The Differential Distribution of RAPTA-T in Non-Invasive and Invasive Breast Cancer Cells Correlates with Its Anti-Invasive and Anti-Metastatic Effects

 ,
,  and
and
Abstract
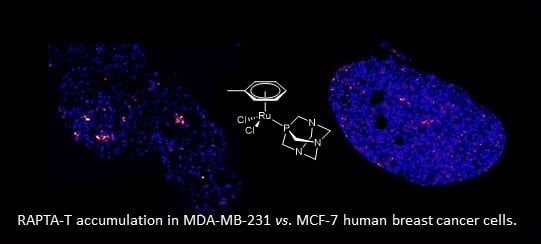
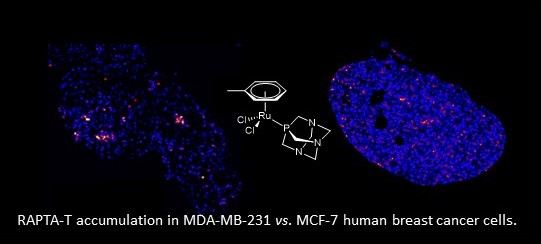
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
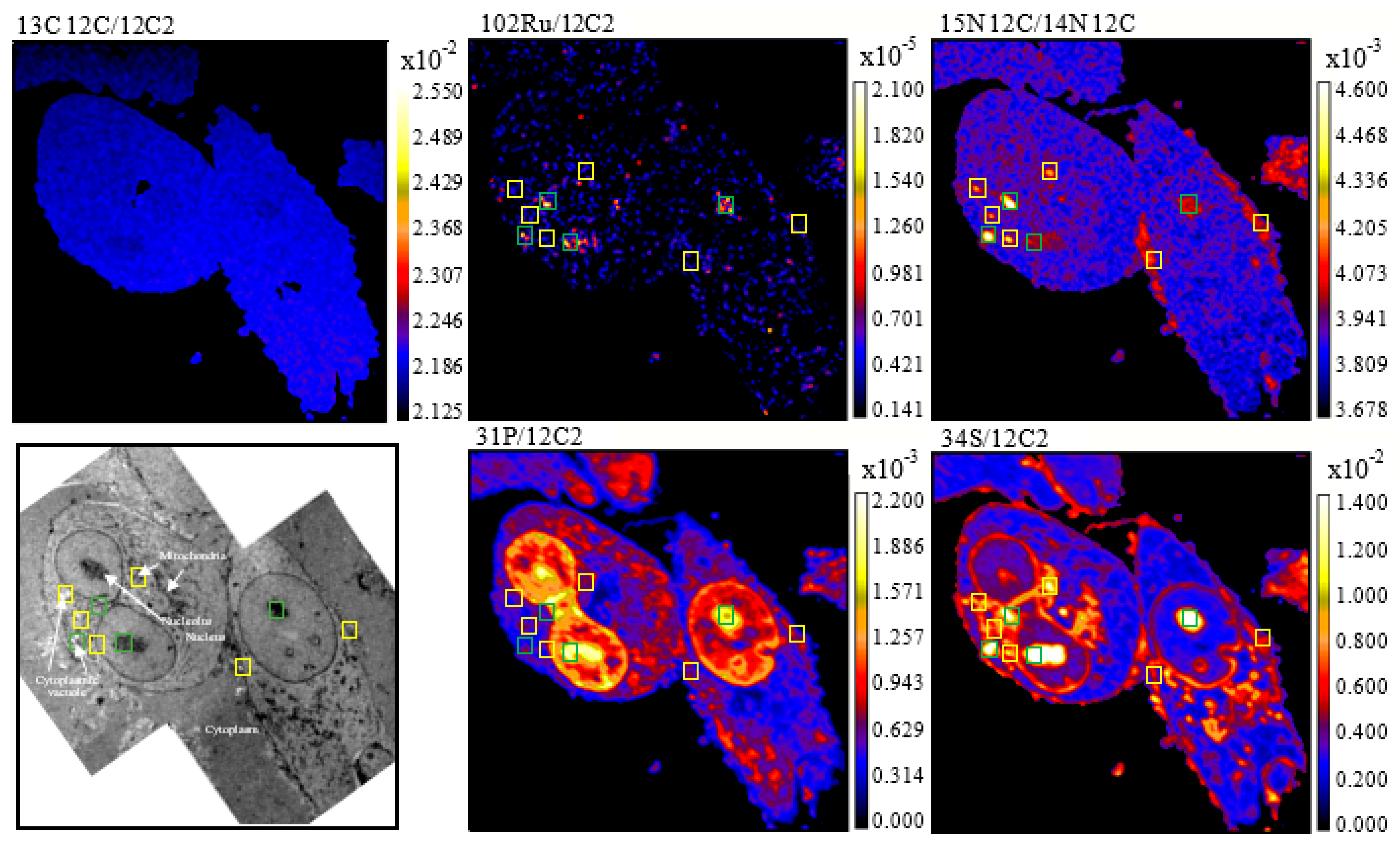
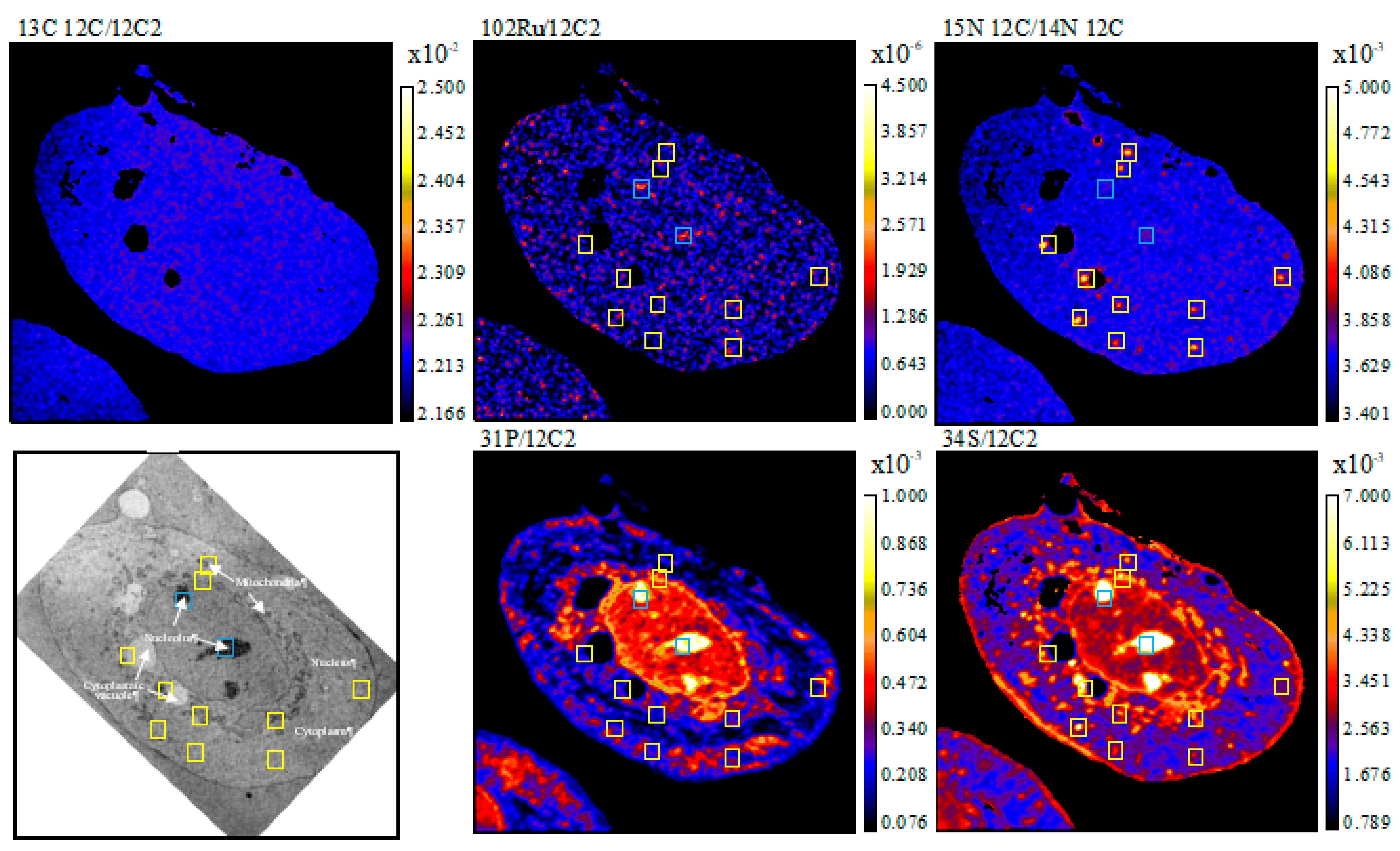
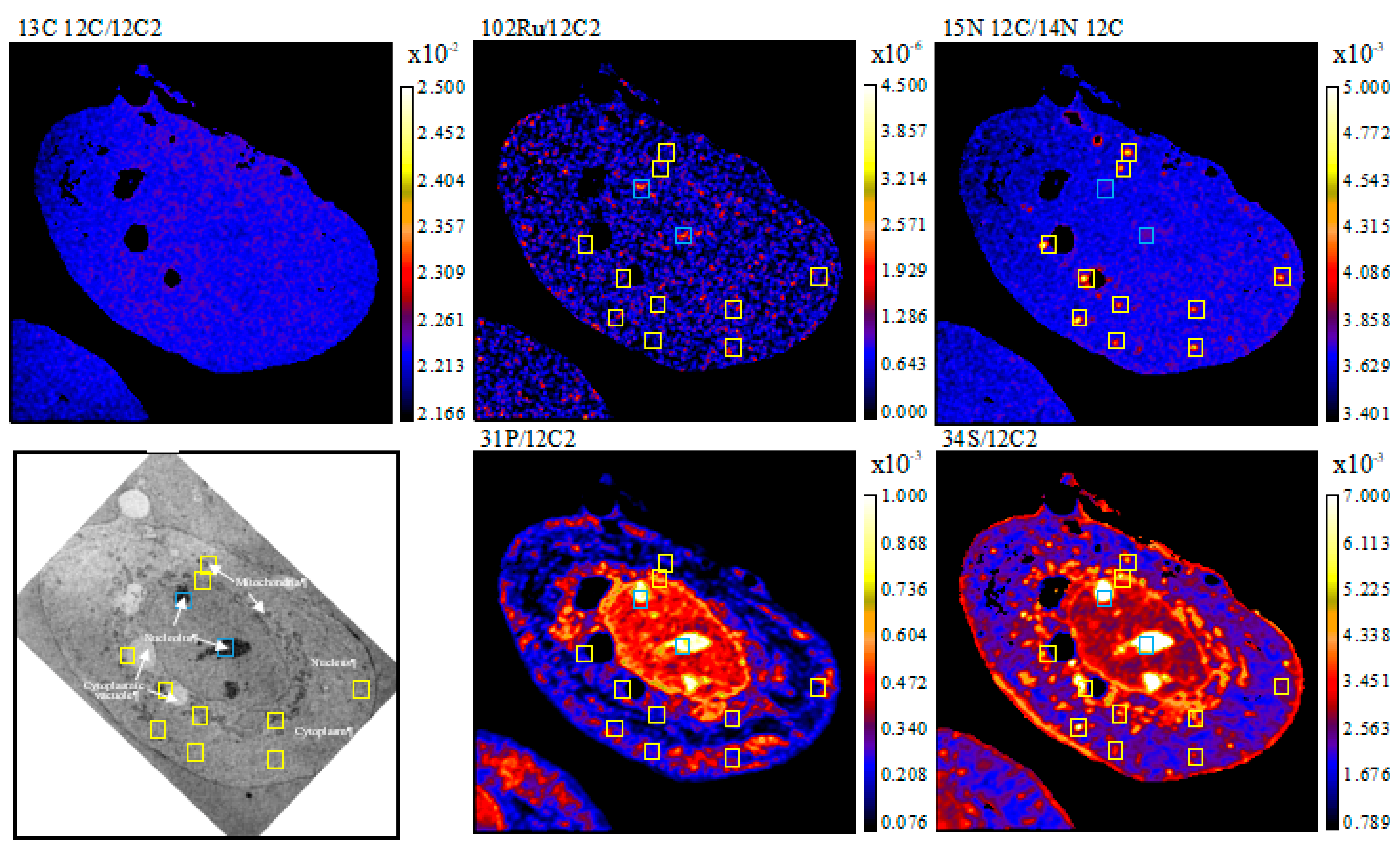
2. Results
3. Discussion
4. Materials and Methods
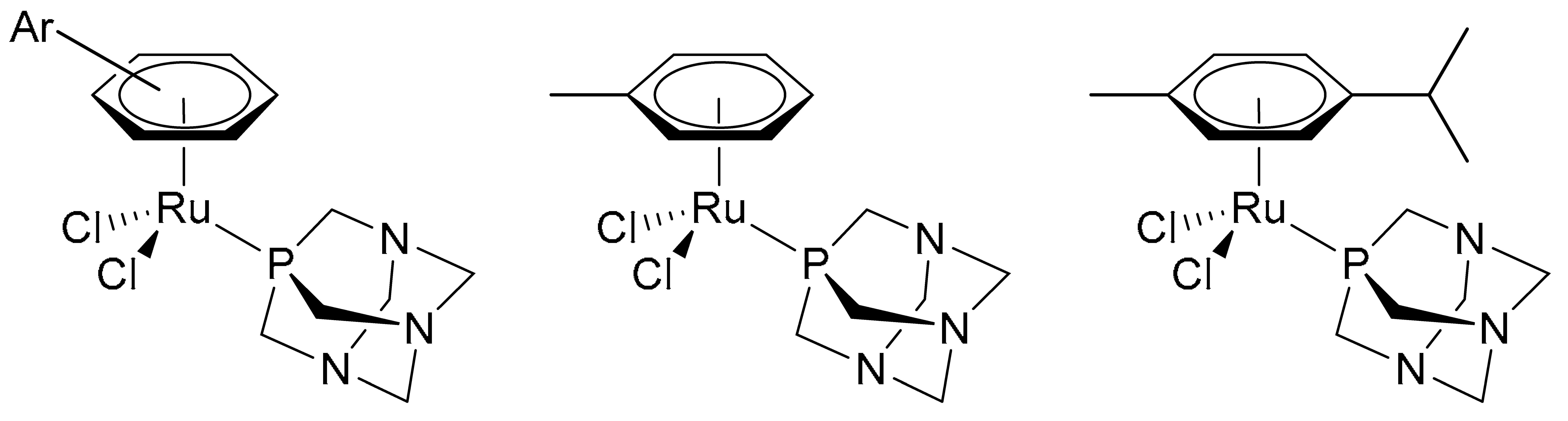
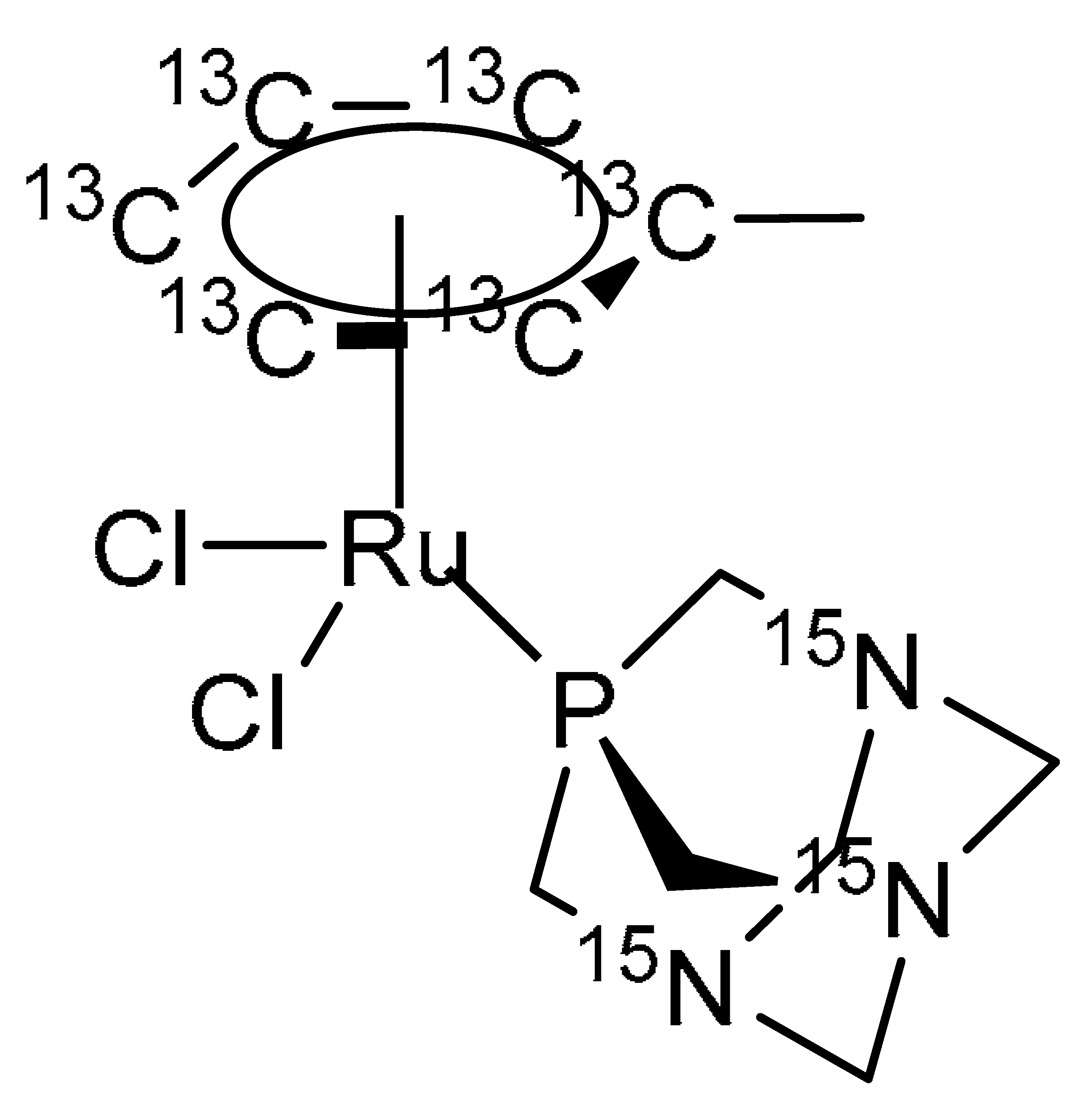
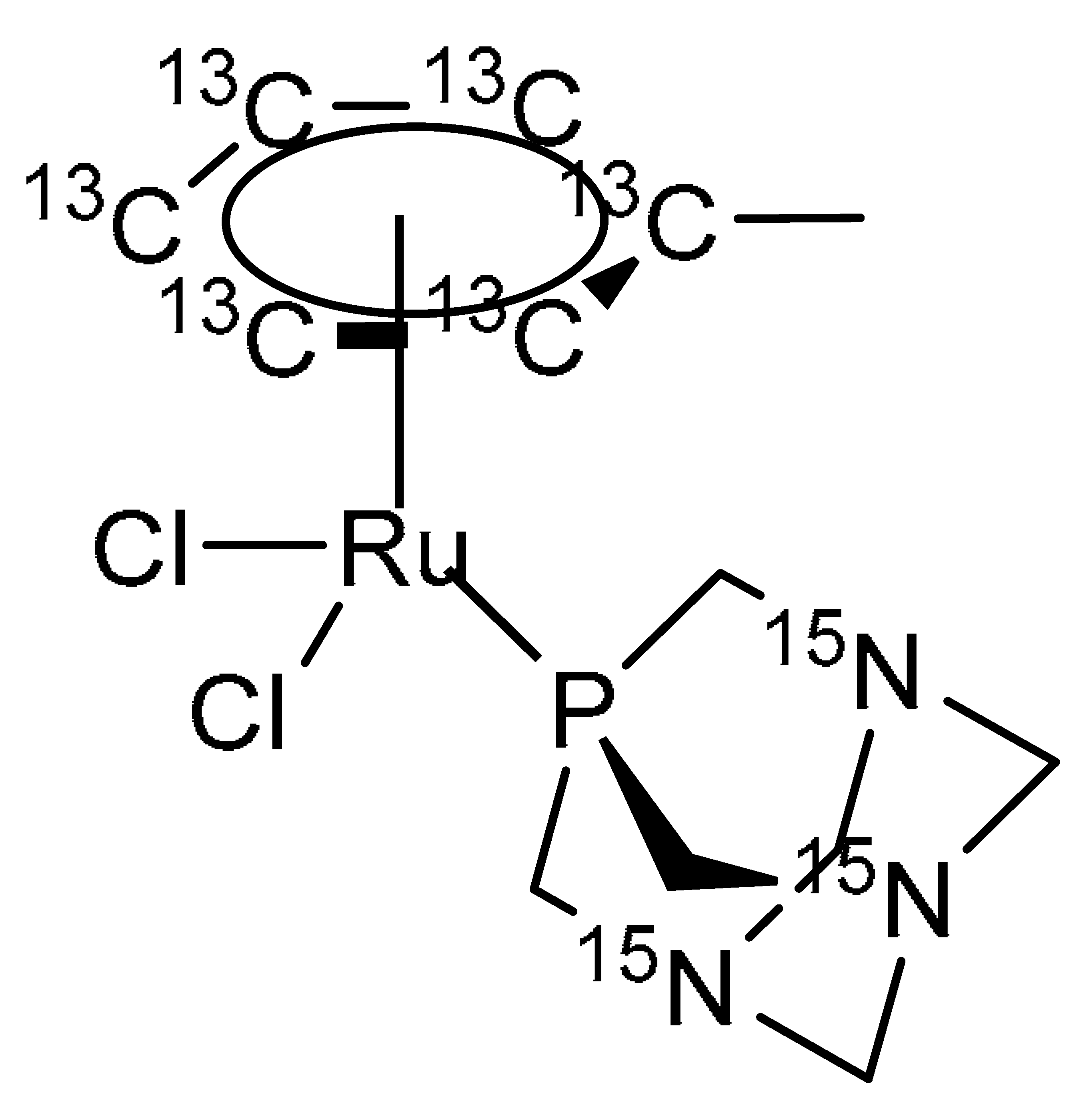
4.1. Synthesis and Characterisation of 13C/15N Labelled RAPTA-T
4.2. Cell Culture
4.3. Cell Preparation
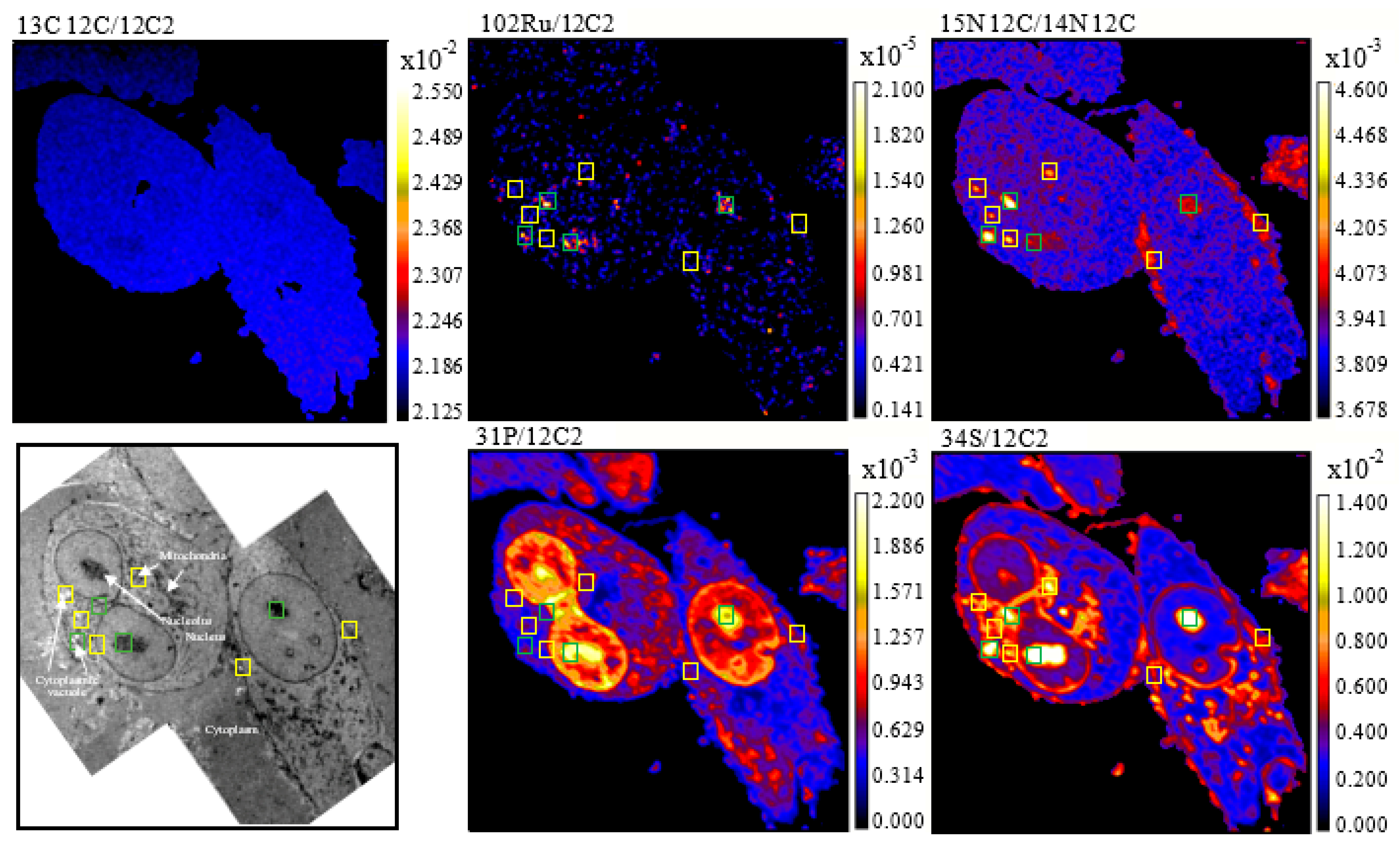
4.4. NanoSIMS Analysis
4.5. Data Extraction and Image Processing
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Galanski, M.; Jakupec, M.A.; Keppler, B.K. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: A personal perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent—preclinical development and results of a clinical Phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Hillard, E.A.; Jaouen, G. Bioorganometallics: Future trends in drug discovery, analytical chemistry, and catalysis. Organometallics 2011, 30, 20–27. [Google Scholar] [CrossRef]
- Hanif, M.; Babak, M.V.; Hartinger, C.G. Development of anticancer agents: Wizardry with osmium. Drug Discov. Today 2014, 19, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Bergamo, A.; Masi, A.; Dyson, P.J.; Sava, G. Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int. J. Oncol. 2008, 33, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium (II)-arene PTA complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]
- Adhireksan, Z.; Palermo, G.; Riedel, T.; Ma, Z.; Muhammad, R.; Rothlisberger, U.; Dyson, P.J.; Davey, C.A. Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nat. Commun. 2017, 8, 14860. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, P.; Cohen, S.; Meibom, A. NanoSIMS: Technical aspects and applications in cosmochemistry and biological geochemistry. Geostand. Geoanal. Res. 2013, 37, 111–154. [Google Scholar] [CrossRef]
- Lee, R.F.S.; Theiner, S.; Meibom, A.; Koellensperger, G.; Keppler, B.K.; Dyson, P.J. Application of imaging mass spectrometry approaches to facilitate metal-based anticancer drug research. Metallomics 2017, 9, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.F.; Escrig, S.; Croisier, M.; Clerc-Rosset, S.; Knott, G.W.; Meibom, A.; Davey, C.A.; Johnsson, K.; Dyson, P.J. NanoSIMS analysis of an isotopically labelled organometallic ruthenium(II) drug to probe its distribution and state in vitro. Chem. Commun. 2015, 51, 16486–16489. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Leclercq, G. Relevance of breast cancer cell lines as models for breast tumours: An update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Kopf, S.H.; McGlynn, S.E.; Green-Saxena, A.; Guan, Y.; Newman, D.K.; Orphan, V.J. Heavy water and 15N labelling with NanoSIMS analysis reveals growth rate-dependent metabolic heterogeneity in chemostats. Environ. Microbiol. 2015, 17, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Dröge, P.; et al. Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity. Nat. Commun. 2014, 5, 3462. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Transformation of cells by rous sarcoma virus: Cytoplasmic vacuolization. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef] [PubMed]
- Aki, T.; Nara, A.; Uemura, K. Cytoplasmic vacuolization in cell death and survival. Cell Biol. Toxicol. 2012, 28, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Groessl, M.; Zava, O.; Dyson, P.J. Cellular uptake and subcellular distribution of ruthenium-based metallodrugs under clinical investigation versus cisplatin. Met. Integr. Biometal Sci. 2011, 3, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wolters, D.A.; Stefanopoulou, M.; Dyson, P.J.; Groessl, M. Combination of metallomics and proteomics to study the effects of the metallodrug RAPTA-T on human cancer cells. Metallomics 2012, 4, 1185. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, R.H.; Weiss, A.; Abdul, U.K.; Wong, T.J.; Meraldi, P.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. Combination of ruthenium(II)-arene complex [Ru(η(6)-p-cymene)Cl2(pta)] (RAPTA-C) and the epidermal growth factor receptor inhibitor erlotinib results in efficient angiostatic and antitumor activity. Sci. Rep. 2017, 7, 43005. [Google Scholar] [CrossRef] [PubMed]
- Pelillo, C.; Mollica, H.; Eble, J.A.; Grosche, J.; Herzog, L.; Codan, B.; Sava, G.; Bergamo, A. Inhibition of adhesion, migration and of α5 β1 integrin in the HCT-116 colorectal cancer cells treated with the ruthenium drug NAMI-A. J. Inorg. Biochem. 2016, 160, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Sava, G. Linking the future of anticancer metal-complexes to the therapy of tumour metastases. Chem. Soc. Rev. 2015, 44, 8818–8835. [Google Scholar] [CrossRef] [PubMed]
- Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Daigle, D.J.; Pepperman, A.B.; Vail, S.L. Synthesis of a monophosphorus analog of hexamethylenetetramine. J. Heterocycl. Chem. 1974, 11, 407–408. [Google Scholar] [CrossRef]
- McDonald, K.L.; Webb, R.I. Freeze substitution in 3 h or less. J. Microsc. 2011, 243, 227–233. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, R.F.S.; Escrig, S.; Maclachlan, C.; Knott, G.W.; Meibom, A.; Sava, G.; Dyson, P.J. The Differential Distribution of RAPTA-T in Non-Invasive and Invasive Breast Cancer Cells Correlates with Its Anti-Invasive and Anti-Metastatic Effects. Int. J. Mol. Sci. 2017, 18, 1869. https://doi.org/10.3390/ijms18091869
Lee RFS, Escrig S, Maclachlan C, Knott GW, Meibom A, Sava G, Dyson PJ. The Differential Distribution of RAPTA-T in Non-Invasive and Invasive Breast Cancer Cells Correlates with Its Anti-Invasive and Anti-Metastatic Effects. International Journal of Molecular Sciences. 2017; 18(9):1869. https://doi.org/10.3390/ijms18091869
Chicago/Turabian StyleLee, Ronald F. S., Stéphane Escrig, Catherine Maclachlan, Graham W. Knott, Anders Meibom, Gianni Sava, and Paul J. Dyson. 2017. "The Differential Distribution of RAPTA-T in Non-Invasive and Invasive Breast Cancer Cells Correlates with Its Anti-Invasive and Anti-Metastatic Effects" International Journal of Molecular Sciences 18, no. 9: 1869. https://doi.org/10.3390/ijms18091869
APA StyleLee, R. F. S., Escrig, S., Maclachlan, C., Knott, G. W., Meibom, A., Sava, G., & Dyson, P. J. (2017). The Differential Distribution of RAPTA-T in Non-Invasive and Invasive Breast Cancer Cells Correlates with Its Anti-Invasive and Anti-Metastatic Effects. International Journal of Molecular Sciences, 18(9), 1869. https://doi.org/10.3390/ijms18091869