Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL

 ,
,
Abstract
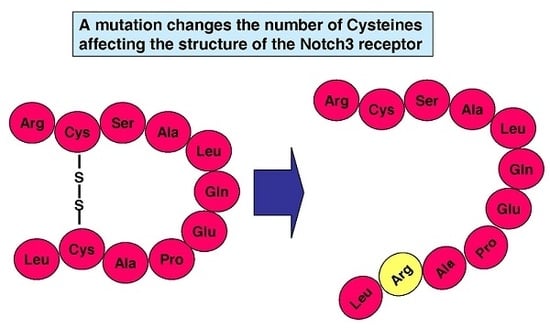
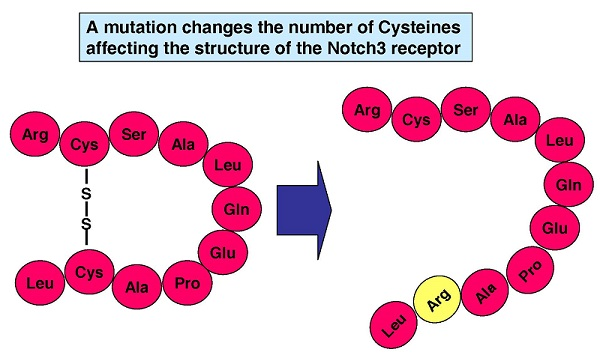
:
1. Introduction
2. Results
2.1. Mutation Features
2.2. Clinical Features
3. Discussion
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CADASIL | Cerebral autosomal dominant arteriopathy with subcortical infarcts and Leukoencephalopathy |
| ECD | Extracellular domain |
| EGF | Epidermal growth factor |
| ICD | Intracellular domain |
| MRI | Magnetic resonance imaging |
| WMH | White matter hyperintensities |
| GOM | Granular osmiophilic material |
| MAF | Minor allele frequency |
| ExAC | Exome aggregation consortium |
References
- Tournier-Lasserve, E.; Joutel, A.; Melki, J.; Weissenbach, J.; Lathrop, G.M.; Chabriat, H.; Mas, J.L.; Cabanis, E.A.; Baudrimont, M.; Maciazek, J.; et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat. Genet. 1993, 3, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cecillion, M.; Marechal, E.; et al. NOTCH3 mutations in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a mendelian condition causing stroke and vascular dementia. Ann. N. Y. Acad. Sci. 1997, 826, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Dziewulska, D.; Lewandowska, E. Pericytes as a new target for pathological processes in CADASIL. Neuropathology 2012, 32, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Karpinska, A.; Rosner, S.; Hopfner, F.; Zechmeister, M.; Peters, N.; Kremmer, E.; Haffner, C.; Giese, A.; Dichgans, M.; et al. Co-aggregate formation of CADASIL-mutant NOTCH3: A single-particle analysis. Hum. Mol. Genet. 2011, 20, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Zhang, X.; Yu, G.; Lee, S.J.; Chen, Y.E.; Prudovsky, I.; Wang, M.M. Biochemical characterization and cellular effects of CADASIL mutants of NOTCH3. PLoS ONE 2012, 7, e44964. [Google Scholar] [CrossRef] [PubMed]
- Opherk, C.; Duering, M.; Peters, N.; Karpinska, A.; Rosner, S.; Schneider, E.; Bader, B.; Giese, A.; Dichgans, M. CADASIL mutations enhance spontaneous multimerization of NOTCH3. Hum. Mol. Genet. 2009, 18, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Cognat, E.; Baron-Menguy, C.; Domenga-Denier, V.; Cleophax, S.; Fouillade, C.; Monet-Leprêtre, M.; Dewerchin, M.; Joutel, A. Archetypal Arg169Cys mutation in NOTCH3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss-of-function mechanism. Stroke 2014, 45, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Rutten, J.W.; Haan, J.; Terwindt, G.M.; van Duinen, S.G. Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert Rev. Mol. Diagn. 2014, 14, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Joutel, A. Transgenic and knock-out mice to probe function and dysfunction of the mutated gene, NOTCH3, in the cerebrovasculature. Bioessays 2011, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Chabriat, H.; Vahedi, K.; Iba-Zizen, M.T.; Joutel, A.; Nibbio, A.; Nagy, T.G.; Krebs, M.O.; Julien, J.; Dubois, B.; Ducrocq, X.; et al. Clinical spectrum of CADASIL: A study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet 1995, 346, 934–939. [Google Scholar] [CrossRef]
- Moreton, F.C.; Razvi, S.S.M.; Davidson, R.; Muir, K.W. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol. Scand. 2014, 130, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Adib-Samii, P.; Brice, G.; Martin, R.J.; Markus, H.S. Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype: Study in 200 consecutively recruited individuals. Stroke 2010, 41, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Cumurciuc, R.; Guichard, J.-P.; Reizine, D.; Gray, F.; Bousser, M.G.; Chabriat, H. Dilation of Virchow-Robin spaces in CADASIL. Eur. J. Neurol. 2006, 13, 187–190. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, M.; Jarosz, J.M.; Martin, R.J.; Deasy, N.; Powell, J.F.; Markus, H.S. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 2001, 56, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.S.; Martin, R.J.; Simpson, M.A.; Dong, Y.B.; Ali, N.; Crosby, A.H.; Powell, J.F. Diagnostic strategies in CADASIL. Neurology 2002, 59, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Ueda, M.; Nagatoshi, A.; Hirano, T.; Ito, T.; Arai, N.; Uyama, E.; Mori, K.; Nakamura, M.; Shinriki, S.; et al. Genotypic and phenotypic spectrum of CADASIL in Japan: The experience at a referral center in Kumamoto University from 1997 to 2014. J. Neurol. 2015, 262, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Ishiko, A.; Shimizu, A.; Nagata, E.; Takahashi, K.; Tabira, T.; Suzuki, N. NOTCH3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol. 2006, 112, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Joutel, A.; Favrole, P.; Labauge, P.; Chabriat, H.; Lescoat, C.; Andreux, F.; Domenga, V.; Cécillon, M.; Vahedi, K.; Ducros, A.; et al. Skin biopsy immunostaining with a NOTCH3 monoclonal antibody for CADASIL diagnosis. Lancet 2001, 358, 2049–2051. [Google Scholar] [CrossRef]
- Shan, S.; He, X.; He, L.; Wang, M.; Liu, C. Coexistence of congenital left ventricular aneurysm and prominent left ventricular trabeculation in a patient with LDB3 mutation: A case report. J. Med. Case Rep. 2017, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Dastsooz, H.; Nemati, H.; Fard, M.A.F.; Fardaei, M.; Faghihi, M.A. Novel mutations in PANK2 and PLA2G6genes in patients with neurodegenerative disorders: Two case reports. BMC Med. Genet. 2017, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Mendioroz, M.; Fernández-Cadenas, I.; del Río-Espinola, A.; Rovira, A.; Solé, E.; Fernández-Figueras, M.T.; Garcia-Patos, V.; Sastre-Garriga, J.; Domingues-Montanari, S.; Alvarez-Sabín, J.; et al. A missense HTRA1 mutation expands CARASIL syndrome to the Caucasian population. Neurology 2010, 75, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Frosch, M.P. A 46-year-old man with migraine, aphasia, and hemiparesis and similarly affected family members. N. Engl. J. Med. 2009, 360, 1656–1665. [Google Scholar]
- Kim, Y.; Choi, E.J.; Choi, C.G.; Kim, G.; Choi, J.H.; Yoo, H.W.; Kim, J.S. Characteristics of CADASIL in Korea: A novel cysteine-sparing NOTCH3 mutation. Neurology 2006, 66, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Muranishi, M.; Torugun, T.; Tango, H.; Nagakane, Y.; Kudeken, T.; Kawabe, K.; Oshima, F.; Yaoi, T.; Itoh, K.; et al. Two Japanese CADASIL families exhibiting NOTCH3 mutation R75P not involving cysteine residue. Intern. Med. 2008, 47, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yuan, Y.; Zhang, W.; Lv, H.; Hong, D.; Chen, B.; Liu, Y.; Luan, X.; Xie, S.; Wu, S. NOTCH3 mutations and clinical features in 33 mainland Chinese families with CADASIL. J. Neurol. Neurosurg. Psychiatry 2011, 82, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Wollenweber, F.A.; Hanecker, P.; Bayer-Karpinska, A.; Malik, R.; Bäzner, H.; Moreton, F.; Muir, K.W.; Müller, S.; Giese, A.; Opherk, C.; et al. Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke 2015, 46, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, C.; Mazzei, R.; Conforti, F.L.; Sprovieri, T.; Servillo, P.; Liguori, M.; Citrigno, L.; Gabriele, A.L.; Magariello, A.; Patitucci, A.; et al. Cadasil: Extended polymorphisms and mutational analysis of the NOTCH3 gene. J. Neurosci. Res. 2009, 87, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Kuang, H.; Wei, B.; Bo, L.; Xu, Z.; Xu, X.; Geng, D.; Sun, M. A novel cysteine-sparing NOTCH3 mutation in a Chinese family with CADASIL. PLoS ONE 2014, 9, e104533. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, I.; Alegre-Abarrategui, J.; Rodal, I.; España, A.; Ros, R.; Sendón, J.L.L.; Galloway, E.G.; Cervelló, A.; Caminero, A.B.; Zabala, A.; et al. On the diagnosis of CADASIL. J. Alzheimers Dis. 2009, 17, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Maksemous, N.; Smith, R.A.; Menon, S.; Davies, G.; Griffiths, L.R. Two novel mutations and a previously unreported intronic polymorphism in the NOTCH3 gene. Mutat. Res. 2012, 732, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Uchino, M.; Hirano, T.; Uyama, Y.H.E. Cerebral Autosomal Dominant Arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and CADASIL-like disorders in Japan. Ann. N. Y. Acad. Sci. 2002, 977, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Santa, Y.; Uyama, E.; Chui, D.H.; Arima, M.; Kotorii, S.; Takahashi, K.; Tabira, T. Genetic, clinical and pathological studies of CADASIL in Japan: A partial contribution of NOTCH3 mutations and implications of smooth muscle cell degeneration for the pathogenesis. J. Neurol. Sci. 2003, 212, 79–84. [Google Scholar] [CrossRef]
- Abramycheva, N.; Stepanova, M.; Kalashnikova, L.; Zakharova, M.; Maximova, M.; Tanashyan, M.; Lagoda, O.; Fedotova, E.; Klyushnikov, S.; Konovalov, R.; et al. New mutations in the NOTCH3 gene in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). J. Neurol. Sci. 2015, 349, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Homer, V.; George, P.M.; Toit, S.; Davidson, J.S.; Wilson, C.J. Novel human pathological mutations. Hum. Genet. 2007, 121, 645–652. [Google Scholar] [PubMed]
- Scheid, R. Cysteine-sparing NOTCH3 mutations: CADASIL or CADASIL variants? Neurology 2008, 71, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Fouillade, C.; Chabriat, H.; Riant, F.; Mine, M.; Arnoud, M.; Magy, L.; Bousser, M.G.; Tournier-Lasserve, E.; Joutel, A. Activating NOTCH3 mutation in a patient with small-vessel-disease of the brain. Hum. Mutat. 2008, 29, 452. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Dotti, M.T.; Candelise, L. Considerations on a mutation in the NOTCH3 gene sparing a cysteine residue: A rare polymorphism rather than a CADASIL variant. Funct. Neurol. 2012, 27, 247–252. [Google Scholar] [PubMed]
- Vlachakis, D.; Tsaniras, S.C.; Ioannidou, K.; Baumann, M.; Kossida, S. A series of NOTCH3 mutations in CADASIL, insights from 3D molecular modelling and evolutionary analyses. J. Mol. Biochem. 2014, 3, 97–105. [Google Scholar]
- Mazzei, R.; Conforti, F.L.; Lanza, P.L.; Sprovieri, T.; Lupo, M.R.; Gallo, O.; Patitucci, A.; Magariello, A.; Caracciolo, M.; Gabriele, A.L.; et al. A novel NOTCH3 gene mutation not involving a cysteine residue in an Italian family with CADASIL. Neurology 2004, 63, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Manent, J.; Lee, J.H.; Tikka, S.; Ospina, C.; Vanderburg, C.R.; Frosch, M.P.; Rodríguez-Falcón, M.; Villen, J.; Gygi, S.; et al. Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc. Natl. Acad. Sci. USA 2011, 108, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cadenas, I.; Andreu, A.L.; Gamez, J.; Gonzalo, R.; Martín, M.A.; Rubio, J.C.; Arenas, J. Splicing mosaic of the myophosphorylase gene due to a silent mutation in McArdle disease. Neurology 2003, 61, 1432–1434. [Google Scholar] [CrossRef] [PubMed]
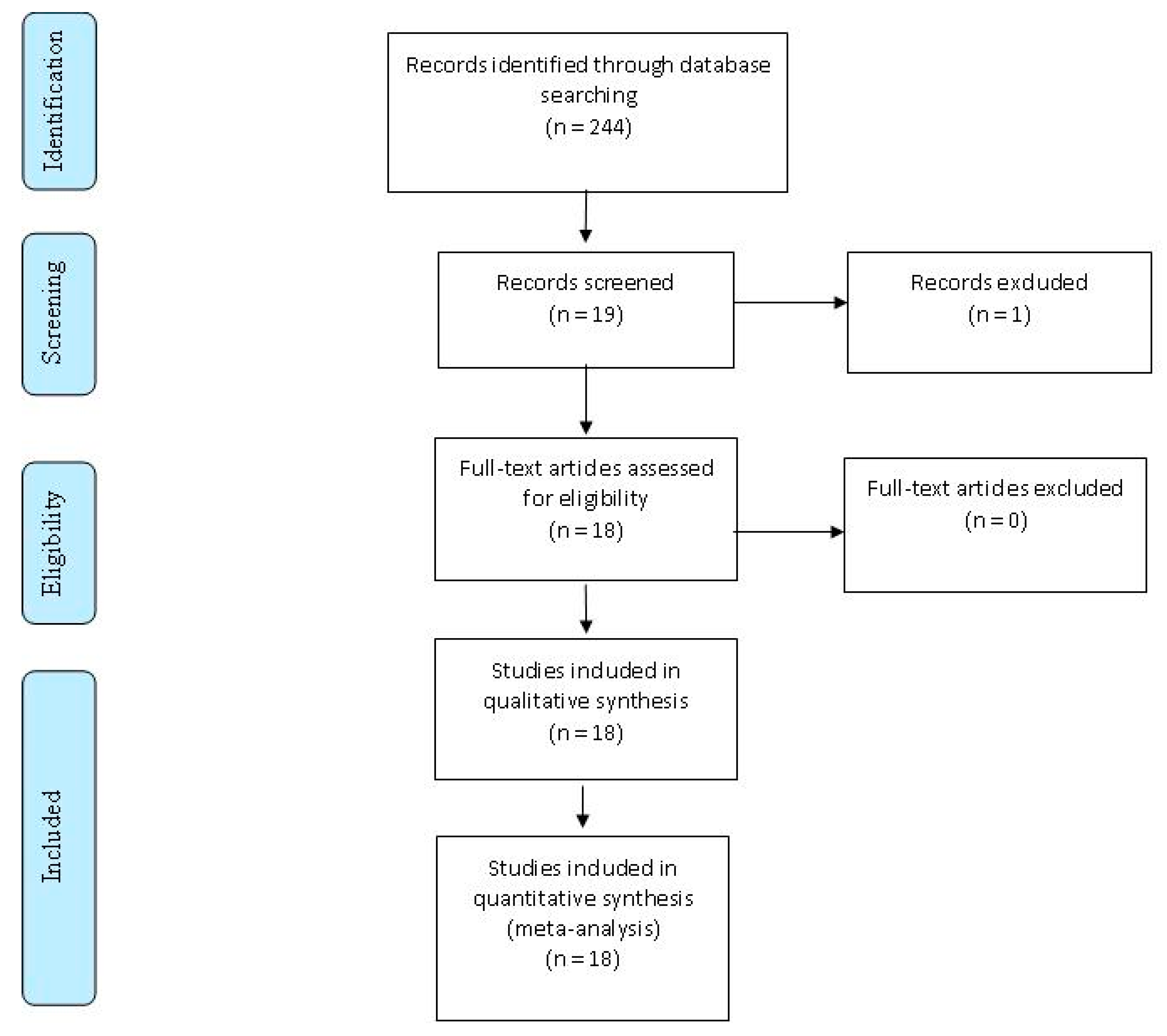

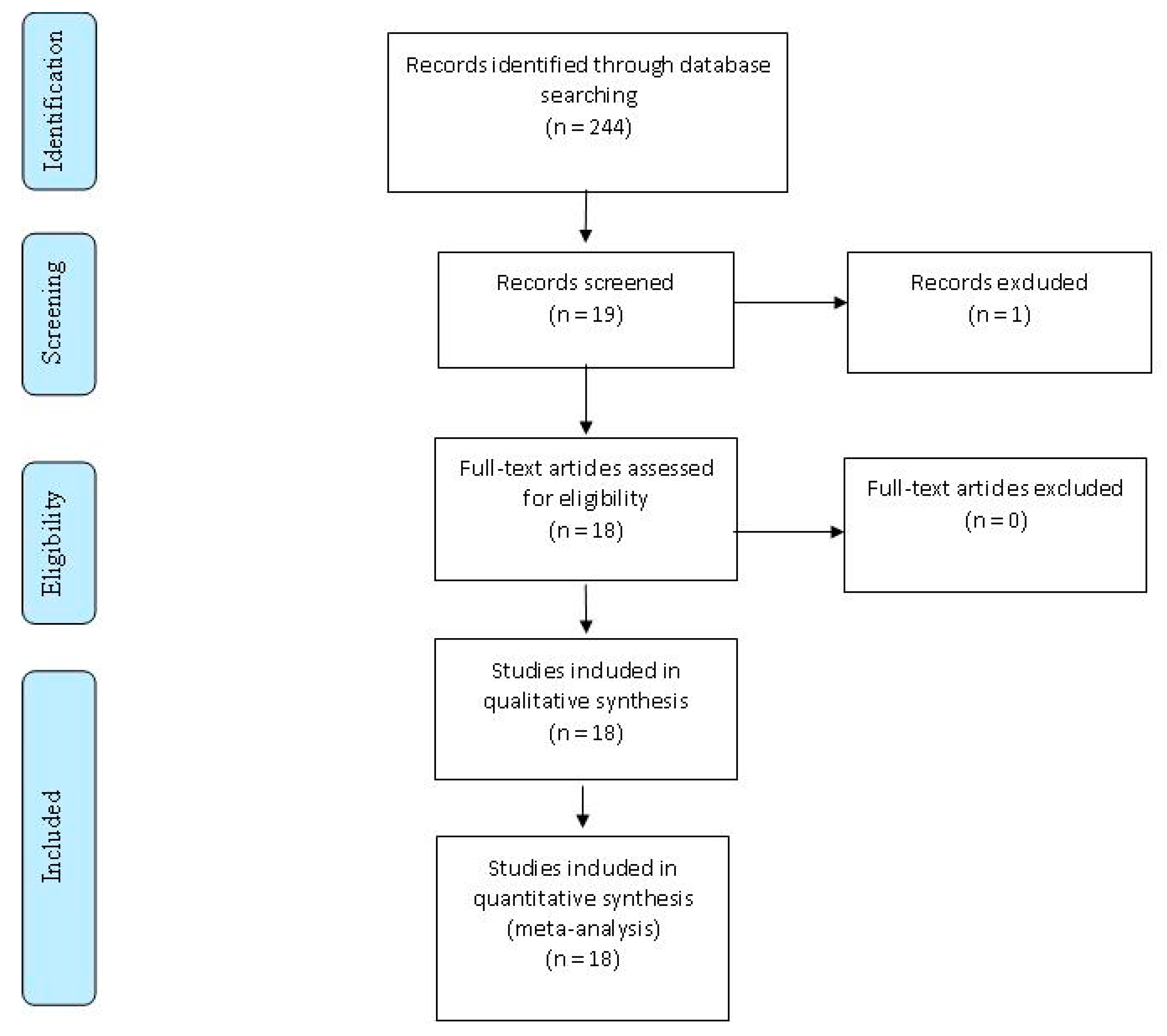
{kind=link}
{kind=link}
| Number | Mutation | Replaced | Substitution | Exon | Sequencing | MAF (ExAC) | MAF (1000 Genomes) | Author |
|---|---|---|---|---|---|---|---|---|
| 1 | p.R61W | Arg | Trp | 2 | 33 exons | 0.00007471 | - | Brass [23] |
| 2 | p.R75P | Arg | Pro | 3 | Exon: 3, 4, 11, 18; intron | 0 | 0 | Kim [24] |
| 3 | p.R75P | Arg | Pro | 3 | Exon: 3, 4, 11, 18; intron | 0 | 0 | Kim [24] |
| 4 | p.R75P | Arg | Pro | 3 | Exon: 3, 4, 11, 18; intron | 0 | 0 | Kim [24] |
| 5 | p.R75P | Arg | Pro | 3 | 33 exons, promotor | 0 | 0 | Mizuno [25] |
| 6 | p.R75P | Arg | Pro | 3 | 33 exons, promotor | 0 | 0 | Mizuno [25] |
| 7 | p.R75P | Arg | Pro | 3 | Exon: 2–24 | 0 | 0 | Wang [26] |
| 8 | p.D80G | Asp | Gly | 3 | 33 exons | 0 | 0 | Wollenweber [27] |
| 9 | p.R107W | Arg | Trp | 3 | Exon: 2–23 | 0.00001750 | - | Ungaro [28] |
| 10 | p.G149V | Gly | Val | 4 | 33 exons, intron | 0 | 0 | Ge [29] |
| 11 | p.Q151E | Gln | Glu | 4 | Exon: 2–23 | 0.00005703 | - | Ungaro [28] |
| 12 | p.Q151E | Gln | Glu | 4 | Exon: 2–6, 8, 11, 14, 18, 19, 22, 23; intron | 0.00005703 | - | Ampuero [30] |
| 13 | p.H170R | His | Arg | 4 | Exon: 2–6, 8, 11, 14, 18, 19, 22, 23; intron | 0.001917 | 0.0014 | Ampuero [30] |
| 14 | p.H170R | His | Arg | 4 | Exon: 2, 3, 4, 11, 18, 19 | 0.001917 | 0.0014 | Roy [31] |
| 15 | p.A198T | Ala | Thr | 4 | Exon: 2–23 | 0.00002513 | - | Ungaro [28] |
| 16 | p.A202V | Ala | Val | 4 | Exon: 2, 3, 4, 11, 18, 19 | 0.00001672 | - | Roy [31] |
| 17 | p.R207H | Arg | His | 4 | Exon: 2–23 | 0.00001664 | 0.0004 | Ungaro [28] |
| 18 | p.R213K | Arg | Lys | 4 | - | 0 | 0 | Uchino [32] |
| 19 | p.R213K | Arg | Lys | 4 | 33 exons | 0 | 0 | Santa [33] |
| 20 | p.V237M | Val | Met | 5 | - | 0.0002239 | 0.0008 | Uchino [32] |
| 21 | p.V252M | Val | Met | 5 | Exon: 2–23; intron | 0.00002493 | - | Abramycheva [34] |
| 22 | p.E309K | Glu | Lys | 6 | Exon: 2–23 | 0 | 0 | Ungaro [28] |
| 23 | p.S497L | Ser | Leu | 9 | Exon: 2–23; intron | 0.01234 | 0.0074 | Abramycheva [34] |
| 24 | p.T577A | Thr | Ala | 11 | - | 0.00001669 | 0 | Ferreira [35] |
| 25 | p.R592S | Arg | Ser | 11 | Exon: 2–23 | 0.00006714 | - | Ungaro [28] |
| 26 | p.V644D | Val | Asp | 12 | Exon: 2–23 | 0.0007013 | 0.0002 | Ungaro [28] |
| 27 | p.S978R | Ser | Arg | 18 | - | 0.0004606 | 0.0004 | Ferreira [35] |
| 28 | p.A1020P | Ala | Pro | 19 | - | 0.07318 | 0.110 | Scheid [36] |
| 29 | p.A1020P | Ala | Pro | 19 | - | 0.07318 | 0.110 | Scheid [36] |
| 30 | p.T1098S | Thr | Ser | 20 | Exon: 2–24 | 0 | 0 | Wang [26] |
| 31 | p.H1133Q | His | Gln | 21 | Exon: 2–23, intron | 0.01022 | 0.0030 | Abramycheva [34] |
| 32 | p.H1235L | His | Leu | 22 | Exon: 2–23 | 0.003990 | 0.0012 | Ungaro [28] |
| 33 | p.L1515P | Leu | Pro | 25 | 33 exons, intron | 0 | 0 | Fouillade [37] |
| 34 | p.V1762M | Val | Met | 29 | 33 exons | 0.0002146 | - | Bersano [38] |
| Numb. | Mutation | Origin | Sex | Smoker (n=7) | HT (n = 15) | DM (n = 13) | Dyslip (n = 11) | Clin. Onset (Years) | Migraine (n = 15) | Stroke (n =17) | Seizure (n = 5) | Psych. Disturb (n = 8) | Pseudb. Palsy (n = 5) | Demen (n = 18) | Gait Distur (n = 4) | FH a (n = 25) | LETP b (n = 11) | GOM (n = 10) | Author |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | p.R61W | USA | - | Yes | No | No | Yes | 20 | Yes | Yes | - | - | - | - | - | Yes | No | * | Brass |
| 2 | p.R75P | Korea | M | - | Yes | No | No | 53 | - | Yes | - | - | - | Yes | - | Yes | - | Yes | Kim |
| 3 | p.R75P | Korea | F | No | No | No | No | 47 | - | Yes | - | - | - | - | - | Yes | - | - | Kim |
| 4 | p.R75P | Korea | M | No | Yes | No | No | 65 | - | Yes | - | - | - | Yes | - | Yes | - | - | Kim |
| 5 | p.R75P | Japan | F | - | - | - | - | - | - | Yes | Yes | Yes | Yes | - | - | Yes | No | - | Mizuno |
| 6 | p.R75P | Japan | F | - | - | - | - | - | - | Yes | Yes | - | Yes | - | - | Yes | No | Yes | Mizuno |
| 7 | p.R75P | China | M | - | - | - | - | 34 | - | Yes | - | Yes | - | - | - | No | No | Yes | Wang |
| 8 | p.D80G | Germany | F | No | No | No | No | - | No | Yes | - | Yes | - | Yes | Yes | Yes | Yes | * | Wollenweber |
| 9 | p.R107W | Germany | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 10 | p.G149V | China | F | No | No | No | No | 39 | - | Yes | - | - | - | - | - | Yes | No | - | Ge |
| 11 | p.Q151E | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 12 | p.Q151E | Spain | - | - | - | - | - | - | - | Yes | - | - | - | - | - | - | - | - | Ampuero |
| 13 | p.H170R | Spain | - | - | - | - | - | - | - | Yes | - | - | - | - | - | - | - | - | Ampuero |
| 14 | p.H170R | Oceania | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Roy |
| 15 | p.A198T | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 16 | p.A202V | Oceania | F | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Roy |
| 17 | p.R207H | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 18 | p.R213K | Japan | M | - | No | No | No | 63 | Yes | Yes | No | - | Yes | Yes | Yes | Yes | - | - | Uchino |
| 19 | p.R213K | Japan | M | - | No | No | No | 10 | Yes | Yes | - | Yes | Yes | Yes | Yes | Yes | - | Yes | Santa |
| 20 | p.V237M | Japan | F | - | No | No | No | 71 | - | Yes | No | - | No | Yes | Yes | Yes | - | - | Uchino |
| 21 | p.V252M | Russia | - | - | No | No | - | - | - | - | - | - | - | - | - | - | - | - | Abramycheva |
| 22 | p.E309K | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 23 | p.S497L | Russia | - | - | No | No | - | - | - | - | - | - | - | - | - | - | - | - | Abramycheva |
| 24 | p.T577A | Portugal | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | Ferreira |
| 25 | p.R592S | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 26 | p.V644D | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 27 | p.S978R | Portugal | F | - | - | - | - | - | - | Yes | Yes | Yes | - | Yes | - | - | - | - | Ferreira |
| 28 | p.A1020P | Germany | F | - | Yes | - | - | Adolesc | Yes | - | - | - | - | Yes | - | Yes | No | Yes | Scheid |
| 29 | p.A1020P | Germany | F | - | Yes | - | - | - | - | - | - | Yes | - | - | - | Yes | No | No | Scheid |
| 30 | p.T1098S | China | M | - | - | - | - | 39 | - | Yes | - | Yes | - | Yes | - | Yes | No | Yes | Wang |
| 31 | p.H1133Q | Russia | - | - | No | No | - | - | - | - | - | - | - | - | - | - | - | - | Abramycheva |
| 32 | p.H1235L | Italy | - | - | - | - | - | - | Yes | - | - | - | - | Yes | - | Yes | - | - | Ungaro |
| 33 | p.L1515P | France | F | No | No | No | No | 35 | Yes | Yes | - | - | - | - | - | Yes | No | No | Fouillade |
| 34 | p.V1762M | Italy | F | Yes | - | - | Yes | Childhd | Yes | - | - | Yes | - | No | - | Yes | No | No | Bersano |
| Perc | - | - | - | 29% | 27% | 0% | 18% | - | 93% | 100% | 60% | 100% | 80% | 94% | 100% | 96% | 9% | 40% | - |
| Number | Mutation | Typical Clinical CADASIL Syndrome | WMH | Whole Exon Analysis | Mutation | GOM | Author |
|---|---|---|---|---|---|---|---|
| 1 | p.R61W | Yes | Yes | Yes | Yes | * | Brass |
| 2 | p.R75P | Yes | Yes | No | Yes | Yes | Kim |
| 3 | p.R75P | Yes | Yes | No | Yes | - | Kim |
| 4 | p.R75P | Yes | Yes | No | Yes | - | Kim |
| 5 | p.R75P | Yes | Yes | Yes | Yes | - | Mizuno |
| 6 | p.R75P | Yes | Yes | Yes | Yes | Yes | Mizuno |
| 7 | p.R75P | Yes | Yes | No | Yes | Yes | Wang |
| 8 | p.D80G | Yes | Yes | Yes | Yes | * | Wollenweber |
| 9 | p.R107W | Yes | Yes | No | Yes | - | Ungaro |
| 10 | p.G149V | Yes | Yes | Yes | Yes | - | Ge |
| 11 | p.Q151E | Yes | Yes | No | Yes | - | Ungaro |
| 12 | p.Q151E | Yes | Yes | No | Yes | - | Ampuero |
| 13 | p.H170R | Yes | Yes | No | No | - | Ampuero |
| 14 | p.H170R | Yes | NS | No | No | - | Roy |
| 15 | p.A198T | Yes | Yes | No | Yes | - | Ungaro |
| 16 | p.A202V | Yes | NS | No | Yes | - | Roy |
| 17 | p.R207H | Yes | Yes | No | Yes | - | Ungaro |
| 18 | p.R213K | Yes | Yes | NS | Yes | - | Uchino |
| 19 | p.R213K | Yes | Yes | Yes | Yes | Yes | Santa |
| 20 | p.V237M | Yes | Yes | NS | Yes | - | Uchino |
| 21 | p.V252M | Yes | Yes | No | Yes | - | Abramycheva |
| 22 | p.E309K | Yes | Yes | No | Yes | - | Ungaro |
| 23 | p.S497L | Yes | NS | No | No | - | Abramycheva |
| 24 | p.T577A | NS | NS | NS | Yes | - | Ferreira |
| 25 | p.R592S | Yes | Yes | No | Yes | - | Ungaro |
| 26 | p.V644D | Yes | Yes | No | Yes | - | Ungaro |
| 27 | p.S978R | Yes | Yes | NS | Yes | - | Ferreira |
| 28 | p.A1020P | Yes | Yes | NS | No | Yes | Scheid |
| 29 | p.A1020P | Yes | Yes | NS | No | No | Scheid |
| 30 | p.T1098S | Yes | Yes | No | Yes | Yes | Wang |
| 31 | p.H1133Q | Yes | NS | No | No | - | Abramycheva |
| 32 | p.H1235L | Yes | Yes | No | No | - | Ungaro |
| 33 | p.L1515P | Yes | Yes | Yes | Yes | No | Fouillade |
| 34 | p.V1762M | Yes | Yes | Yes | Yes | No | Bersano |
| Mutation | Score | Confidence | Domain | Prediction |
|---|---|---|---|---|
| R61W | 0.773 | Sensitivity: 0.76 | EGF-like 1 | Possibly damaging |
| Especificity: 0.86 | ||||
| R75P | 0.884 | Sensitivity: 0.71 | EGF-like 1 | Possibly damaging |
| Especificity: 0.89 | ||||
| D80G | 0.694 | Sensitivity: 0.78 | EGF-like 2 | Possibly damaging |
| Especificity: 0.85 | ||||
| R213K | 0.171 | Sensitivity: 0.89 | EGF-like 5 | Benign |
| Especificity: 0.72 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muiño, E.; Gallego-Fabrega, C.; Cullell, N.; Carrera, C.; Torres, N.; Krupinski, J.; Roquer, J.; Montaner, J.; Fernández-Cadenas, I. Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. Int. J. Mol. Sci. 2017, 18, 1964. https://doi.org/10.3390/ijms18091964
Muiño E, Gallego-Fabrega C, Cullell N, Carrera C, Torres N, Krupinski J, Roquer J, Montaner J, Fernández-Cadenas I. Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. International Journal of Molecular Sciences. 2017; 18(9):1964. https://doi.org/10.3390/ijms18091964
Chicago/Turabian StyleMuiño, Elena, Cristina Gallego-Fabrega, Natalia Cullell, Caty Carrera, Nuria Torres, Jurek Krupinski, Jaume Roquer, Joan Montaner, and Israel Fernández-Cadenas. 2017. "Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL" International Journal of Molecular Sciences 18, no. 9: 1964. https://doi.org/10.3390/ijms18091964
APA StyleMuiño, E., Gallego-Fabrega, C., Cullell, N., Carrera, C., Torres, N., Krupinski, J., Roquer, J., Montaner, J., & Fernández-Cadenas, I. (2017). Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. International Journal of Molecular Sciences, 18(9), 1964. https://doi.org/10.3390/ijms18091964