Unfolding Role of a Danger Molecule Adenosine Signaling in Modulation of Microbial Infection and Host Cell Response


Abstract
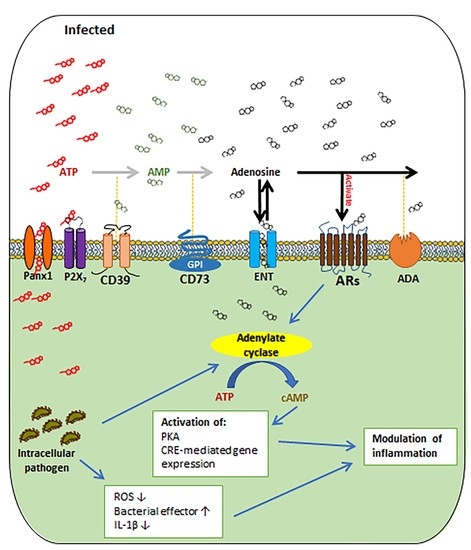
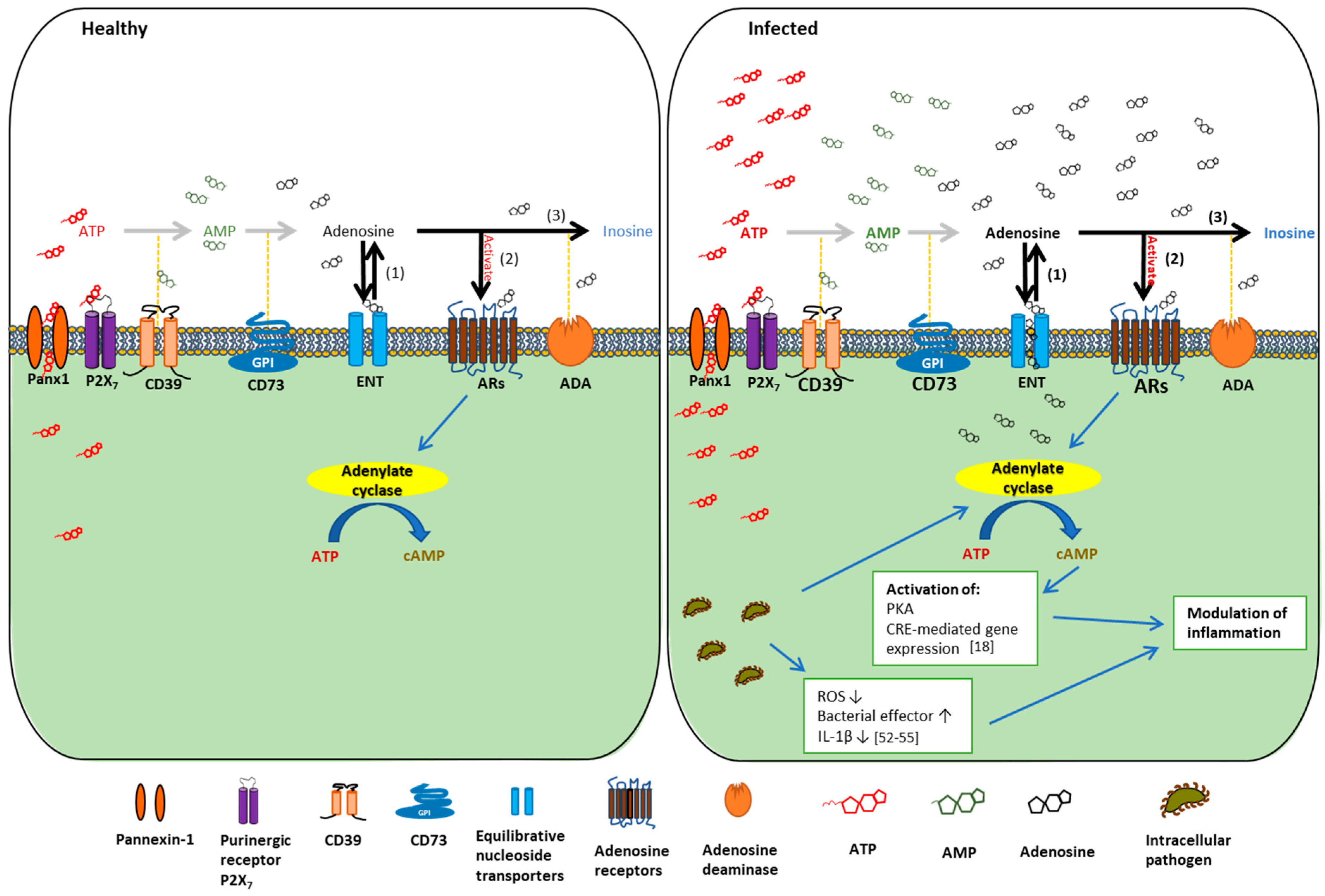
1. Introduction
2. The Basics of Extracellular Adenosine Metabolism and Signaling
3. The Roles of the Main Regulators of Extracellular Adenosine Signaling in Regulating Microbial Infection and Inflammation
3.1. CD39 and CD73
3.2. Adenosine Receptors
4. Diagnostic and Therapeutic Potential of Adenosine Receptor Signaling in Treating Chronic Inflammatory and Infectious Diseases
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, S.L.; Urey, H.C. Organic Compound Synthesis on the Primitive Earth. Science 1959, 130, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Burnstock, G. The Double Life of ATP. Sci. Am. 2009, 301, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.; Verkhratsky, A. Purines—80 Years and Very Much Alive. Acta Physiol. 2010, 199, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Riegel, A.K.; Eltzschig, H.K. Extracellular Nucleotide and Nucleoside Signaling in Vascular and Blood Disease. Blood 2014, 124, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.P.; Thompson, L.; Takedachi, M. The Role of CD73 in the Generation of Extracellular Adenosine for Adenosine Receptor Signaling. In Adenosine Receptors; CRC Press: Boca Raton, FL, USA, 2006; pp. 39–48. [Google Scholar]
- Mariathasan, S.; Monack, D.M. Inflammasome Adaptors and Sensors: Intracellular Regulators of Infection and Inflammation. Nat. Rev. Immunol. 2007, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Lee, K.L. The Inflammasome and Danger Molecule Signaling: At the Crossroads of Inflammation and Pathogen Persistence in the Oral Cavity. Periodontology 2000 2015, 69, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Yilmaz, O. Modulation of Inflammasome Activity by Porphyromonas Gingivalis in Periodontitis and Associated Systemic Diseases. J. Oral Microbiol. 2016, 8, 30385. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine Is Required for Sustained Inflammasome Activation Via the A(2)a Receptor and the HIF-1α Pathway. Nat. Commun. 2013, 4, 2909. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Cronstein, B.N. Adenosine: An Endogenous Regulator of Innate Immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.; DeGuzman, J.; Lee, K.L.; Yilmaz, O. Danger Signal Adenosine Via Adenosine 2a Receptor Stimulates Growth of Porphyromonas Gingivalis in Primary Gingival Epithelial Cells. Mol. Oral Microbiol. 2014, 29, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, L.; Gorini, S.; Rosano, G.; la Sala, A. Immunoregulation through Extracellular Nucleotides. Blood 2012, 120, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Bours, M.J.; Swennen, E.L.; di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5′-Triphosphate and Adenosine as Endogenous Signaling Molecules in Immunity and Inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP Drives Systemic Inflammation, Tissue Damage and Mortality. Cell. Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef] [PubMed]
- Karmouty-Quintana, H.; Xia, Y.; Blackburn, M.R. Adenosine Signaling during Acute and Chronic Disease States. J. Mol. Med. 2013, 91, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Schneider, D.J.; Morschl, E.; Song, L.; Pedroza, M.; Karmouty-Quintana, H.; Le, T.; Sun, C.X.; Blackburn, M.R. Distinct Roles for the A2b Adenosine Receptor in Acute and Chronic Stages of Bleomycin-Induced Lung Injury. J. Immunol. 2011, 186, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Linden, J. Purinergic Regulation of the Immune System. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.G. Adenosine Receptors as Therapeutic Targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A Potent Suppressor of Antitumor Immune Responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative Stress Controls Regulatory T Cell Apoptosis and Suppressor Activity and PD-L1-Blockade Resistance in Tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Seminario-Vidal, L.; Lazarowski, E.R.; Okada, S.F. Assessment of Extracellular ATP Concentrations. Methods Mol. Biol. 2009, 574, 25–36. [Google Scholar] [PubMed]
- Blume, J.; Douglas, S.D.; Evans, D.L. Immune Suppression and Immune Activation in Depression. Brain Behav. Immun. 2011, 25, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Drygiannakis, I.; Ernst, P.B.; Lowe, D.; Glomski, I.J. Immunological Alterations Mediated by Adenosine during Host-Microbial Interactions. Immunol. Res. 2011, 50, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Hamidzadeh, K.; Mosser, D.M. Purinergic Signaling to Terminate TLR Responses in Macrophages. Front. Immunol. 2016, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Decking, U.K.; Schlieper, G.; Kroll, K.; Schrader, J. Hypoxia-Induced Inhibition of Adenosine Kinase Potentiates Cardiac Adenosine Release. Circ. Res. 1997, 81, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Lovatt, D.; Xu, Q.; Liu, W.; Takano, T.; Smith, N.A.; Schnermann, J.; Tieu, K.; Nedergaard, M. Neuronal Adenosine Release, and Not Astrocytic ATP Release, Mediates Feedback Inhibition of Excitatory Activity. Proc. Natl. Acad. Sci. USA 2012, 109, 6265–6270. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, S.; Matzinger, P. Danger Signals: SOS to the Immune System. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Johnston-Cox, H.A.; Ravid, K. Adenosine and Blood Platelets. Purinergic Signal. 2011, 7, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Gorzolla, I.C.; Schittenhelm, J.; Robson, S.C.; Eltzschig, H.K. SP1-Dependent Induction of CD39 Facilitates Hepatic Ischemic Preconditioning. J. Immunol. 2010, 184, 4017–4024. [Google Scholar] [CrossRef] [PubMed]
- Reutershan, J.; Vollmer, I.; Stark, S.; Wagner, R.; Ngamsri, K.C.; Eltzschig, H.K. Adenosine and Inflammation: CD39 and CD73 Are Critical Mediators in LPS-Induced PMN Trafficking into the Lungs. FASEB J. 2009, 23, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Grenz, A.; Zhang, H.; Hermes, M.; Eckle, T.; Klingel, K.; Huang, D.Y.; Muller, C.E.; Robson, S.C.; Osswald, H.; Eltzschig, H.K. Contribution of E-NTPDase1 (CD39) to Renal Protection from Ischemia-Reperfusion Injury. FASEB J. 2007, 21, 2863–2873. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Weissmuller, T.; Mager, A.; Eckle, T. Nucleotide Metabolism and Cell-Cell Interactions. Methods Mol. Biol. 2006, 341, 73–87. [Google Scholar] [PubMed]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP Release from Activated Neutrophils Occurs via Connexin 43 and Modulates Adenosine-Dependent Endothelial Cell Function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. 5′-Nucleotidase: Molecular Structure and Functional Aspects. Biochem. J. 1992, 285, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Sevigny, J.; Zimmermann, H. The E-NTPDase Family of Ectonucleotidases: Structure Function Relationships and Pathophysiological Significance. Purinergic Signal. 2006, 2, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Wollner, A.; Wollner, S.J.; Smith, B. Acting via A2 Receptors, Adenosine Inhibits the Upregulation of Mac-1 (Cd11b/Cd18) Expression on FMLP-Stimulated Neutrophils. Am. J. Respir. Cell Mol. Biol. 1993, 9, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Olah, M.E.; Stiles, G.L. Adenosine Receptor Subtypes: Characterization and Therapeutic Regulation. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 581–606. [Google Scholar] [CrossRef] [PubMed]
- Hashikawa, T.; Takedachi, M.; Terakura, M.; Yamada, S.; Thompson, L.F.; Shimabukuro, Y.; Murakami, S. Activation of Adenosine Receptor on Gingival Fibroblasts. J. Dent. Res. 2006, 85, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K. Adenosine: An Old Drug Newly Discovered. Anesthesiology 2009, 111, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Grenz, A.; Laucher, S.; Eltzschig, H.K. A2b Adenosine Receptor Signaling Attenuates Acute Lung Injury by Enhancing Alveolar Fluid Clearance in Mice. J. Clin. Investig. 2008, 118, 3301–3315. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated Adenine Nucleotide Phosphohydrolysis and Nucleoside Signaling in Posthypoxic Endothelium: Role of Ectonucleotidases and Adenosine A2b Receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine Receptors: Therapeutic Aspects for Inflammatory and Immune Diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B. Adenosine, an Endogenous Distress Signal, Modulates Tissue Damage and Repair. Cell. Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Yang, C.; Sit, A.S.; Lin, S.Y.; Ho, E.Y.; Leung, G.P. Physiological and Pharmacological Roles of Vascular Nucleoside Transporters. J. Cardiovasc. Pharmacol. 2012, 59, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Morote-Garcia, J.C.; Rosenberger, P.; Nivillac, N.M.; Coe, I.R.; Eltzschig, H.K. Hypoxia-Inducible Factor-Dependent Repression of Equilibrative Nucleoside Transporter 2 Attenuates Mucosal Inflammation during Intestinal Hypoxia. Gastroenterology 2009, 136, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Loffler, M.; Morote-Garcia, J.C.; Eltzschig, S.A.; Coe, I.R.; Eltzschig, H.K. Physiological Roles of Vascular Nucleoside Transporters. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Abdulla, P.; Hoffman, E.; Hamilton, K.E.; Daniels, D.; Schonfeld, C.; Loffler, M.; Reyes, G.; Duszenko, M.; Karhausen, J.; et al. HIF-1-Dependent Repression of Equilibrative Nucleoside Transporter (ENT) in Hypoxia. J. Exp. Med. 2005, 202, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.A.; Tak, E.; Ehrentraut, S.F.; Kaplan, M.; Giebler, A.; Weng, T.; Choi, D.S.; Blackburn, M.R.; Kam, I.; Eltzschig, H.K.; et al. Equilibrative Nucleoside Transporter (ENT)-1-Dependent Elevation of Extracellular Adenosine Protects the Liver during Ischemia and Reperfusion. Hepatology 2013, 58, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, K.; Keresztes, T.; Fekete, I.; Molnar, L. Effect of I.V. Dipyridamole on Cerebral Blood Flow, Blood Pressure, Plasma Adenosine and cAMP Levels in Rabbits. J. Neurol. Sci. 1997, 148, 153–161. [Google Scholar] [CrossRef]
- Yilmaz, O.; Sater, A.A.; Yao, L.; Koutouzis, T.; Pettengill, M.; Ojcius, D.M. ATP-Dependent Activation of an Inflammasome in Primary Gingival Epithelial Cells Infected by Porphyromonas Gingivalis. Cell. Microbiol. 2010, 12, 188–198. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Marques-da-Silva, E.; de Oliveira, J.C.; Figueiredo, A.B.; Junior, D.D.S.L.; Carneiro, C.M.; Fietto, J.L.R.; Afonso, L.C.C. Extracellular Nucleotide Metabolism in Leishmania: Influence of Adenosine in the Establishment of Infection. Microbes Infect. 2008, 10, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas Gingivalis-Nucleoside-Diphosphate-Kinase Inhibits ATP-Induced Reactive-Oxygen-Species via P2X7 Receptor/NADPH-Oxidase Signalling and Contributes to Persistence. Cell. Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Atanasova, K.R.; Bui, P.Q.; Lee, J.; Hung, S.C.; Yilmaz, O.; Ojcius, D.M. Porphyromonas Gingivalis Attenuates ATP-Mediated Inflammasome Activation and HMGB1 Release through Expression of a Nucleoside-Diphosphate Kinase. Microbes Infect. 2015, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, K.; Lee, J.; Roberts, J.; Lee, K.; Ojcius, D.M.; Yilmaz, O. Nucleoside-Diphosphate-Kinase of P. gingivalis Is Secreted from Epithelial Cells in the Absence of a Leader Sequence through a Pannexin-1 Interactome. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.J.; Atanasova, K.R.; Lee, J.; Diamond, G.; Deguzman, J.; Choi, C.H.; Yilmaz, Ö. Opportunistic Pathogen Porphyromonas Gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Front. Cell. Infect. Microbiol. 2017, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, S.; Eugenin, E.A. Role of Pannexin-1 Hemichannels and Purinergic Receptors in the Pathogenesis of Human Diseases. Front. Physiol. 2014, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Kuo, J.L.; Ernst, P.B.; Derr-Castillo, V.; Pereira, M.; Gaines, D.; Costales, M.; Bigley, E.; Williams, K. Ecto-5′-Nucleotidase (CD73) Regulates Host Inflammatory Responses and Exacerbates Murine Salmonellosis. Sci. Rep. 2014, 4, 4486. [Google Scholar] [CrossRef] [PubMed]
- Francois, V.; Shehade, H.; Acolty, V.; Preyat, N.; Delree, P.; Moser, M.; Oldenhove, G. Intestinal Immunopathology Is Associated with Decreased CD73-Generated Adenosine during Lethal Infection. Mucosal Immunol. 2015, 8, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Telesford, K.M.; Ochoa-Reparaz, J.; Haque-Begum, S.; Christy, M.; Kasper, E.J.; Wang, L.; Wu, Y.; Robson, S.C.; Kasper, D.L.; et al. An Intestinal Commensal Symbiosis Factor Controls Neuroinflammation via TLR2-Mediated CD39 Signalling. Nat. Commun. 2014, 5, 4432. [Google Scholar] [CrossRef] [PubMed]
- Boer, M.C.; van Meijgaarden, K.E.; Bastid, J.; Ottenhoff, T.H.; Joosten, S.A. CD39 Is Involved in Mediating Suppression by Mycobacterium Bovis BCG-Activated Human CD8(+) CD39(+) Regulatory T Cells. Eur. J. Immunol. 2013, 43, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Theatre, E.; Frederix, K.; Guilmain, W.; Delierneux, C.; Lecut, C.; Bettendorff, L.; Bours, V.; Oury, C. Overexpression of CD39 in Mouse Airways Promotes Bacteria-Induced Inflammation. J. Immunol. 2012, 189, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Kurtz, C.C.; Rowlett, R.M.; Reuter, B.K.; Wiznerowicz, E.; Das, S.; Linden, J.; Crowe, S.E.; Ernst, P.B. CD73 Is Expressed by Human Regulatory T Helper Cells and Suppresses Proinflammatory Cytokine Production and Helicobacter Felis-Induced Gastritis in Mice. J. Infect. Dis. 2009, 199, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.; Canton, R.; Campo, P.; Baquero, F.; Blazquez, J. High Frequency of Hypermutable Pseudomonas Aeruginosa in Cystic Fibrosis Lung Infection. Science 2000, 288, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Travis, S.M.; Greenberg, E.P.; Welsh, M.J. Cystic Fibrosis Airway Epithelia Fail to Kill Bacteria Because of Abnormal Airway Surface Fluid. Cell 1996, 85, 229–236. [Google Scholar] [CrossRef]
- Joosten, S.A.; Ottenhoff, T.H. Human CD4 and CD8 Regulatory T Cells in Infectious Diseases and Vaccination. Hum. Immunol. 2008, 69, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y. Regulatory T Cells and Infection: A Dangerous Necessity. Nat. Rev. Immunol. 2007, 7, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Ottenhoff, T.H. New Pathways of Protective and Pathological Host Defense to Mycobacteria. Trends Microbiol. 2012, 20, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Scott-Browne, J.P.; Shafiani, S.; Tucker-Heard, G.; Ishida-Tsubota, K.; Fontenot, J.D.; Rudensky, A.Y.; Bevan, M.J.; Urdahl, K.B. Expansion and Function of Foxp3-Expressing T Regulatory Cells During Tuberculosis. J. Exp. Med. 2007, 204, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Mandapathil, M.; Lang, S.; Gorelik, E.; Whiteside, T.L. Isolation of Functional Human Regulatory T Cells (Treg) from the Peripheral Blood Based on the CD39 Expression. J. Immunol. Methods 2009, 346, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bastid, J.; Cottalorda-Regairaz, A.; Alberici, G.; Bonnefoy, N.; Eliaou, J.F.; Bensussan, A. ENTPD1/CD39 Is a Promising Therapeutic Target in Oncology. Oncogene 2013, 32, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Cairns, B.; Levashova, Z.; Chinn, L.; Perez, M.; Theunissen, J.W.; Liao-Chan, S.; Bermudez, A.; Flory, M.R.; Schweighofer, K.J.; et al. CD39 Is a Promising Therapeutic Antibody Target for the Treatment of Soft Tissue Sarcoma. Am. J. Transl. Res. 2015, 7, 1181–1188. [Google Scholar] [PubMed]
- Bonnefoy, N.; Bastid, J.; Alberici, G.; Bensussan, A.; Eliaou, J.F. CD39: A Complementary Target to Immune Checkpoints to Counteract Tumor-Mediated Immunosuppression. Oncoimmunology 2015, 4, e1003015. [Google Scholar] [CrossRef] [PubMed]
- Aliagas, E.; Vidal, A.; Texido, L.; Ponce, J.; Condom, E.; Martin-Satue, M. High Expression of Ecto-Nucleotidases CD39 and CD73 in Human Endometrial Tumors. Mediators Inflamm. 2014, 2014, 509027. [Google Scholar] [CrossRef] [PubMed]
- Bastid, J.; Regairaz, A.; Bonnefoy, N.; Dejou, C.; Giustiniani, J.; Laheurte, C.; Cochaud, S.; Laprevotte, E.; Funck-Brentano, E.; Hemon, P.; et al. Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol. Res. 2015, 3, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.; Islam, A.; Kabir, I.; Jones, P.K. Patterns of Morbidity and Mortality in Typhoid Fever Dependent on Age and Gender: Review of 552 Hospitalized Patients with Diarrhea. Rev. Infect. Dis. 1991, 13, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Geddes, K.; Rubino, S.J.; Magalhaes, J.G.; Streutker, C.; le Bourhis, L.; Cho, J.H.; Robertson, S.J.; Kim, C.J.; Kaul, R.; Philpott, D.J. Identification of an Innate T Helper Type 17 Response to Intestinal Bacterial Pathogens. Nat. Med. 2011, 17, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Ladel, C.; Miko, D.; Kaufmann, S.H. Salmonella Typhimurium aroA- Infection in Gene-Targeted Immunodeficient Mice: Major Role of CD4+ TCR-Alpha Beta Cells and IFN-Gamma in Bacterial Clearance Independent of Intracellular Location. J. Immunol. 1996, 156, 3321–3326. [Google Scholar] [PubMed]
- Johanns, T.M.; Ertelt, J.M.; Rowe, J.H.; Way, S.S. Regulatory T Cell Suppressive Potency Dictates the Balance between Bacterial Proliferation and Clearance During Persistent Salmonella Infection. PLoS Pathog. 2010, 6, e1001043. [Google Scholar] [CrossRef] [PubMed]
- Bou Ghanem, E.N.; Clark, S.; Roggensack, S.E.; McIver, S.R.; Alcaide, P.; Haydon, P.G.; Leong, J.M. Extracellular Adenosine Protects against Streptococcus Pneumoniae Lung Infection by Regulating Pulmonary Neutrophil Recruitment. PLoS Pathog. 2015, 11, e1005126. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Costales, M.G.; Cavanaugh, C.; Williams, K. Extracellular Adenosine Generation in the Regulation of Pro-Inflammatory Responses and Pathogen Colonization. Biomolecules 2015, 5, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.R. Too Much of a Good Thing: Adenosine Overload in Adenosine-Deaminase-Deficient Mice. Trends Pharmacol. Sci. 2003, 24, 66–70. [Google Scholar] [CrossRef]
- Cronstein, B.N. Adenosine, an Endogenous Anti-Inflammatory Agent. J. Appl. Physiol. 1994, 76, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Cronstein, B. Regulation of Inflammation by Adenosine. Front. Immunol. 2013, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Townsend-Nicholson, A.; Baker, E.; Schofield, P.R.; Sutherland, G.R. Localization of the Adenosine A1 Receptor Subtype Gene (ADORA1) to Chromosome 1q32.1. Genomics 1995, 26, 423–425. [Google Scholar] [CrossRef]
- Livingston, M.; Heaney, L.G.; Ennis, M. Adenosine, Inflammation and Asthma—A Review. Inflamm. Res. 2004, 53, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection from Tissue Damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A. Adenosine A3 Receptors: Novel Ligands and Paradoxical Effects. Trends Pharmacol. Sci. 1998, 19, 184–191. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Brambilla, R.; Ceruti, S.; Kim, H.O.; von Lubitz, D.K.; Jacobson, K.A.; Cattabeni, F.G. Protein-Dependent Activation of Phospholipase C by Adenosine A3 Receptors in Rat Brain. Mol. Pharmacol. 1995, 48, 1038–1045. [Google Scholar] [CrossRef]
- Cieslak, M.; Komoszynski, M.; Wojtczak, A. Adenosine A(2a) Receptors in Parkinson’s Disease Treatment. Purinergic Signal. 2008, 4, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.R.; Vance, C.O.; Morschl, E.; Wilson, C.N. Adenosine Receptors and Inflammation. Handb. Exp. Pharmacol. 2009, 193, 215–269. [Google Scholar]
- Strohmeier, G.R.; Reppert, S.M.; Lencer, W.I.; Madara, J.L. The A2b Adenosine Receptor Mediates cAMP Responses to Adenosine Receptor Agonists in Human Intestinal Epithelia. J. Biol. Chem. 1995, 270, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Hashikawa, T.; Saho, T.; Takedachi, M.; Nozaki, T.; Shimabukuro, Y.; Okada, H. Adenosine Regulates the IL-1β-Induced Cellular Functions of Human Gingival Fibroblasts. Int. Immunol. 2001, 13, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Fazzi, D.; Stefanelli, A.; Varani, K.; Borea, P.A. Adenosine Receptor Targeting in Health and Disease. Expert. Opin. Investig. Drugs 2011, 20, 1591–1609. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Baraldi, P.G.; Moorman, A.R.; Borea, P.A.; Varani, K. History and Perspectives of A2a Adenosine Receptor Antagonists as Potential Therapeutic Agents. Med. Res. Rev. 2015, 35, 790–848. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Padovan, M.; Govoni, M.; Vincenzi, F.; Trotta, F.; Borea, P.A. The Role of Adenosine Receptors in Rheumatoid Arthritis. Autoimmun. Rev. 2010, 10, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine Receptors and Cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. Selective Deletion of the A1 Adenosine Receptor Abolishes Heart-Rate Slowing Effects of Intravascular Adenosine in Vivo. PLoS ONE 2009, 4, e6784. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Daguma, L.; Nichols, D.; Hutchison, A.J.; Williams, M. The Adenosine/Neutrophil Paradox Resolved: Human Neutrophils Possess Both A1 and A2 Receptors That Promote Chemotaxis and Inhibit O2 Generation, Respectively. J. Clin. Investig. 1990, 85, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.H.; Peterson-Yantorno, K.; Carre, D.A.; McGlinn, A.M.; Coca-Prados, M.; Stone, R.A.; Civan, M.M. A3 Adenosine Receptors Regulate Cl- Channels of Nonpigmented Ciliary Epithelial Cells. Am. J. Physiol. 1999, 276 Pt 1, C659–C666. [Google Scholar] [CrossRef] [PubMed]
- Odashima, M.; Bamias, G.; Rivera-Nieves, J.; Linden, J.; Nast, C.C.; Moskaluk, C.A.; Marini, M.; Sugawara, K.; Kozaiwa, K.; Otaka, M.; et al. Activation of A2a Adenosine Receptor Attenuates Intestinal Inflammation in Animal Models of Inflammatory Bowel Disease. Gastroenterology 2005, 129, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.A.; Li, Y.; Calabrese, G.M.; Freire, R.S.; Zaja-Milatovic, S.; van Opstal, E.; Figler, R.A.; Linden, J.; Guerrant, R.L. Contribution of Adenosine A(2b) Receptors in Clostridium Difficile Intoxication and Infection. Infect. Immun. 2012, 80, 4463–4473. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.K.; Olson, R.A.; Jones, H.M.; Duffey, M.E. Release of ATP During Host Cell Killing by Enteropathogenic E. Coli and Its Role as a Secretory Mediator. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G74–G86. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Hart, A.; Batt, R.M.; McDougall, C.; McLean, L. Ultrastructural and Biochemical Changes in Human Jejunal Mucosa Associated with Enteropathogenic Escherichia coli (0111) Infection. J. Pediatr. Gastroenterol. Nutr. 1986, 5, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.A.; Mathers, D.A.; Yan, H.; Baimbridge, K.G.; Finlay, B.B. Enteropathogenic Escherichia coli Markedly Decreases the Resting Membrane Potential of Caco-2 and Hela Human Epithelial Cells. Infect. Immun. 1996, 64, 4820–4825. [Google Scholar] [PubMed]
- Crane, J.K.; Shulgina, I. Feedback Effects of Host-Derived Adenosine on Enteropathogenic Escherichia coli. FEMS Immunol. Med. Microbiol. 2009, 57, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D. The Bacterial Etiology of Destructive Periodontal Disease: Current Concepts. J. Periodontol. 1992, 63 (Suppl. 4), 322–331. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, K.R.; Yilmaz, O. Prelude to Oral Microbes and Chronic Diseases: Past, Present and Future. Microbes Infect. 2015, 17, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, K.R.; Yilmaz, O. Looking in the Porphyromonas Gingivalis Cabinet of Curiosities: The Microbium, the Host and Cancer Association. Mol. Oral Microbiol. 2014, 29, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Verbeke, P.; Lamont, R.J.; Ojcius, D.M. Intercellular Spreading of Porphyromonas Gingivalis Infection in Primary Gingival Epithelial Cells. Infect. Immun. 2006, 74, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Yao, L.; Maeda, K.; Rose, T.M.; Lewis, E.L.; Duman, M.; Lamont, R.J.; Ojcius, D.M. ATP Scavenging by the Intracellular Pathogen Porphyromonas Gingivalis Inhibits P2X7-Mediated Host-Cell Apoptosis. Cell. Microbiol. 2008, 10, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Gerbase, C.A.; Rowley, J.T.; Mertens, T.E. Global Epidemiology of Sexually Transmitted Diseases. Lancet 1998, 351 (Suppl. 3), 2–4. [Google Scholar] [CrossRef]
- Pettengill, A.M.; Lam, V.W.; Ojcius, D.M. The Danger Signal Adenosine Induces Persistence of Chlamydial Infection through Stimulation of A2b Receptors. PLoS ONE 2009, 4, e8299. [Google Scholar] [CrossRef] [PubMed]
- Barletta, K.E.; Cagnina, R.E.; Burdick, M.D.; Linden, J.; Mehrad, B. Adenosine A(2b) Receptor Deficiency Promotes Host Defenses against Gram-Negative Bacterial Pneumonia. Am. J. Respir. Crit. Care Med. 2012, 186, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Schingnitz, U.; Hartmann, K.; Macmanus, C.F.; Eckle, T.; Zug, S.; Colgan, S.P.; Eltzschig, H.K. Signaling through the A2b Adenosine Receptor Dampens Endotoxin-Induced Acute Lung Injury. J. Immunol. 2010, 184, 5271–5279. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.W.; Wang, H.P.; Lin, F.; Wang, X.; Long, M.; Zhang, H.Z.; Dong, K. CD73 Promotes Proliferation and Migration of Human Cervical Cancer Cells Independent of Its Enzyme Activity. BMC Cancer 2017, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Kern, J.W.; Missiakas, D.M.; Schneewind, O. Staphylococcus Aureus Synthesizes Adenosine to Escape Host Immune Responses. J. Exp. Med. 2009, 206, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling During Inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.E.; Jacobson, K.A. Recent Developments in Adenosine Receptor Ligands and Their Potential as Novel Drugs. Biochim. Biophys. Acta 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- Press, N.J.; Fozard, J.R. Progress towards Novel Adenosine Receptor Therapeutics Gleaned from the Recent Patent Literature. Expert. Opin. Ther. Pat. 2010, 20, 987–1005. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Varani, K.; Tringali, G.; Polosa, R. Adenosine and Adenosine Receptors: Their Contribution to Airway Inflammation and Therapeutic Potential in Asthma. Curr. Med. Chem. 2009, 16, 3875–3885. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Tabrizi, M.A.; Gessi, S.; Borea, P.A. Adenosine Receptor Antagonists: Translating Medicinal Chemistry and Pharmacology into Clinical Utility. Chem. Rev. 2008, 108, 238–263. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Gao, Z.G.; Jacobson, K.A.; Spalluto, G. Progress in the Pursuit of Therapeutic Adenosine Receptor Antagonists. Med. Res. Rev. 2006, 26, 131–159. [Google Scholar] [CrossRef] [PubMed]
- Trevethick, M.A.; Mantell, S.J.; Stuart, E.F.; Barnard, A.; Wright, K.N.; Yeadon, M. Treating Lung Inflammation with Agonists of the Adenosine A2a Receptor: Promises, Problems and Potential Solutions. Br. J. Pharmacol. 2008, 155, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Kurtz, C.C.; Wilson, J.M.; Burnette, B.R.; Wiznerowicz, E.B.; Ross, W.G.; Rieger, J.M.; Figler, R.A.; Linden, J.; Crowe, S.E.; et al. A2a Adenosine Receptor (AR) Activation Inhibits Pro-Inflammatory Cytokine Production by Human CD4+ Helper T Cells and Regulates Helicobacter-Induced Gastritis and Bacterial Persistence. Mucosal. Immunol. 2009, 2, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.M.; Trevethick, M.A. Suppression of Inflammatory and Immune Responses by the A(2a) Adenosine Receptor: An Introduction. Br. J. Pharmacol. 2008, 153, S27–S34. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine Receptors as Drug Targets—What Are the Challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed]
- El-Tayeb, A.; Iqbal, J.; Behrenswerth, A.; Romio, M.; Schneider, M.; Zimmermann, H.; Schrader, J.; Muller, C.E. Nucleoside-5′-Monophosphates as Prodrugs of Adenosine A2a Receptor Agonists Activated by Ecto-5′-Nucleotidase. J. Med. Chem. 2009, 52, 7669–7677. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, F.; Sun, F.; Huang, P. Adenosine Promotes IL-6 Release in Airway Epithelia. J. Immunol. 2008, 180, 4173–4181. [Google Scholar] [CrossRef] [PubMed]
- Picher, M.; Burch, L.H.; Hirsh, A.J.; Spychala, J.; Boucher, R.C. Ecto 5′-Nucleotidase and Nonspecific Alkaline Phosphatase. Two AMP-Hydrolyzing Ectoenzymes with Distinct Roles in Human Airways. J. Biol. Chem. 2003, 278, 13468–13479. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Fullbier, L.; Wehrmann, M.; Khoury, J.; Mittelbronn, M.; Ibla, J.; Rosenberger, P.; Eltzschig, H.K. Identification of Ectonucleotidases CD39 and CD73 in Innate Protection During Acute Lung Injury. J. Immunol. 2007, 178, 8127–8137. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chowdhury, N.; Roberts, J.S.; Yilmaz, O. Ectonucleotidase CD73 Promotes Porphyromonas gingivalis Perstistence in the Oral Epithelium. Unpublished Work.
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a Extracellular Adenosine Receptor-Mediated Signaling in Adenosine-Mediated Inhibition of T-Cell Activation and Expansion. Blood 1997, 90, 1600–1610. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
| Cell locations | Cell Types | CD39 Expression | CD73 Expression | Expressed Adenosine Receptor | Relevant Microbe | Adenosine Signaling in Infection | References | |
|---|---|---|---|---|---|---|---|---|
| Inflammatory Response | Other Immune Response | |||||||
| Epithelial tissue | Airway/Bronchi | Expressed | Expressed | A1 A2A A2B A3 | Pseudomonas aeruginosa Streptococcus pneumoniae | Pro-inflammatory cytokine levels | Infiltration of m macrophages and neutrophils Pathogen killing of PMNs | [61,79,128,129,130] |
| Gingiva | Expressed * | Expressed * | A1 A2A A2B A3 | Porphyromonas gingivalis | Anti-inflammatory cytokine levels Inflammasome activation | Modulation of NADPH oxidase signaling and cAMP generation | [11,53,54,55,107,108,109,110,111,131] | |
| Colon (T84) | Expressed | Expressed | A2A A2B | Helicobacter pylori | Pro-inflammatory cytokine levels | cAMP generation | [57,91,124] | |
| Cervix (HeLa 229) | Highly expressed | Not known | A2B | Chlamydia trachomatis | Not known | cAMP generation | [112] | |
| Immune tissue | Regulatory T cells | Highly expressed | Highly expressed | A2A A2B | Mycobacterium tuberculosis | Not known | CD4+ T helper-1 cell responses | [60,67] |
| Splenocytes | Expressed | Expressed | A2A | Salmonella spp. | Pro-inflammatory and anti-inflammatory cytokine levels | Pathogen clearance ability of host | [57,75,132] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.S.; Yilmaz, Ö. Unfolding Role of a Danger Molecule Adenosine Signaling in Modulation of Microbial Infection and Host Cell Response. Int. J. Mol. Sci. 2018, 19, 199. https://doi.org/10.3390/ijms19010199
Lee JS, Yilmaz Ö. Unfolding Role of a Danger Molecule Adenosine Signaling in Modulation of Microbial Infection and Host Cell Response. International Journal of Molecular Sciences. 2018; 19(1):199. https://doi.org/10.3390/ijms19010199
Chicago/Turabian StyleLee, Jaden S., and Özlem Yilmaz. 2018. "Unfolding Role of a Danger Molecule Adenosine Signaling in Modulation of Microbial Infection and Host Cell Response" International Journal of Molecular Sciences 19, no. 1: 199. https://doi.org/10.3390/ijms19010199
APA StyleLee, J. S., & Yilmaz, Ö. (2018). Unfolding Role of a Danger Molecule Adenosine Signaling in Modulation of Microbial Infection and Host Cell Response. International Journal of Molecular Sciences, 19(1), 199. https://doi.org/10.3390/ijms19010199