Rosiglitazone as a Modulator of TLR4 and TLR3 Signaling Pathways in Rat Primary Neurons and Astrocytes

 , ,
, ,
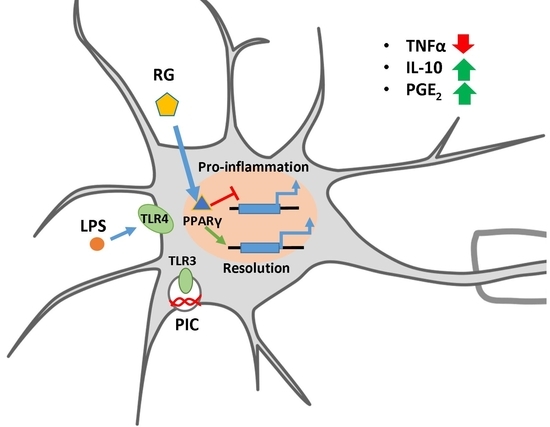
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
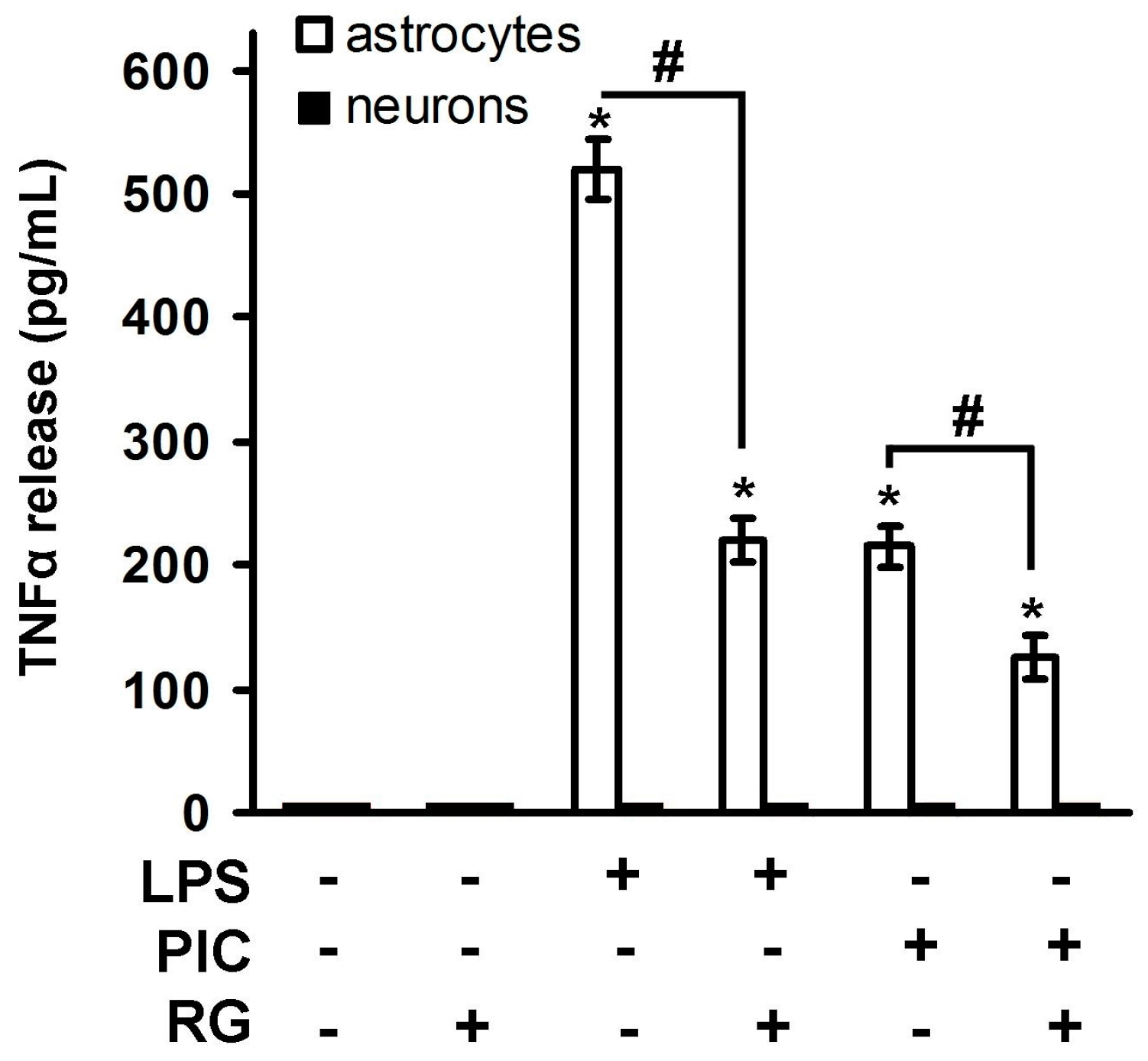
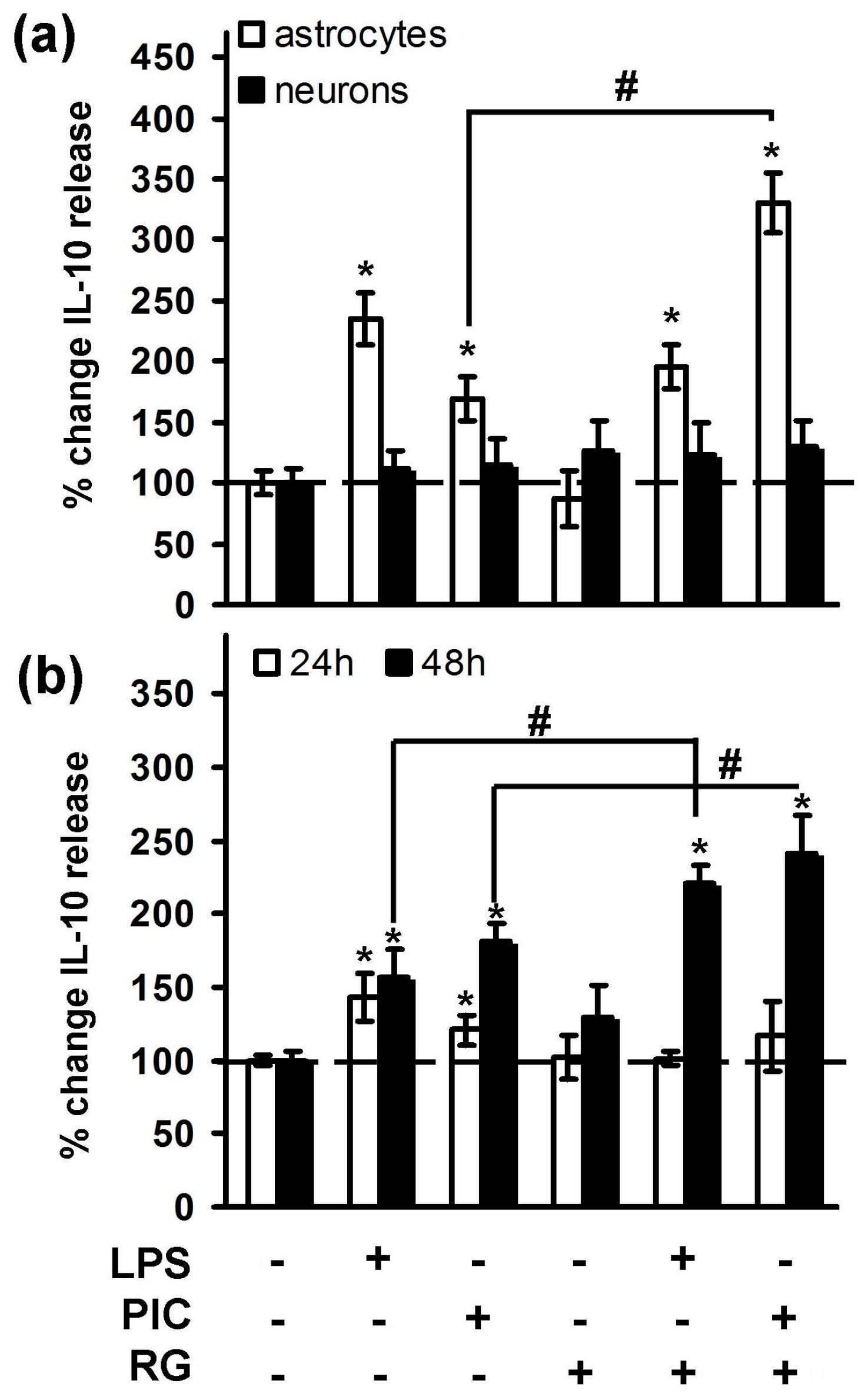
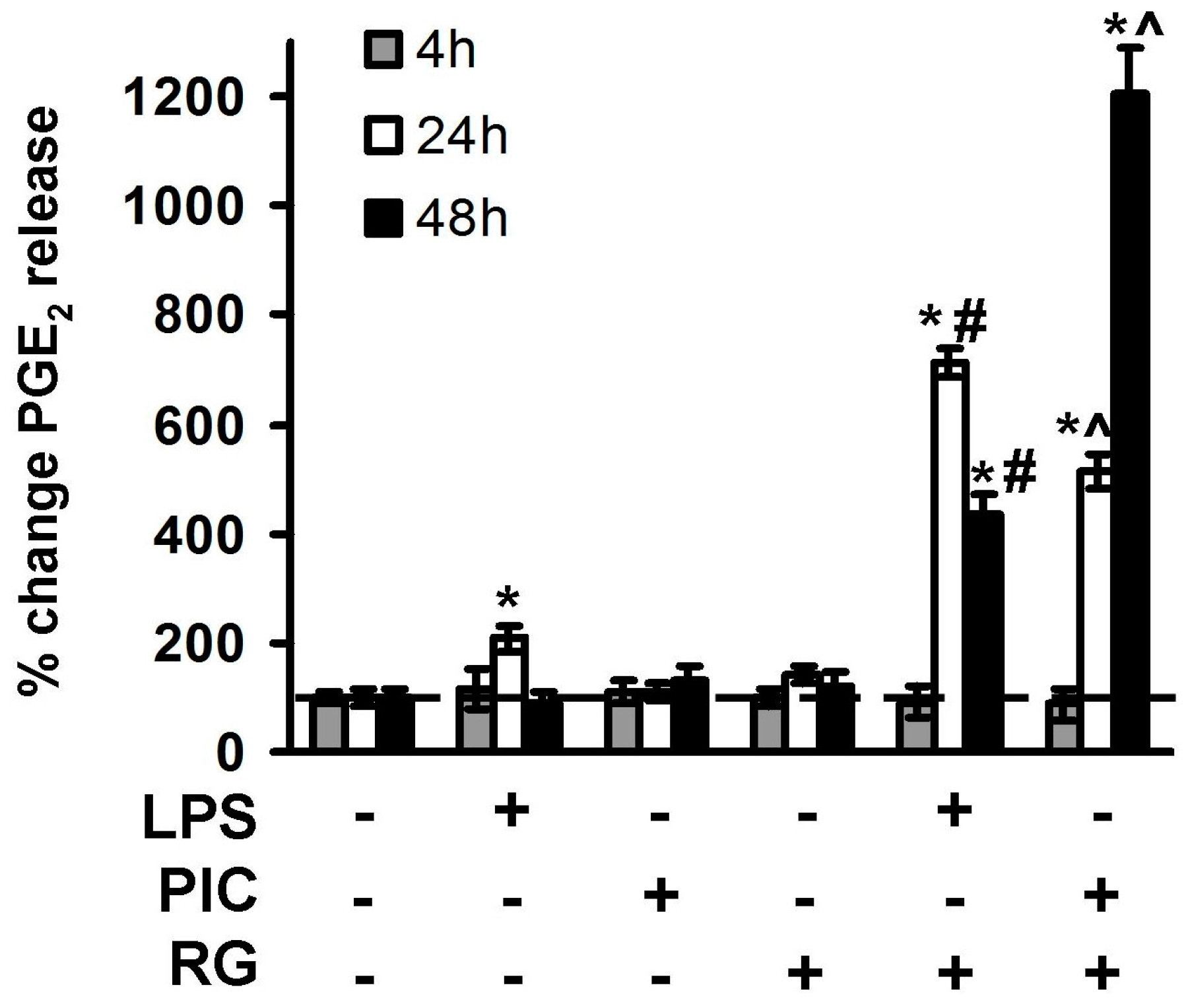
2. Results
2.1. Rosiglitazone Regulation of IL-10 Expression upon Activation of TLR3 and TLR4 Receptor in Astrocytes and Neurons
2.2. The Effect of Rosiglitazone on PGE2 Release upon Activation of TLR3 and TLR4 in Astrocytes and Neurons
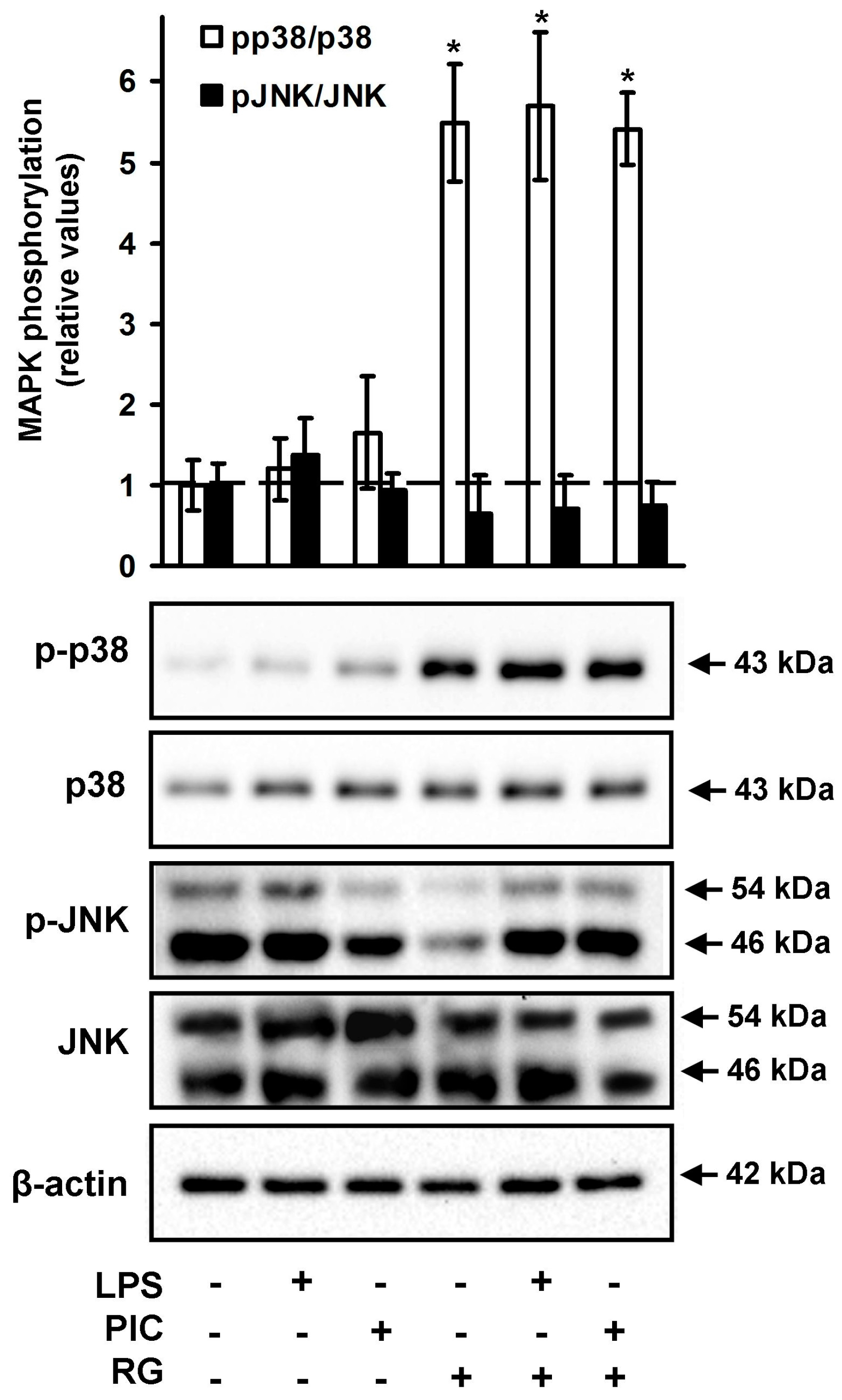
2.3. The Effect of Rosiglitazone on the Phosphorylation of JNK and p38 MAPK in Neurons
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Astrocytes Primary Cell Culture
4.3. Primary Cultures of Rat Neuron Cortical Cells
4.4. Western Blotting
4.5. Determination of TNFα, IL-10 and PGE2 by Enzyme-Linked Immunoassay
4.6. Experimental Data Analysis and Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TLR | toll-like receptor |
| PIC | poly I:C |
| LPS | lipopolysaccharide |
| TNFα | tumor necrosis factor alpha |
| MAPK | mitogen-activated kinase |
| RG | rosiglitazone |
| IL-10 | interleukin-10 |
References
- Ransohoff, R.M.; Schafer, D.; Vincent, A.; Blachère, N.E.; Bar-Or, A. Neuroinflammation: Ways in Which the Immune System Affects the Brain. Neurotherapeutics 2015, 12, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Kerschensteiner, M.; Meinl, E. Dual role of inflammation in CNS disease. Neurology 2007, 68, S58–S63. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2015, 109, 14.12.1–14.12.10. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Filous, A.R.; Silver, J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of microglia in CNS inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Rietdijk, C.D.; Van Wezel, R.J.A.; Garssen, J.; Kraneveld, A.D. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol. Neuroinflamm. 2016, 3, 27–37. [Google Scholar] [CrossRef]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Normando, E.M.; Davis, B.M.; De Groef, L.; Nizari, S.; Turner, L.A.; Ravindran, N.; Pahlitzsch, M.; Brenton, J.; Malaguarnera, G.; Guo, L.; et al. The retina as an early biomarker of neurodegeneration in a rotenone-induced model of Parkinson’s disease: Evidence for a neuroprotective effect of rosiglitazone in the eye and brain. Acta Neuropathol. Commun. 2016, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Albertz, J.; Guo, Z.; Peng, Q.; Rudow, G.; Troncoso, J.C.; Ross, C.A.; Duan, W. Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J. Neurochem. 2013, 125, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Okami, N.; Narasimhan, P.; Yoshioka, H.; Sakata, H.; Kim, G.S.; Jung, J.E.; Maier, C.M.; Chan, P.H. Prevention of JNK phosphorylation as a mechanism for rosiglitazone in neuroprotection after transient cerebral ischemia: activation of dual specificity phosphatase. J. Cereb. Blood Flow Metab. 2013, 33, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, R.; Yi, J.H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Luna-Medina, R.; Cortes-Canteli, M.; Alonso, M.; Santos, A.; Martinez, A.; Perez-Castillo, A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2005, 280, 21453–21462. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Grabeklis, S.; Hanck, T.; Sergeeva, M.; Reiser, G. Peroxisome proliferator-activated receptor (PPAR)-positively controls and PPAR negatively controls cyclooxygenase-2 expression in rat brain astrocytes through a convergence on PPAR/via mutual control of PPAR expression levels. Mol. Pharmacol. 2009, 76, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists suppress the production of IL-12 family cytokines by activated glia. J. Immunol. 2007, 178, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, L.; Meng, H.; Wang, X.; Liu, S. Rosiglitazone protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity through inhibition of microglia activation. Int. J. Neurosci. 2012, 122, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Young, K.C.; Bai, C.H.; Yu, B.C.; Ma, C.T.; Chien, Y.C.; Chiang, C.L.; Liao, C.S.; Lai, H.W.; Tsao, C.W. Rosiglitazone regulates anti-inflammation and growth inhibition via PTEN. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Pankevich, E.V.; Astakhova, A.A.; Chistyakov, D.V.; Sergeeva, M.G. Antiinflammatory effect of rosiglitazone via modulation of mRNA stability of interleukin 10 and cyclooxygenase 2 in astrocytes. Biochemistry 2017, 82, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Li, C.; Yang, H.; Shen, A.; Wu, X.; Yuan, Q.; Kang, L.; Liu, Z.; Zhang, G.; Lu, X.; et al. The relationship between Src-suppressed C kinase substrate and β-1,4 galactosyltransferase-I in the process of lipopolysaccharide-induced TNF-α secretion in rat primary astrocytes. Cell. Mol. Neurobiol. 2011, 31, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.A.; Getts, M.T.; Miller, S.D. Pro-inflammatory functions of astrocytes correlate with viral clearance and strain-dependent protection from TMEV-induced demyelinating disease. Virology 2008, 375, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.V.; Aleshin, S.; Sergeeva, M.G.; Reiser, G. Regulation of peroxisome proliferator-activated receptor β/δ expression and activity levels by toll-like receptor agonists and MAP kinase inhibitors in rat astrocytes. J. Neurochem. 2014, 130, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Renauld, A.E.; Spengler, R.N. Tumor necrosis factor expressed by primary hippocampal neurons and SH-SY5Y cells is regulated by α2-adrenergic receptor activation. J. Neurosci. Res. 2002, 67, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Clark, R.K.; McDonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Terrando, N.; Monaco, C.; Ma, D.; Foxwell, B.M.J.; Feldmann, M.; Maze, M. Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc. Natl. Acad. Sci. USA 2010, 107, 20518–20522. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Silva, D.; Carriche, G.M.; Castro, A.G.; Roque, S.; Saraiva, M. Balancing the immune response in the brain: IL-10 and its regulation. J. Neuroinflamm. 2016, 13, 297. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H.; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Peng, X.; Insolera, R.; Fink, D.J.; Mata, M. IL-10 promotes neuronal survival following spinal cord injury. Exp. Neurol. 2009, 220, 183–190. [Google Scholar] [CrossRef] [PubMed]
- García-Bueno, B.; Caso, J.R.; Leza, J.C. Stress as a neuroinflammatory condition in brain: Damaging and protective mechanisms. Neurosci. Biobehav. Rev. 2008, 32, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Hos, D.; Bucher, F.; Regenfuss, B.; Dreisow, M.L.; Bock, F.; Heindl, L.M.; Eming, S.A.; Cursiefen, C. IL-10 Indirectly Regulates Corneal Lymphangiogenesis and Resolution of Inflammation via Macrophages. Am. J. Pathol. 2016, 186, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rose, M.E.; Culver, S.; Ma, X.; Dixon, C.E.; Graham, S.H. Rosiglitazone attenuates inflammation and CA3 neuronal loss following traumatic brain injury in rats. Biochem. Biophys. Res. Commun. 2016, 472, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Uchimura, K.; Zhu, R.L.; Nagayama, T.; Rose, M.E.; Stetler, R.A.; Isakson, P.C.; Chen, J.; Graham, S.H. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc. Natl. Acad. Sci. USA 1998, 95, 10954–10959. [Google Scholar] [CrossRef] [PubMed]
- Nogawa, S.; Zhang, F.; Ross, M.E.; Iadecola, C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J. Neurosci. 1997, 17, 2746–2755. [Google Scholar] [PubMed]
- McCullough, L. Neuroprotective Function of the PGE2 EP2 Receptor in Cerebral Ischemia. J. Neurosci. 2004, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kwon, K.J.; Park, J.Y.; Lee, S.H.; Moon, C.H.; Baik, E.J. Neuroprotective effects of prostaglandin E2 or cAMP against microglial and neuronal free radical mediated toxicity associated with inflammation. J. Neurosci. Res. 2002, 70, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Kozicky, L.K.; Zhao, Z.Y.; Menzies, S.C.; Fidanza, M.; Reid, G.S.D.; Wilhelmsen, K.; Hellman, J.; Hotte, N.; Madsen, K.L.; Sly, L.M. Intravenous immunoglobulin skews macrophages to an anti-inflammatory, IL-10-producing activation state. J. Leukoc. Biol. 2015, 98, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rao, Y.-H.; Inoue, M.; Hao, R.; Lai, C.-H.; Chen, D.; McDonald, S.L.; Choi, M.-C.; Wang, Q.; Shinohara, M.L.; et al. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat. Commun. 2014, 5, 3479. [Google Scholar] [CrossRef] [PubMed]
- Lopachev, A.V.; Lopacheva, O.M.; Abaimov, D.A.; Koroleva, O.V.; Vladychenskaya, E.A.; Erukhimovich, A.A.; Fedorova, T.N. Neuroprotective Effect of Carnosine on Primary Culture of Rat Cerebellar Cells under Oxidative Stress. Biochemistry 2016, 81, 511–520. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chistyakov, D.V.; Azbukina, N.V.; Lopachev, A.V.; Kulichenkova, K.N.; Astakhova, A.A.; Sergeeva, M.G. Rosiglitazone as a Modulator of TLR4 and TLR3 Signaling Pathways in Rat Primary Neurons and Astrocytes. Int. J. Mol. Sci. 2018, 19, 113. https://doi.org/10.3390/ijms19010113
Chistyakov DV, Azbukina NV, Lopachev AV, Kulichenkova KN, Astakhova AA, Sergeeva MG. Rosiglitazone as a Modulator of TLR4 and TLR3 Signaling Pathways in Rat Primary Neurons and Astrocytes. International Journal of Molecular Sciences. 2018; 19(1):113. https://doi.org/10.3390/ijms19010113
Chicago/Turabian StyleChistyakov, Dmitry V., Nadezda V. Azbukina, Alexandr V. Lopachev, Ksenia N. Kulichenkova, Alina A. Astakhova, and Marina G. Sergeeva. 2018. "Rosiglitazone as a Modulator of TLR4 and TLR3 Signaling Pathways in Rat Primary Neurons and Astrocytes" International Journal of Molecular Sciences 19, no. 1: 113. https://doi.org/10.3390/ijms19010113
APA StyleChistyakov, D. V., Azbukina, N. V., Lopachev, A. V., Kulichenkova, K. N., Astakhova, A. A., & Sergeeva, M. G. (2018). Rosiglitazone as a Modulator of TLR4 and TLR3 Signaling Pathways in Rat Primary Neurons and Astrocytes. International Journal of Molecular Sciences, 19(1), 113. https://doi.org/10.3390/ijms19010113