang, H.; Fan, D.F.; Feng, J.; Lu, S.Y.; Liu, J.L.; et al. Metformin’s antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. J. Cell. Mol. Med. 2018, 22, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Guo, J.; Zhu, Y.; Huang, Z.; Liu, T.; Cai, J.; Yu, L.; Wang, Z. Metformin inhibits ovarian cancer via decreasing H3K27 trimethylation. Int. J. Oncol. 2018, 52, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, V.; Gordon, L.I.; Venugopal, P.; Borgia, J.A.; Karmali, R. Metabolic changes associated with metformin potentiates Bcl-2 inhibitor, Venetoclax, and CDK9 inhibitor, BAY1143572 and reduces viability of lymphoma cells. Oncotarget 2018, 9, 21166–21181. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Hindupur, S.K.; Betz, C.; Lane, H.A.; El-Shemerly, M.Y.; Lu, M.; Quagliata, L.; Terracciano, L.; Moes, S.; et al. Syrosingopine sensitizes cancer cells to killing by metformin. Sci. Adv. 2016, 2, e1601756. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Corominas-Faja, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Dorca, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Menendez, J.A. Metformin-induced preferential killing of breast cancer initiating CD44(+)CD24(−/low) cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012, 3, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-Initiating Cell Elimination by Metformin Activation of FOXO3 via AMPK. Stem. Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Meyer, A.L.; Renner, K.; Leidgens, V.; Moeckel, S.; Jachnik, B.; Dettmer, K.; Tischler, U.; Gerthofer, V.; Rauer, L.; et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-beta2. Cell Cycle 2016, 15, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zhao, L.; Zhao, G. Metformin inhibits TGF-beta1-induced epithelial-to-mesenchymal transition-like process and stem-like properties in GBM via AKT/mTOR/ZEB1 pathway. Oncotarget 2018, 9, 7023–7035. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yuan, W.; Tong, D.; Liu, G.; Lan, W.; Zhang, D.; Xiao, H.; Zhang, Y.; Huang, Z.; Yang, J.; et al. Metformin represses bladder cancer progression by inhibiting stem cell repopulation via COX2/PGE2/STAT3 axis. Oncotarget 2016, 7, 28235–28246. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Wa Cheng, K.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Lin, C.T.; Chen, C.N.; Chang, S.F.; Chang, H.I.; Lee, K.C. Metformin increases the cytotoxicity of oxaliplatin in human DLD-1 colorectal cancer cells through down-regulating HMGB1 expression. J. Cell. Biochem. 2018, 119, 6943–6952. [Google Scholar] [CrossRef] [PubMed]
- Bizjak, M.; Malavasic, P.; Dolinar, K.; Pohar, J.; Pirkmajer, S.; Pavlin, M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci. Rep. 2017, 7, 1761. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Dong, W.; Qu, X.; Shen, H.; Xu, J.; Zhu, L.; Liu, Q.; Du, J. Metformin Enhances the Therapy Effects of Anti-IGF-1R mAb Figitumumab to NSCLC. Sci. Rep. 2016, 6, 31072. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Y.; Lin, C.; Lu, C.; Han, R.; Jiao, L.; Li, L.; He, Y. Vorinostat and metformin sensitize EGFR-TKI resistant NSCLC cells via BIM-dependent apoptosis induction. Oncotarget 2017, 8, 93825–93838. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lim, J.H.; Hong, Y.K.; Yang, S.H. High-Dose Metformin Plus Temozolomide Shows Increased Anti-tumor Effects in Glioblastoma In Vitro and In Vivo Compared with Monotherapy. J. Korean Cancer Assoc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, S.; Dico, A.L.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Darko, K.O.; Tao, T.; Huang, Y.; Su, Q.; He, C.; Yin, T.; Liu, Z.; Yang, X. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat. Rev. 2017, 54, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gong, F.; Liu, P.; Miao, Q. Metformin combined with quercetin synergistically repressed prostate cancer cells via inhibition of VEGF/PI3K/Akt signaling pathway. Gene 2018, 664, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, S.; Zarghami, N. Synergistic Growth Inhibitory Effects of Chrysin and Metformin Combination on Breast Cancer Cells through hTERT and Cyclin D1 Suppression. Asian Pac. J. Cancer Prev. 2018, 19, 977–982. [Google Scholar] [PubMed]
- Zheng, Z.; Zhu, W.; Yang, B.; Chai, R.; Liu, T.; Li, F.; Ren, G.; Ji, S.; Liu, S.; Li, G. The co-treatment of metformin with flavone synergistically induces apoptosis through inhibition of PI3K/AKT pathway in breast cancer cells. Oncol. Lett. 2018, 15, 5952–5958. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Zheng, X.; Lin, Y.; Yang, C.S.; Xu, Q.; Carpizo, D.; Huang, H.; DiPaola, R.S.; Tan, X.L. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget 2015, 6, 21208–21224. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Doyen, J. Metformin for non-small cell lung cancer patients: Opportunities and pitfalls. Crit. Rev. Oncol. Hematol. 2018, 125, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Farajzadeh, R.; Pilehvar-Soltanahmadi, Y.; Dadashpour, M.; Javidfar, S.; Lotfi-Attari, J.; Sadeghzadeh, H.; Shafiei-Irannejad, V.; Zarghami, N. Nano-encapsulated metformin-curcumin in PLGA/PEG inhibits synergistically growth and hTERT gene expression in human breast cancer cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Shafiei-Irannejad, V.; Samadi, N.; Salehi, R.; Yousefi, B.; Rahimi, M.; Akbarzadeh, A.; Zarghami, N. Reversion of Multidrug Resistance by Co-Encapsulation of Doxorubicin and Metformin in Poly(lactide-co-glycolide)-d-alpha-tocopheryl Polyethylene Glycol 1000 Succinate Nanoparticles. Pharm. Res. 2018, 35, 119. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Singla, A.; Mathieu, C.; Montanya, E.; Pfeiffer, A.F.H.; Johnsson, E.; Zhao, J.; Iqbal, N.; Bailey, C. Metformin extended-release versus immediate-release: An international, randomized, double-blind, head-to-head trial in pharmacotherapy-naive patients with type 2 diabetes. Diabetes Obes. Metab. 2018, 20, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Baldassari, S.; Solari, A.; Zuccari, G.; Drava, G.; Pastorino, S.; Fucile, C.; Marini, V.; Daga, A.; Pattarozzi, A.; Ratto, A.; et al. Development of an Injectable Slow-Release Metformin Formulation and Evaluation of Its Potential Antitumor Effects. Sci. Rep. 2018, 8, 3929. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Lubet, R.A.; McCormick, D.L.; Clapper, M.L.; Bode, A.M.; Juliana, M.M.; Moeinpour, F.; Grubbs, C.J. Lack of chemopreventive efficacy of metformin in rodent models of urinary bladder, head and neck, and colon/intestine cancer. Oncol. Lett. 2017, 14, 3480–3486. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Barron, M.; Aubry, M.C.; Wigle, D.A. Patient- and Cell Type-Specific Heterogeneity of Metformin Response. Basic Clin. Pharmacol. Toxicol. 2018, 122, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Mao, S.; Lu, G.; Li, L.; Lan, X.; Huang, Z.; Chen, Y.; Zhao, M.; Zhao, Y.; Xia, Q. Valproic acid sensitizes metformin-resistant human renal cell carcinoma cells by upregulating H3 acetylation and EMT reversal. BMC Cancer 2018, 18, 434. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci. Rep. 2018, 8, 409. [Google Scholar] [CrossRef] [PubMed]
- Hodeib, M.; Ogrodzinski, M.P.; Vergnes, L.; Reue, K.; Karlan, B.Y.; Lunt, S.Y.; Aspuria, P.J.P. Metformin induces distinct bioenergetic and metabolic profiles in sensitive versus resistant high grade serous ovarian cancer and normal fallopian tube secretory epithelial cells. Oncotarget 2018, 9, 4044–4060. [Google Scholar] [CrossRef] [PubMed]
- Scherbakov, A.M.; Sorokin, D.V.; Tatarskiy, V.V., Jr.; Prokhorov, N.S.; Semina, S.E.; Berstein, L.M.; Krasil’nikov, M.A. The phenomenon of acquired resistance to metformin in breast cancer cells: The interaction of growth pathways and estrogen receptor signaling. IUBMB Life 2016, 68, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Brown, C.; Castro, R.; Shi, R.; Lin, E.; Owen, R.; Sheardown, S.; Yue, L.; Burchard, E.; Brett, C.; et al. Effect of Genetic Variation in the Organic Cation Transporter 1, OCT1, on Metformin Pharmacokinetics. Clin. Pharmacol. Ther. 2008, 83, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Yee, S.W.; Seiser, E.L.; van Leeuwen, N.; Tavendale, R.; Bennett, A.J.; Groves, C.J.; Coleman, R.L.; van der Heijden, A.A.; Beulens, J.W.; et al. Variation in the Glucose Transporter gene SLC2A2 is associated with glycaemic response to metformin. Nat. Genet. 2016, 48, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Rotroff, D.M.; Yee, S.W.; Zhou, K.; Marvel, S.W.; Shah, H.S.; Jack, J.R.; Havener, T.M.; Hedderson, M.M.; Kubo, M.; Herman, M.A.; et al. Genetic Variants in CPA6 and PRPF31 Are Associated With Variation in Response to Metformin in Individuals With Type 2 Diabetes. Diabetes 2018, 67, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.; Dufay Wojcicki, A.; d’Hayer, B.; Boudy, V. Metformin: An anti-diabetic drug to fight cancer. Pharmacol. Res. 2016, 113, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Tsilidis, K.K.; Capothanassi, D.; Allen, N.E.; Rizos, E.C.; Lopez, D.S.; van Veldhoven, K.; Sacerdote, C.; Ashby, D.; Vineis, P.; Tzoulaki, I.; et al. Metformin does not affect cancer risk: A cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care 2014, 37, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Landman, G.W.D.; Kleefstra, N.; van Hateren, K.J.J.; Groenier, K.H.; Gans, R.O.B.; Bilo, H.J.G. Metformin Associated With Lower Cancer Mortality in Type 2 Diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.; Chun, K.H.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kim, Y.S.; Woo, J.T.; Nam, M.S.; Baik, S.H.; et al. Metformin reduces the risk of cancer in patients with type 2 diabetes: An analysis based on the Korean National Diabetes Program Cohort. Medicine (Baltimore) 2018, 97, e0036. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Hsu, C.C.; Wahlqvist, M.L.; Tsai, H.N.; Chang, Y.H.; Huang, Y.C. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: A representative population prospective cohort study of 800,000 individuals. BMC Cancer 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Tsai, H.L.; Kung, Y.T.; Yeh, Y.S.; Huang, C.W.; Ma, C.J.; Chiu, H.C.; Wang, J.Y. Dose-Dependent Relationship Between Metformin and Colorectal Cancer Occurrence Among Patients with Type 2 Diabetes-A Nationwide Cohort Study. Transl. Oncol. 2018, 11, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.C.; Ferrara, A.; Achacoso, N.; Ehrlich, S.F.; Quesenberry, C.P., Jr.; Habel, L.A. A Cohort Study of Metformin and Colorectal Cancer Risk among Patients with Diabetes Mellitus. Cancer Epidemiol. Biomark. Prev. 2018, 27, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Margel, D.; Urbach, D.R.; Lipscombe, L.L.; Bell, C.M.; Kulkarni, G.; Austin, P.C.; Fleshner, N. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J. Clin. Oncol. 2013, 31, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Häggström, C.; Van Hemelrijck, M.; Zethelius, B.; Robinson, D.; Grundmark, B.; Holmberg, L.; Gudbjörnsdottir, S.; Garmo, H.; Stattin, P. Prospective study of Type 2 diabetes mellitus, anti-diabetic drugs and risk of prostate cancer. Int. J. Cancer 2017, 140, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Rastmanesh, R.; Hejazi, J.; Marotta, F.; Hara, N. Type 2 diabetes: A protective factor for prostate cancer? An overview of proposed mechanisms. Clin. Genitourin Cancer 2014, 12, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Azoulay, L. Metformin and the risk of cancer: Time-related biases in observational studies. Diabetes Care 2012, 35, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
- Farmer, R.E.; Ford, D.; Forbes, H.J.; Chaturvedi, N.; Kaplan, R.; Smeeth, L.; Bhaskaran, K. Metformin and cancer in type 2 diabetes: A systematic review and comprehensive bias evaluation. Int. J. Epidemiol. 2017, 46, 728–744. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zheng, L.; Mei, Z.; Xu, C.; Liu, C.; Chu, X.; Hao, B. The impact of metformin use on survival in prostate cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 100449–100458. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, C.; Matteoni, S.; Signore, M.; Cardone, L.; Nath, K.; Glickson, J.D.; Paggi, M.G. Drug repurposing for the treatment of glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2017, 36, 169. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Agbor-Tarh, D.; Bradbury, I.; Di Cosimo, S.; Azim, H.A.; Fumagalli, D.; Sarp, S.; Wolff, A.C.; Andersson, M.; Kroep, J.; et al. Impact of Diabetes, Insulin, and Metformin Use on the Outcome of Patients With Human Epidermal Growth Factor Receptor 2–Positive Primary Breast Cancer: Analysis From the ALTTO Phase III Randomized Trial. J. Clin. Oncol. 2017, 35, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and Pathologic Complete Responses to Neoadjuvant Chemotherapy in Diabetic Patients With Breast Cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Peled, Y.; Lavee, J.; Raichlin, E.; Katz, M.; Arad, M.; Kassif, Y.; Peled, A.; Asher, E.; Elian, D.; Har-Zahav, Y.; et al. Metformin therapy reduces the risk of malignancy after heart transplantation. J. Heart Lung Transplant. 2017, 36, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Khawaja, M.R.; Nick, A.M.; Madhusudanannair, V.; Fu, S.; Hong, D.; McQuinn, L.M.; Ng, C.S.; Piha-Paul, S.A.; Janku, F.; Subbiah, V.; et al. Phase I dose escalation study of temsirolimus in combination with metformin in patients with advanced/refractory cancers. Cancer Chemother Pharmacol. 2016, 77, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Vernieri, C.; Di Maio, M.; Marconcini, R.; Spada, F.; Massironi, S.; Ibrahim, T.; Brizzi, M.P.; Campana, D.; Faggiano, A.; et al. Metformin Use Is Associated With Longer Progression-Free Survival of Patients With Diabetes and Pancreatic Neuroendocrine Tumors Receiving Everolimus and/or Somatostatin Analogues. Gastroenterology 2018, 155, 479–489.e7. [Google Scholar] [CrossRef] [PubMed]
- Klubo-Gwiezdzinska, J.; Costello, J., Jr.; Patel, A.; Bauer, A.; Jensen, K.; Mete, M.; Burman, K.D.; Wartofsky, L.; Vasko, V. Treatment with metformin is associated with higher remission rate in diabetic patients with thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, 3269–3279. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, B.; Puntoni, M.; Cazzaniga, M.; Pruneri, G.; Serrano, D.; Guerrieri-Gonzaga, A.; Gennari, A.; Trabacca, M.S.; Galimberti, V.; Veronesi, P.; et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 2012, 30, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Niraula, S.; Dowling, R.J.; Ennis, M.; Chang, M.C.; Done, S.J.; Hood, N.; Escallon, J.; Leong, W.L.; McCready, D.R.; Reedijk, M.; et al. Metformin in early breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat. 2012, 135, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Parulekar, W.R.; Gelmon, K.A.; Shepherd, L.E.; Ligibel, J.A.; Hershman, D.L.; Rastogi, P.; Mayer, I.A.; Hobday, T.J.; Lemieux, J.; et al. Effect of Metformin vs Placebo on Weight and Metabolic Factors in NCIC CTG MA.32. JNCI 2015, 107, djv006. [Google Scholar] [CrossRef] [PubMed]
- Schuler, K.M.; Rambally, B.S.; DiFurio, M.J.; Sampey, B.P.; Gehrig, P.A.; Makowski, L.; Bae-Jump, V.L. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015, 4, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Kiyokawa, T.; Sato, Y.; Shozu, M. Effects of metformin on endometrial cancer cell growth in vivo: A preoperative prospective trial. Cancer 2014, 120, 2986–2995. [Google Scholar] [CrossRef] [PubMed]
- Sivalingam, V.N.; Kitson, S.; McVey, R.; Roberts, C.; Pemberton, P.; Gilmour, K.; Ali, S.; Renehan, A.G.; Kitchener, H.C.; Crosbie, E.J. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br. J. Cancer 2016, 114, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Soliman, P.T.; Zhang, Q.; Broaddus, R.R.; Westin, S.N.; Iglesias, D.; Munsell, M.F.; Schmandt, R.; Yates, M.; Ramondetta, L.; Lu, K.H. Prospective evaluation of the molecular effects of metformin on the endometrium in women with newly diagnosed endometrial cancer: A window of opportunity study. Gynecol. Oncol. 2016, 143, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wu, Y.; Wang, J.; Liu, K.; Wang, X. The Effect of Metformin on Mortality Among Diabetic Cancer Patients: A Systematic Review and Meta-analysis. JNCI Cancer Spectrum. 2017, 1, pkx007. [Google Scholar] [CrossRef]
- Zhou, X.L.; Xue, W.H.; Ding, X.F.; Li, L.F.; Dou, M.M.; Zhang, W.J.; Lv, Z.; Fan, Z.R.; Zhao, J.; Wang, L.X. Association between metformin and the risk of gastric cancer in patients with type 2 diabetes mellitus: A meta-analysis of cohort studies. Oncotarget 2017, 8, 55622–55631. [Google Scholar] [CrossRef] [PubMed]
- Mansourian, M.; Karimi, R.; Vaseghi, G. Different effects of metformin and insulin on primary and secondary chemoprevention of colorectal adenoma in diabetes type 2: Traditional and Bayesian meta-analysis. EXCLI J. 2018, 17, 45–56. [Google Scholar] [PubMed]
- Tian, S.; Lei, H.B.; Liu, Y.L.; Chen, Y.; Dong, W.G. The association between metformin use and colorectal cancer survival among patients with diabetes mellitus: An updated meta-analysis. Chronic Dis. Transl. Med. 2017, 3, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zheng, Y.; Xiao, Y.; Zhou, P.; Tan, H. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients. Medicine (Baltimore) 2017, 96, e6888. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.X.; Fang, L.; Fang, Q.L.; Zheng, X.W.; Ding, H.Y.; Huang, P. Effect of hypoglycemic agents on survival outcomes of lung cancer patients with diabetes mellitus: A meta-analysis. Medicine (Baltimore) 2018, 97, e0035. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schunemann, H.J.; Muti, P. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2018, 27, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.L.; Zhu, L.Y.; Li, Y.; Yu, J.; Wang, J.; Zeng, X.X.; Hu, K.X.; Liu, J.Y.; Xu, J.X. Metformin Use Is Associated with Reduced Incidence and Improved Survival of Endometrial Cancer: A Meta-Analysis. Biomed. Res. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, K.; An, M.; Zhao, Y. Metformin and endometrial cancer survival: A quantitative synthesis of observational studies. Oncotarget 2017, 8, 66169–66177. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, T.; Liu, Z.; Gou, S.; Wang, C. The effect of metformin on survival of patients with pancreatic cancer: A meta-analysis. Sci. Rep. 2017, 7, 5825. [Google Scholar] [CrossRef] [PubMed]
- Barua, R.; Templeton, A.J.; Seruga, B.; Ocana, A.; Amir, E.; Ethier, J.L. Hyperglycaemia and Survival in Solid Tumours: A Systematic Review and Meta-analysis. Clin. Oncol. R. Coll. Radiol. 2018, 30, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Marinac, C.R.; Sears, D.D.; Kerr, J.; Hartman, S.J.; Cadmus-Bertram, L.; Villaseñor, A.; Flatt, S.W.; Godbole, S.; Li, H.; et al. The Effects of Metformin and Weight Loss on Biomarkers Associated With Breast Cancer Outcomes. JNCI 2018. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Tumor Site | Study Design | Cancer Incidence | CSS/DFS/PFS/RFS | OS/ACM | Number of Participants | Reference |
|---|---|---|---|---|---|---|
| Gastric cancer | 7 cohort | HR 0.763, 95% CI 0.642–0.905 | >100,000 | [166] | ||
| Colorectal adenoma | 4 retrospective 1 retrospective cross-sectional 2 retrospective case-control | OR 0.86, 95% CI 0.66–1.12, p = 0.274 | 10,000–100,000 | [167] | ||
| Advanced colorectal adenoma | 3 retrospective | OR 0.51, 95% CI 0.41–0.63, p < 0.001 | <10,000 | [167] | ||
| Colorectal cancer | 8 retrospective cohort (OS) 3 retrospective cohort (CSS) | HR 0.84, 95% CI 0.69–1.02, p = 0.079 (CSS) | HR 0.82, 95% CI 0.77–0.87, p = 0.000 (OS) | 10,000–100,000 | [168] | |
| Liver cancer | 10 cohort 9 case-control 2 RCTs | OR 0.52, 95% CI 0.40–0.68, p < 0.001 | >100,000 | [169] | ||
| Lung cancer | 13 cohort, 2 case-control (OS) 5 cohort (DFS) | HR 0.50, 95% CI 0.39–0.64, p < 0.0001 (DFS) | HR 0.77, 95% CI 0.68–0.86, p < 0.0001 (OS) | 10,000–100,000 | [170] | |
| Breast cancer | 10 retrospective cohort, 1 prospective cohort, 1 nested case-control (cancer incidence) 11 retrospective (ACM) | OR 0.93, 95% CI 0.85–1.03 | HR 0.55, 95% CI 0.44–0.70 (ACM) | 10,000–100,000 | [171] | |
| Endometrial cancer | 3 retrospective cohort 1 case-control 1 nested case-control | RR 0.87, 95% CI 0.80–0.95, p = 0.006 | >100,000 | [172] | ||
| Endometrial cancer | 9 observational (OS) 5 observational (PFS) | HR 0.61; 95% CI 0.49–0.76 (PFS) | HR 0.58; 95% CI 0.45–0.76 (OS) | <10,000 | [173] | |
| Prostate cancer | 7 retrospective cohort, 1 prospective cohort (OS) 6 retrospective cohort (CSS) 5 retrospective cohort (RFS) | HR 0.76, 95% CI 0.57–1.02 (CSS) HR 0.74, 95% CI 0.58–0.95 (RFS) | HR 0.79, 95% CI 0.63–0.98 (OS) | 10,000–100,000 | [148] | |
| Pancreatic cancer | 9 retrospective cohort 2 RCTs | HR 0.86, 95% CI 0.76–0.97, p = 0.01 (OS) | <10,000 | [174] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulten, H.-J. Pleiotropic Effects of Metformin on Cancer. Int. J. Mol. Sci. 2018, 19, 2850. https://doi.org/10.3390/ijms19102850
Schulten H-J. Pleiotropic Effects of Metformin on Cancer. International Journal of Molecular Sciences. 2018; 19(10):2850. https://doi.org/10.3390/ijms19102850
Chicago/Turabian StyleSchulten, Hans-Juergen. 2018. "Pleiotropic Effects of Metformin on Cancer" International Journal of Molecular Sciences 19, no. 10: 2850. https://doi.org/10.3390/ijms19102850
APA StyleSchulten, H.-J. (2018). Pleiotropic Effects of Metformin on Cancer. International Journal of Molecular Sciences, 19(10), 2850. https://doi.org/10.3390/ijms19102850