PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation

Abstract
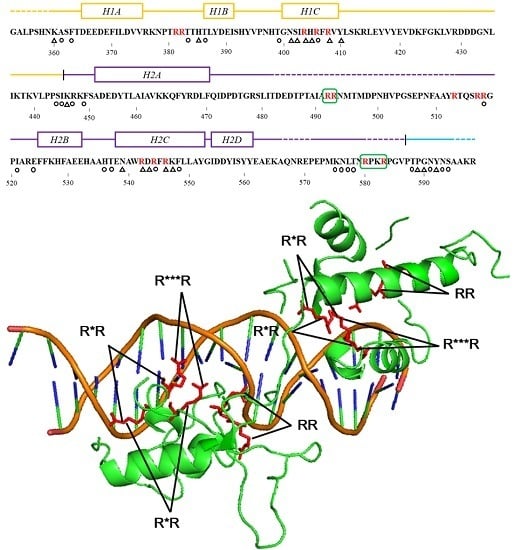
:
1. Introduction
2. Result and Discussion
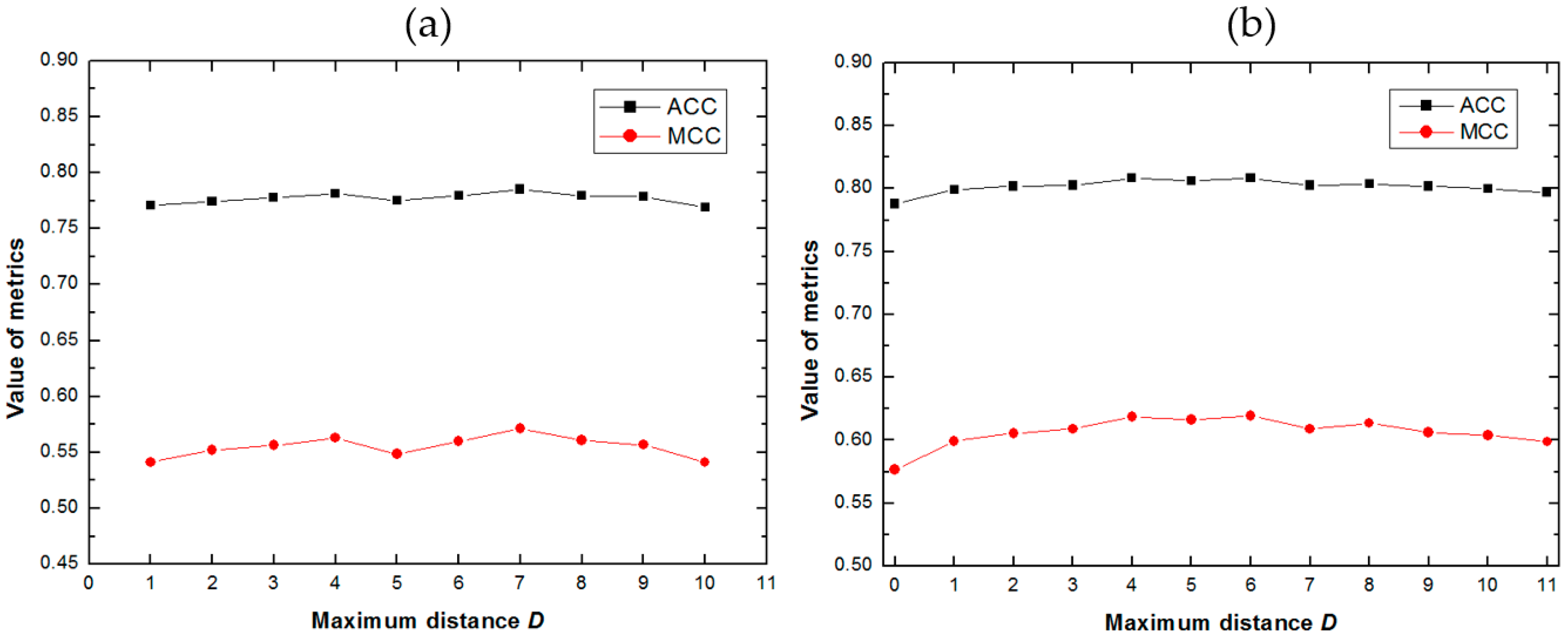
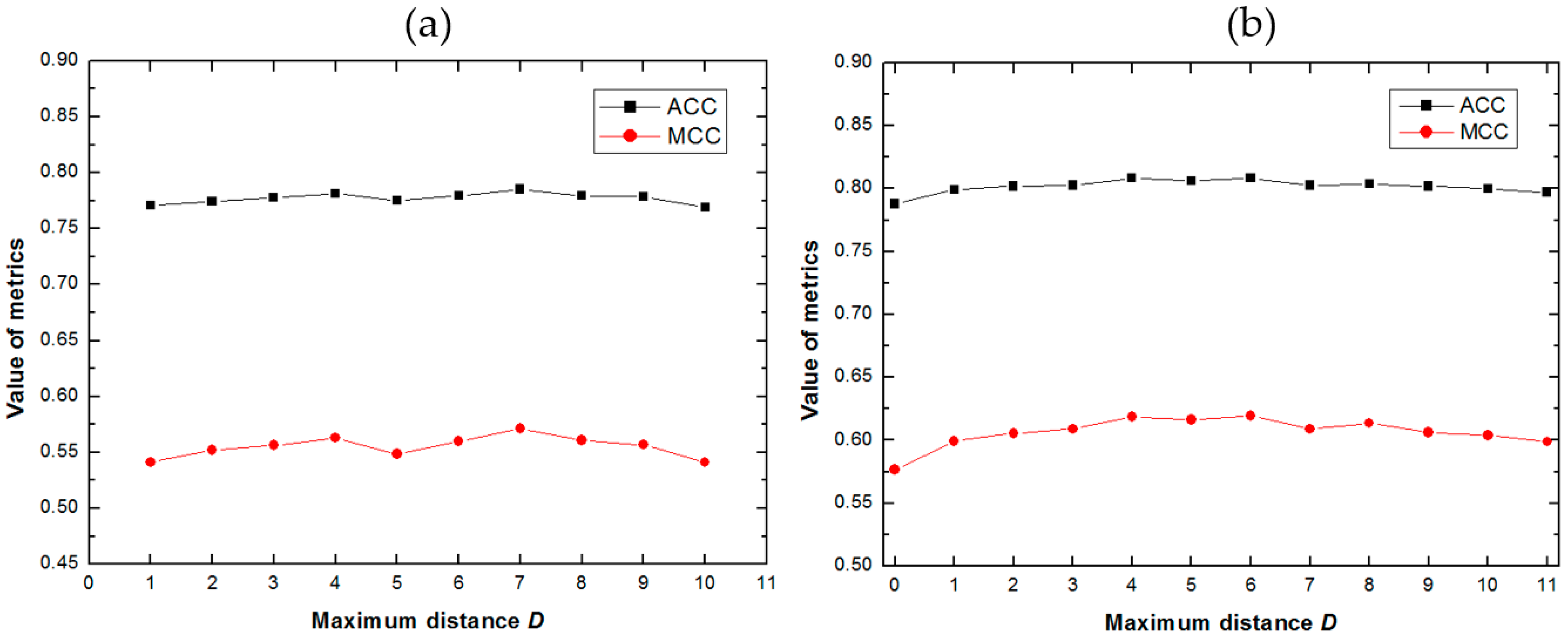
2.1. Impact of the Maximum Distance D
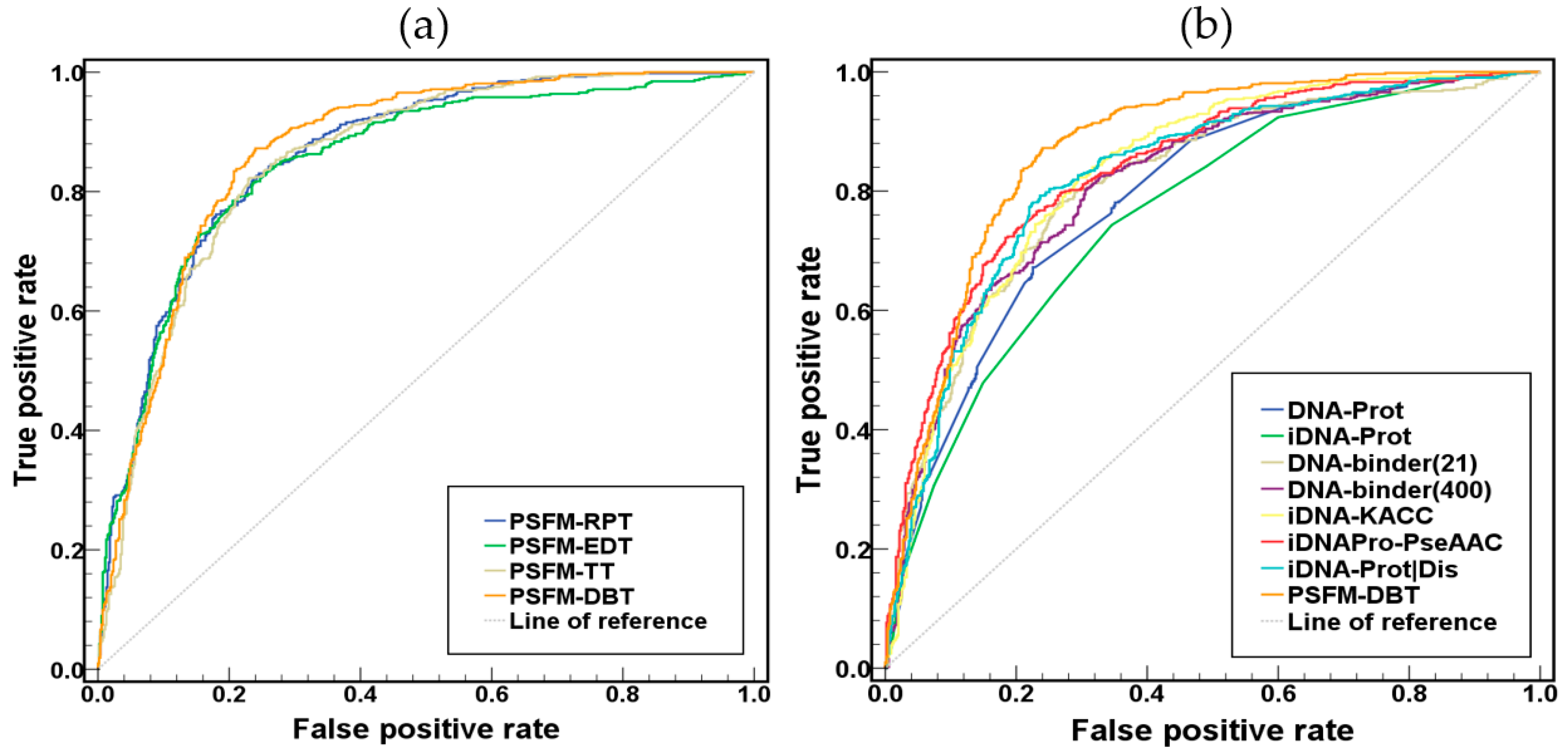
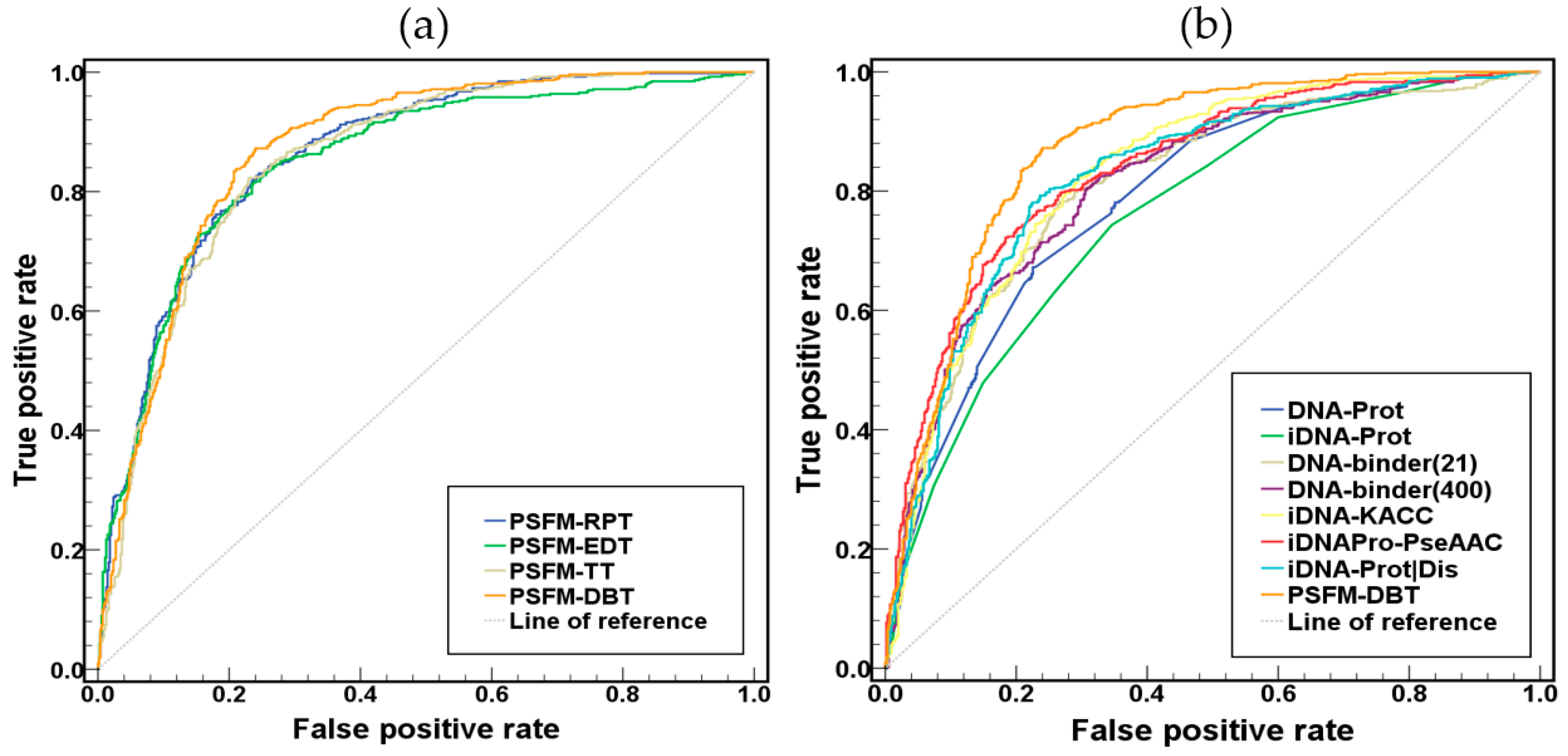
2.2. Comparison of the Four PSFM-Based Methods
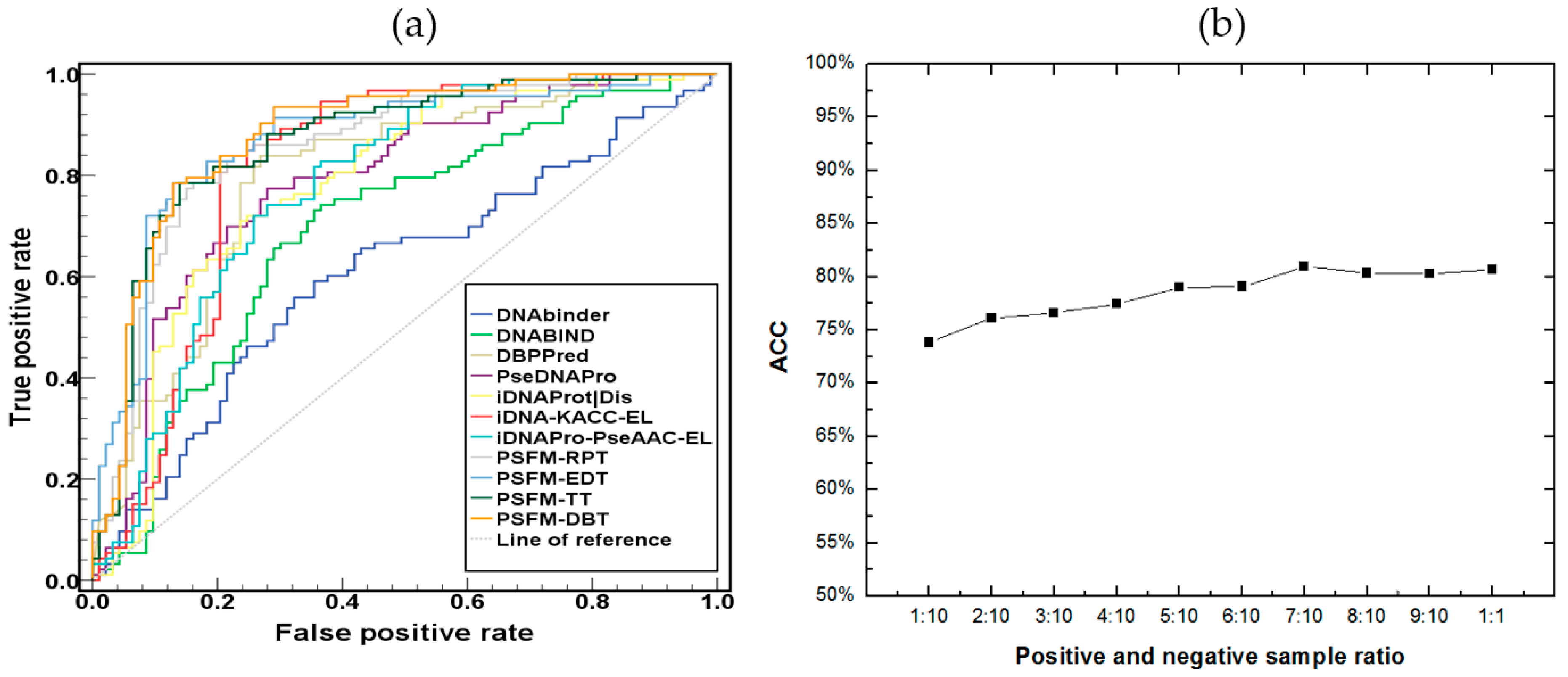
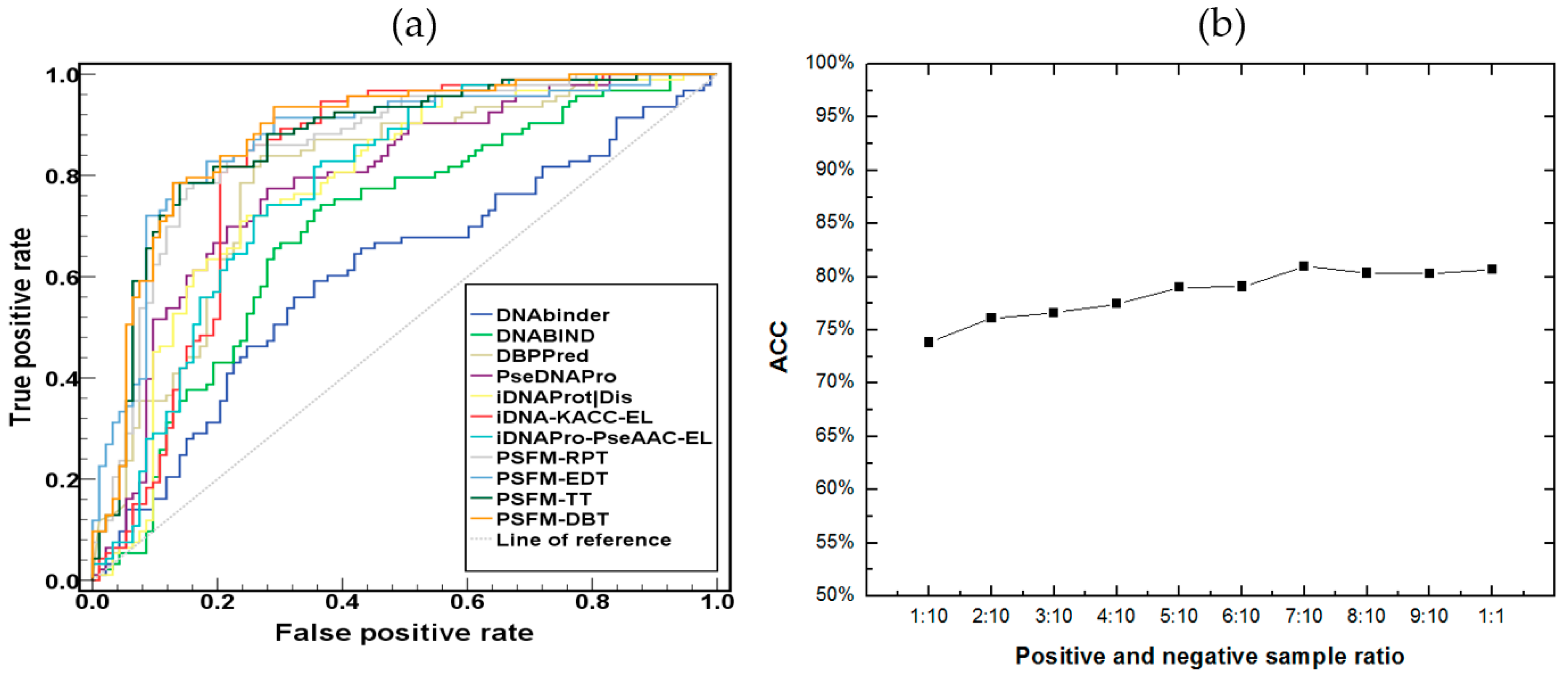
2.3. Comparison with Existing Methods
2.4. Independent Test
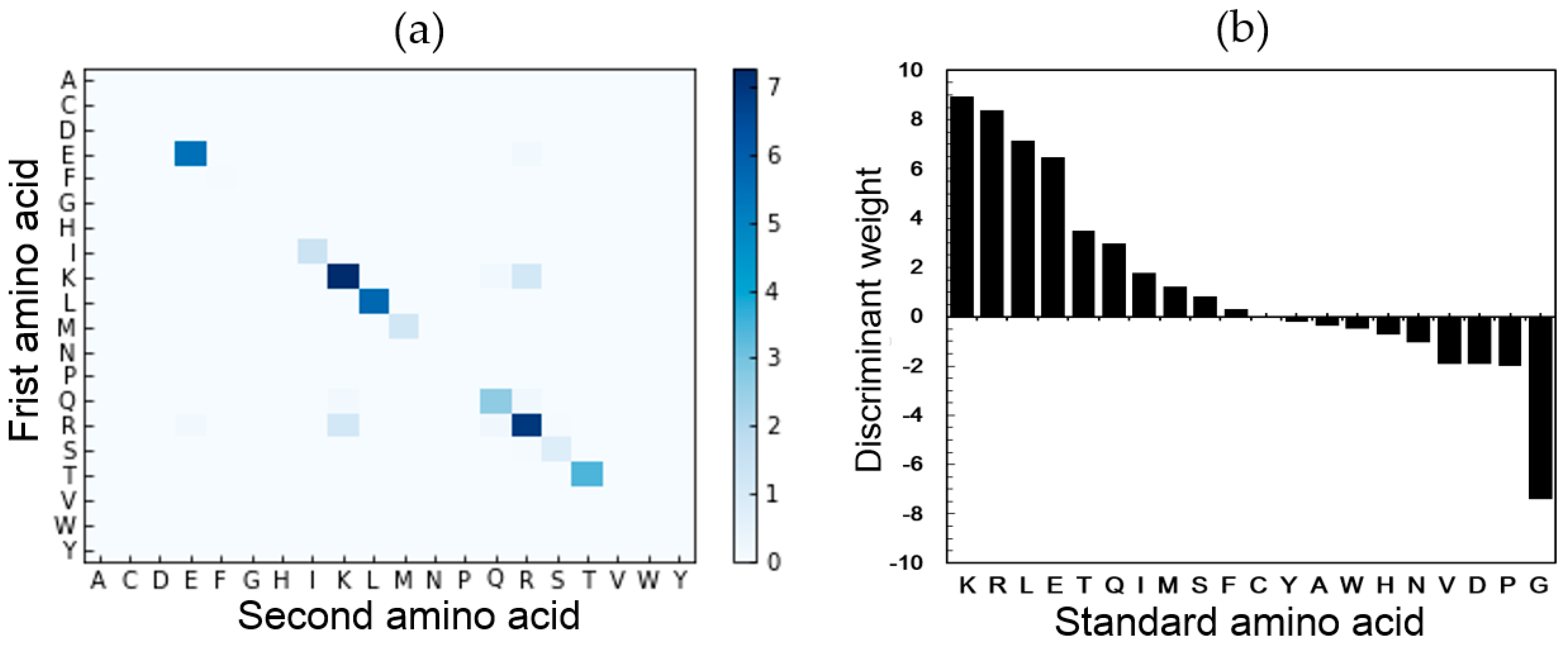
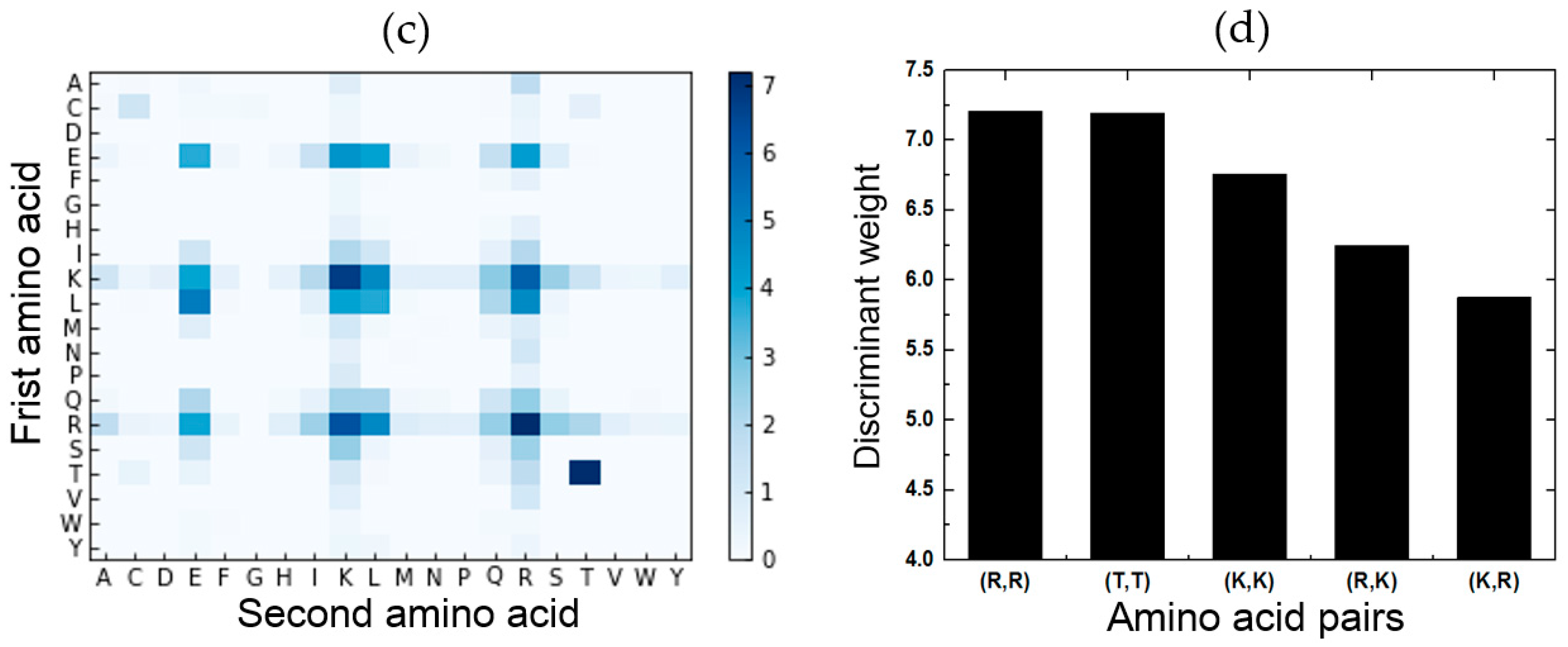
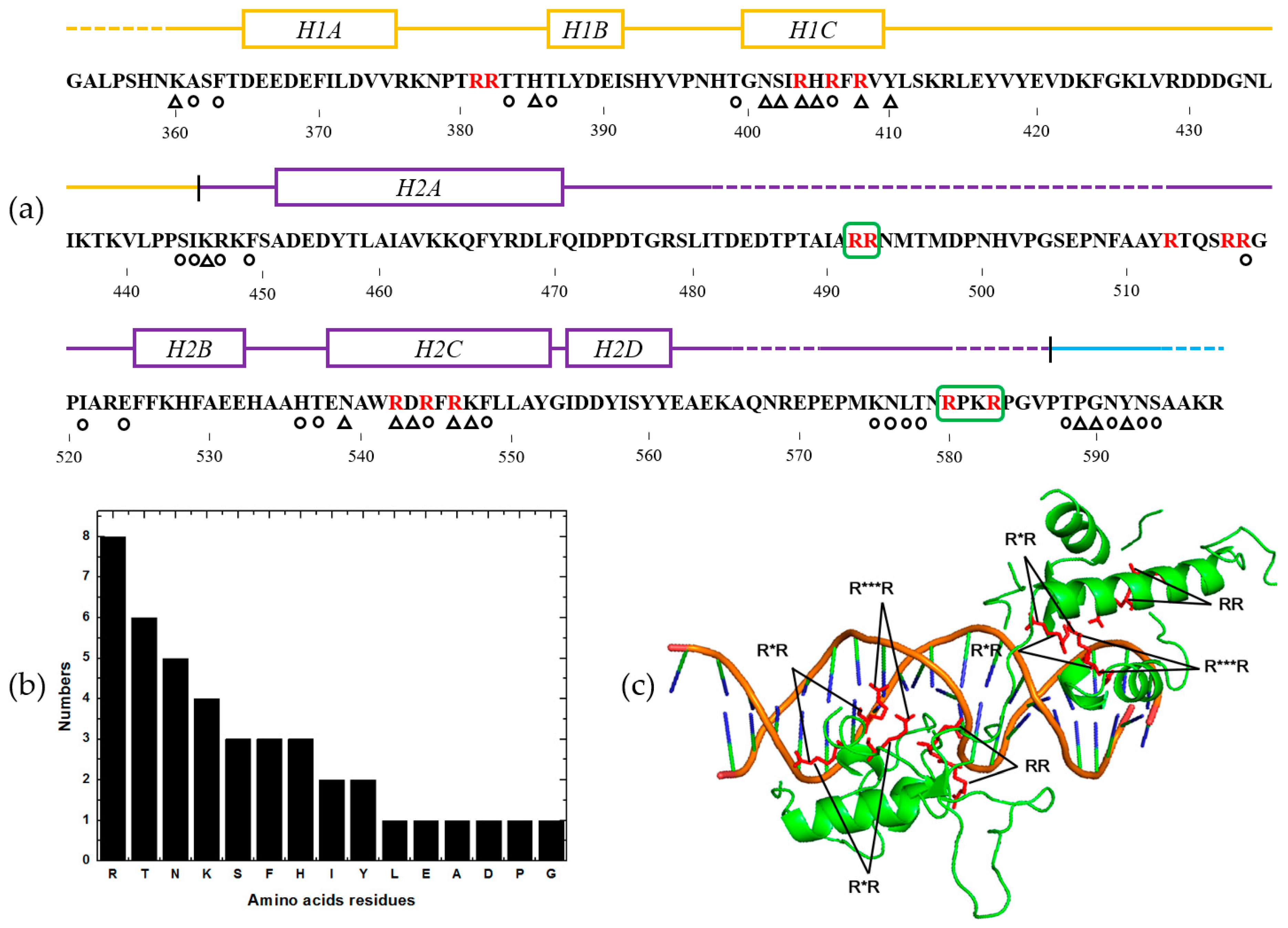
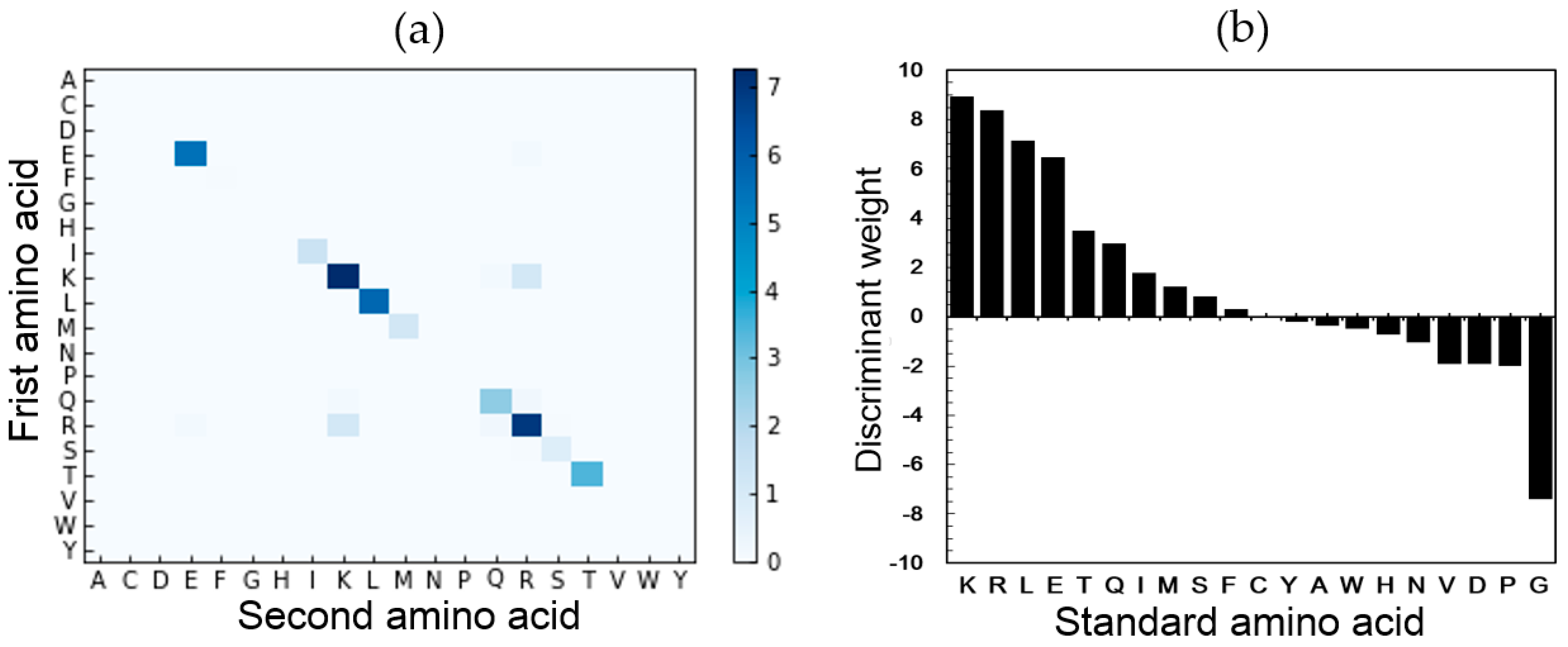
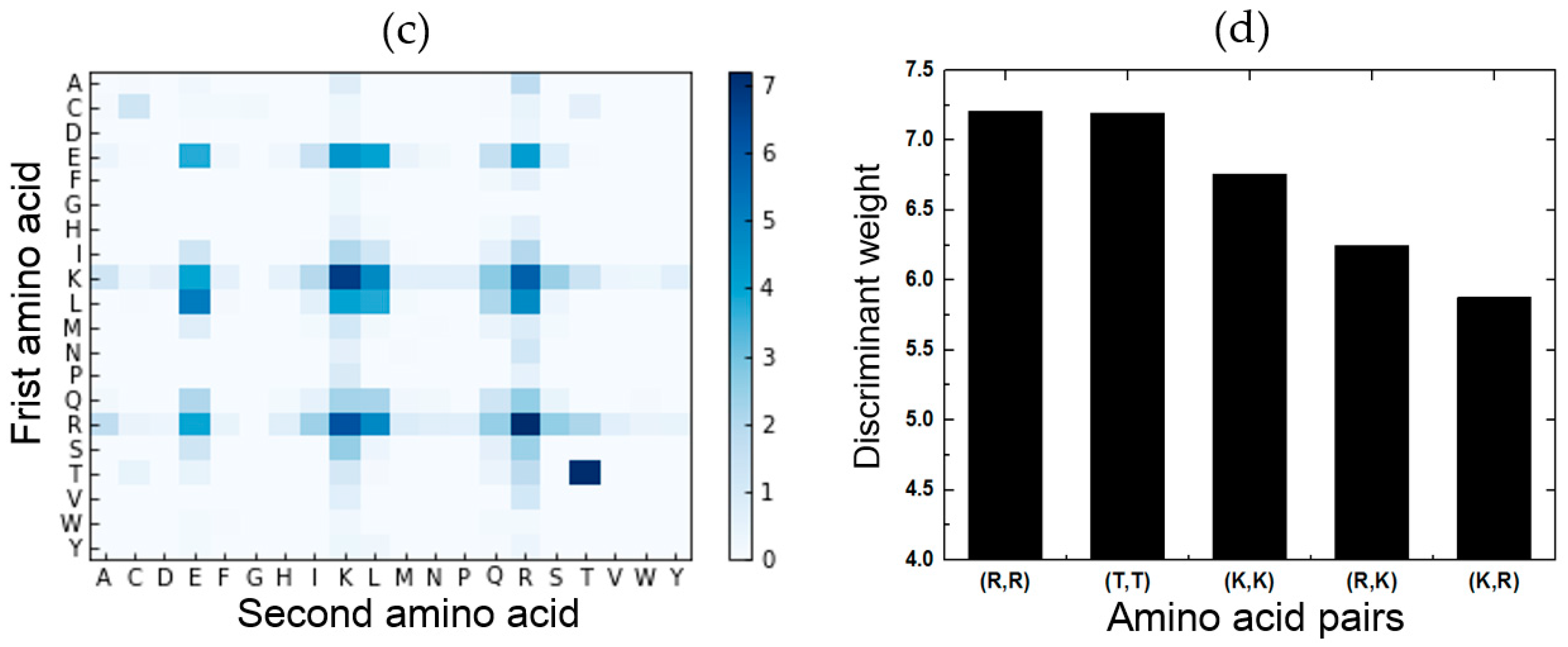
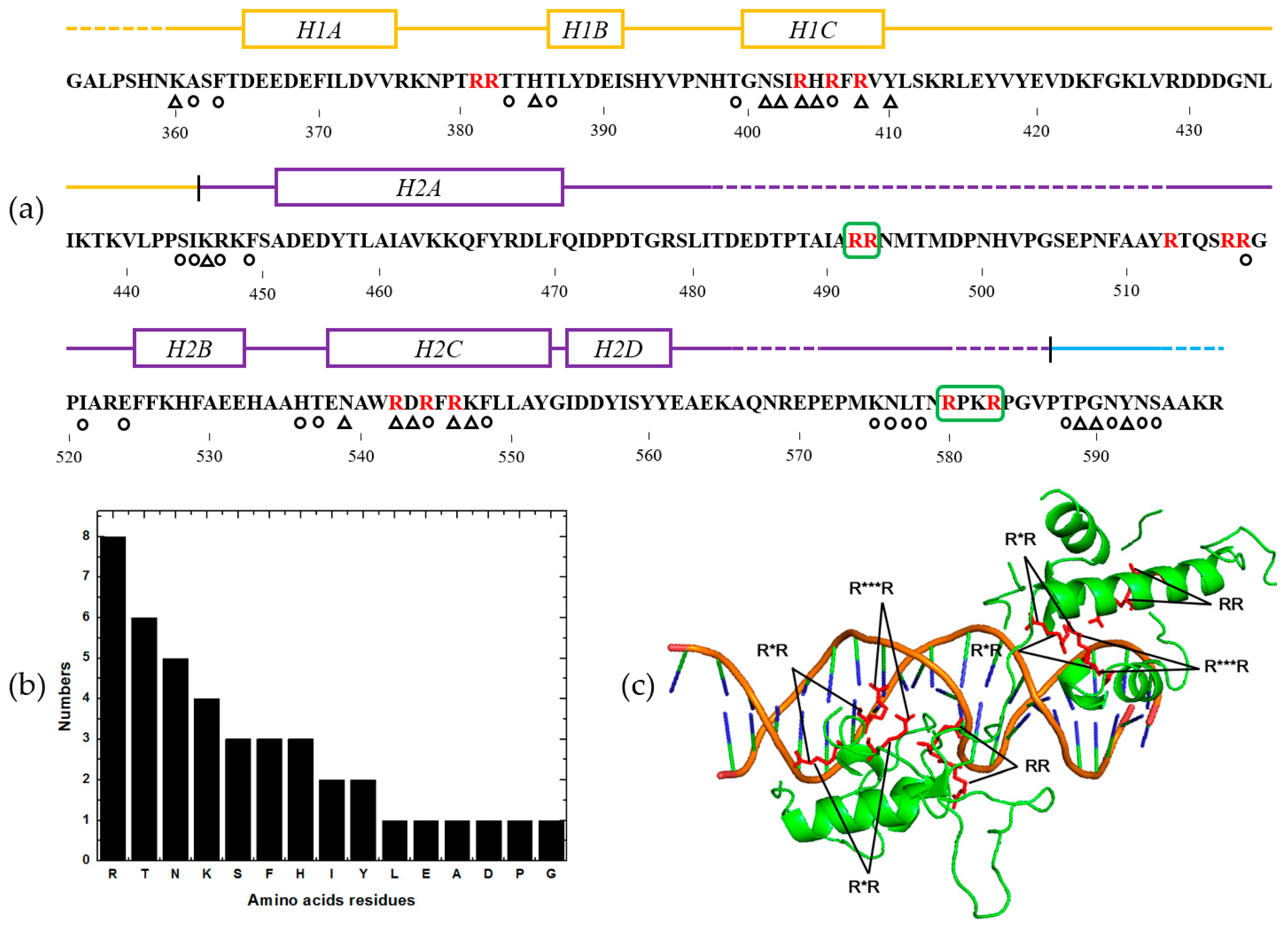
2.5. Feature Analysis
2.6. Web-Server Guide
3. Methods and Materials
3.1. Dataset
3.2. Protein Representation
3.2.1. Position Specific Frequency Matrix
3.2.2. Residue Probing Transformation
3.2.3. Evolutionary Difference Transformation
3.2.4. Distance-Bigram Transformation
3.2.5. Trigram Transformation
3.3. Support Vector Machine
3.4. Evaluation of Performance
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lou, W.; Wang, X.; Chen, F.; Chen, Y.; Jiang, B.; Zhang, H. Sequence Based Prediction of DNA-Binding Proteins Based on Hybrid Feature Selection Using Random Forest and Gaussian Naive Bayes. PLoS ONE 2014, 9, e86703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, Y.; Zhou, Y. Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function. Bioinforma 2010, 26, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, X.; Kong, L. Predict protein structural class for low-similarity sequences by evolutionary difference information into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2014, 355, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cao, J.; Cai, Y.; Shi, T.; Li, Y. Predicting rRNA-, RNA-, and DNA-binding proteins from primary structure with support vector machines. J. Theor. Biol. 2006, 240, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhao, X.; Huang, D. Predicting protein-protein interactions from protein sequences using meta predictor. Amino Acids 2010, 39, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Tjong, H.; Zhou, H. DISPLAR: An accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007, 35, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Stawiski, E.W.; Gregoret, L.M.; Mandelgutfreund, Y. Annotating Nucleic Acid-Binding Function Based on Protein Structure. J. Mol. Biol. 2003, 326, 1065–1079. [Google Scholar] [CrossRef]
- Shanahan, H.P.; Garcia, M.A.; Jones, S.; Thornton, J.M. Identifying DNA-binding proteins using structural motifs and the electrostatic potential. Nucleic Acids Res. 2004, 32, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Nimrod, G.; Schushan, M.; Szilagyi, A.; Leslie, C.; Bental, N. iDBPs: A web server for the identification of DNA binding proteins. Bioinformatics 2010, 26, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, D.; Zeng, X.; Wu, Y.; Guo, L.; Zou, Q. nDNA-prot: Identification of DNA-binding Proteins Based on Unbalanced Classification. BMC Bioinform. 2014, 15, 298. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Langlois, R.; Zhao, G.; Lu, H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005, 33, 6486–6493. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, G.; Chou, K.-C. Support Vector Machines for Predicting Membrane Protein Types by Using Functional Domain Composition. Biophys. J. 2003, 84, 3257–3263. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of protein cellular attributes using pseudo amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.K.; Pugalenthi, G.; Suganthan, P.N. DNA-Prot: Identification of DNA binding proteins from protein sequence information using random forest. J. Biomol. Struct. Dyn. 2009, 26, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Guo, Y.; Feng, Y.; Li, M. Predicting DNA-binding proteins: Approached from Chou’s pseudo amino acid composition and other specific sequence features. Amino Acids 2007, 34, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinform. 2007, 8, 463. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou’s PseAAC and Physicochemical Distance Transformation. Mol. Inform. 2015, 34, 8–17. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2016, 384, 135–144. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, B.; Chai, H.; Ma, Z.; Yang, G. Identification of DNA-binding proteins using multi-features fusion and binary firefly optimization algorithm. BMC Bioinform. 2016, 17, 323. [Google Scholar] [CrossRef] [PubMed]
- Waris, M.; Ahmad, K.; Kabir, M.; Hayat, M. Identification of DNA binding proteins using evolutionary profiles position specific scoring matrix. Neurocomputing 2016, 199, 154–162. [Google Scholar] [CrossRef]
- Liu, S.; Wang, S.; Ding, H. Protein sub-nuclear location by fusing AAC and PSSM features based on sequence information. In Proceedings of the International Conference on Electronics Information and Emergency Communication, Beijing, China, 14 May 2015. [Google Scholar]
- Jeong, J.C.; Lin, X.; Chen, X.-W. On Position-Specific Scoring Matrix for Protein Function Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Lan, X.; Xu, R.; Zhou, J.; Wang, X.; Chou, K.-C. iDNA-Prot|dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. PLoS ONE 2014, 9, e106691. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.; Raicar, G.; Lal, S.P.; Dehzangi, A.; Imoto, S.; Sharma, A. Protein Fold Recognition Using Genetic Algorithm Optimized Voting Scheme and Profile Bigram. J. Softw. 2016, 11, 756–767. [Google Scholar] [CrossRef]
- Paliwal, K.K.; Sharma, A.; Lyons, J.; Dehzangi, A. A tri-gram based feature extraction technique using linear probabilities of position specific scoring matrix for protein fold recognition. IEEE Trans. Nanobiosci. 2014, 13, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Fang, J.; Xiao, X.; Chou, K.-C. iDNA-Prot: Identification of DNA binding proteins using random forest with grey model. PLoS ONE 2011, 6, e24756. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Dong, Q.; Li, S.; Liu, X. Identification of DNA-binding proteins by combining auto-cross covariance transformation and ensemble learning. IEEE Trans. NanoBiosci. 2016, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Wang, X. DNA binding protein identifcation by combining pseudo amino acid composition and profle-based protein representation. Sci. Rep. 2015, 5, 15497. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.-C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, X.; Lin, L.; Dong, Q.; Wang, X. A Discriminative Method for Protein Remote Homology Detection and Fold Recognition Combining Top-n-grams and Latent Semantic Analysis. BMC Bioinform. 2008, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Mandelgutfreund, Y.; Schueler, O.; Margalit, H. Comprehensive Analysis of Hydrogen Bonds in Regulatory Protein DNA-Complexes: In Search of Common Principles. J. Mol. Biol. 1995, 253, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Van Heyningen, P.; Berman, H.M.; Thornton, J.M. Protein-DNA interactions: A structural analysis. J. Mol. Biol. 1999, 287, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nureki, O.; Kurumizaka, H.; Fukai, S.; Kawaguchi, S.; Ikuta, M.; Iwahara, J.; Okazaki, T.; Yokoyama, S. Crystal structure of the CENP-B protein–DNA complex: The DNA-binding domains of CENP-B induce kinks in the CENP-B box DNA. EMBO J. 2001, 20, 6612–6618. [Google Scholar] [CrossRef] [PubMed]
- Szabóová, A.; Kuželka, O.; Železný, F.; Tolar, J. Prediction of DNA-binding propensity of proteins by the ball-histogram method using automatic template search. BMC Bioinform. 2012, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Konig, P.; Giraldo, R.; Chapman, L.; Rhodes, D. The crystal structure of the DNA-binding domain of yeast RAP1 in complex with telomeric DNA. Cell 1996, 85, 125. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L. PISCES: Recent improvements to a PDB sequence culling server. Nucleic Acids Res. 2005, 33, W94–W98. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.-C. repRNA: A web server for generating various feature vectors of RNA sequences. Mol. Genet. Genom. 2016, 291, 473–481. [Google Scholar] [CrossRef]
- Zhu, L.; Deng, S.-P.; Huang, D.-S. A Two-Stage Geometric Method for Pruning Unreliable Links in Protein-Protein Networks. IEEE Trans. Nanobiosci. 2015, 14, 528–534. [Google Scholar]
- Deng, S.-P.; Huang, D.-S. SFAPS: An R package for structure/function analysis of protein sequences based on informational spectrum method. Methods 2014, 69, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-Q.; Huang, D.-S.; Sun, B.-Y. Human face recognition based on multi-features using neural networks committee. Pattern Recognit. Lett. 2004, 25, 1351–1358. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Chen, Q.; Dong, Q.; Lan, X. Using Amino Acid Physicochemical Distance Transformation for Fast Protein Remote Homology Detection. PLoS ONE 2012, 7, e46633. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, P.; Huang, D.-S.; Li, J.-J.; Lok, T.-M.; Lyu, M.R. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. FEBS Lett. 2006, 580, 380–384. [Google Scholar] [CrossRef]
- Holm, L.; Sander, C. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 1998, 14, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Li, D.; Ju, Y.; Zou, Q. Protein Folds Prediction with Hierarchical Structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Guo, J.; Ju, Y.; Wu, M.; Zeng, X.; Hong, Z. Improving tRNAscan-SE annotation results via ensemble classifiers. Mol. Inform. 2015, 34, 761–770. [Google Scholar] [CrossRef]
- Zhu, L.; You, Z.-H.; Huang, D.-S. Increasing the reliability of protein–protein interaction networks via non-convex semantic embedding. Neurocomputing 2013, 121, 99–107. [Google Scholar] [CrossRef]
- Chang, C.; Lin, C. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Sonego, P.; Kocsor, A.; Pongor, S. ROC analysis: Applications to the classification of biological sequences and 3D structures. Brief. Bioinform. 2008, 9, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.-S. Radial basis probabilistic neural networks: Model and application. Int. J. Pattern Recognit. Artif. Int. 1999, 13, 1083–1101. [Google Scholar] [CrossRef]
- Huang, D.S.; Du, J.-X. A constructive hybrid structure optimization methodology for radial basis probabilistic neural networks. IEEE Trans. Neural Netw. 2008, 19, 2099–2115. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | ACC (%) | MCC | AUC (%) | SN (%) | SP (%) |
|---|---|---|---|---|---|
| PSFM-RPT a | 78.88 | 0.5785 | 86.35 | 80.76 | 77.09 |
| PSFM-EDT b | 79.35 | 0.5868 | 84.49 | 78.86 | 79.82 |
| PSFM-DBT c | 81.02 | 0.6224 | 87.12 | 84.19 | 78.00 |
| PSFM-TT d | 79.16 | 0.5840 | 85.54 | 80.95 | 77.45 |
| Method | ACC (%) | MCC | AUC (%) | SN (%) | SP (%) |
|---|---|---|---|---|---|
| DNA-Prot | 72.55 | 0.44 | 78.90 | 82.67 | 59.75 |
| iDNA-Prot | 75.40 | 0.50 | 76.10 | 83.81 | 64.73 |
| DNAbinder (dimension 400) | 73.58 | 0.47 | 81.50 | 66.47 | 80.36 |
| DNAbinder (dimension 21) | 73.95 | 0.48 | 81.40 | 68.57 | 79.09 |
| PseDNA-Pro | 76.55 | 0.53 | N/A | 79.61 | 73.63 |
| iDNA-Prot|dis | 77.30 | 0.54 | 82.60 | 79.40 | 75.27 |
| iDNAPro-PseAAC | 76.56 | 0.53 | 83.92 | 75.62 | 77.45 |
| iDNA-KACC | 75.16 | 0.50 | 83.00 | 77.52 | 72.90 |
| PSSM-DT | 79.96 | 0.62 | 86.50 | 78.00 | 81.91 |
| Local-DPP | 79.10 | 0.59 | N/A | 84.80 | 73.60 |
| PSFM-DBT a | 81.02 | 0.62 | 87.12 | 84.19 | 78.00 |
| Method | ACC (%) | MCC | AUC (%) | SN (%) | SP (%) |
|---|---|---|---|---|---|
| DNA-Prot | 61.80 | 0.240 | N/A | 69.90 | 53.80 |
| iDNA-Prot | 67.20 | 0.344 | N/A | 67.70 | 66.70 |
| DNAbinder | 60.80 | 0.216 | 60.70 | 57.00 | 64.50 |
| DNABIND | 67.70 | 0.355 | 69.40 | 66.70 | 68.80 |
| DBPPred | 76.90 | 0.538 | 79.10 | 79.60 | 74.20 |
| iDNA-Prot|dis | 72.00 | 0.445 | 78.60 | 79.50 | 64.50 |
| iDNAPro-PseAAC-EL | 71.50 | 0.442 | 77.80 | 82.80 | 60.2 |
| iDNA-KACC-EL | 79.03 | 0.611 | 81.40 | 94.62 | 63.44 |
| PSSM-DT | 80.00 | 0.647 | 87.40 | 87.09 | 72.83 |
| Local-DPP | 79.00 | 0.625 | N/A | 92.50 | 65.60 |
| PSFM-TT | 78.49 | 0.580 | 86.63 | 88.17 | 68.82 |
| PSFM-RPT | 79.57 | 0.594 | 85.67 | 84.95 | 74.19 |
| PSFM-EDT | 79.57 | 0.600 | 86.88 | 88.17 | 70.97 |
| PSFM-DBT | 80.65 | 0.624 | 88.03 | 90.32 | 70.97 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Liu, B. PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation. Int. J. Mol. Sci. 2017, 18, 1856. https://doi.org/10.3390/ijms18091856
Zhang J, Liu B. PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation. International Journal of Molecular Sciences. 2017; 18(9):1856. https://doi.org/10.3390/ijms18091856
Chicago/Turabian StyleZhang, Jun, and Bin Liu. 2017. "PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation" International Journal of Molecular Sciences 18, no. 9: 1856. https://doi.org/10.3390/ijms18091856
APA StyleZhang, J., & Liu, B. (2017). PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation. International Journal of Molecular Sciences, 18(9), 1856. https://doi.org/10.3390/ijms18091856