Impact of Platelet-Rich Plasma on Viability and Proliferation in Wound Healing Processes after External Radiation


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of Irradiation on Specific Surface Markers Using Flow Cytometry of Mono- and Co-Cultured Cells
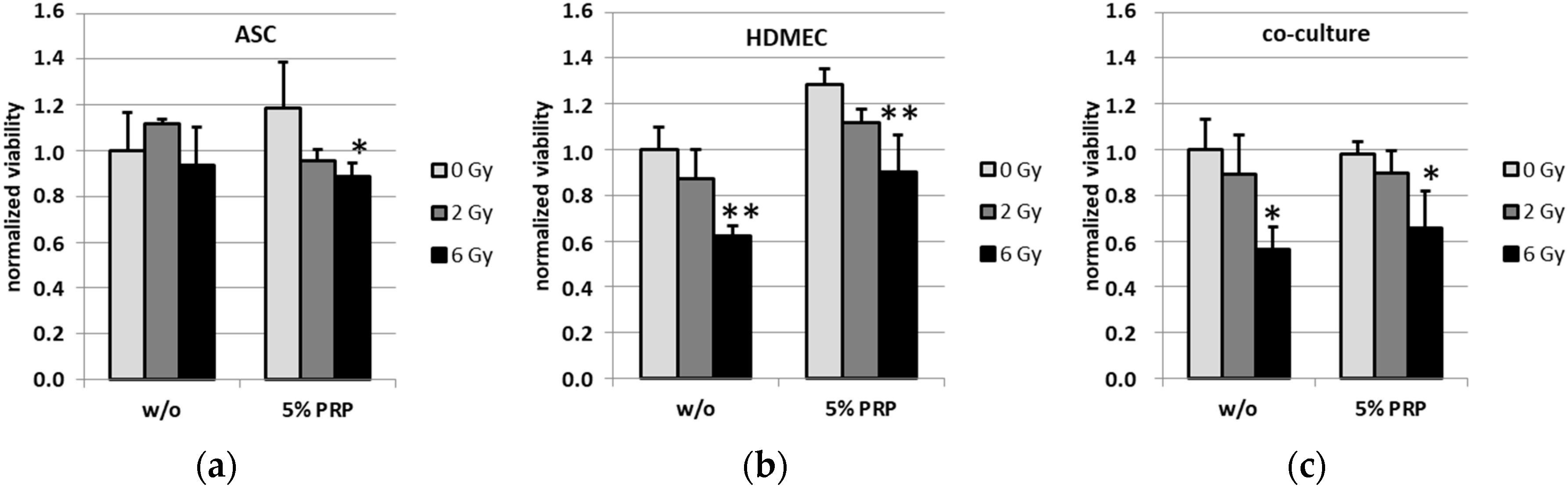
2.2. Effect of Irradiation on Cell Viability
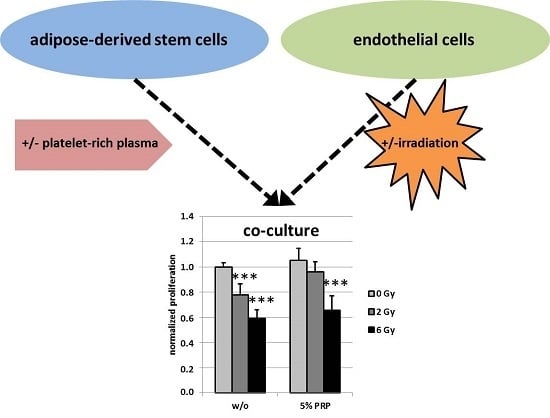
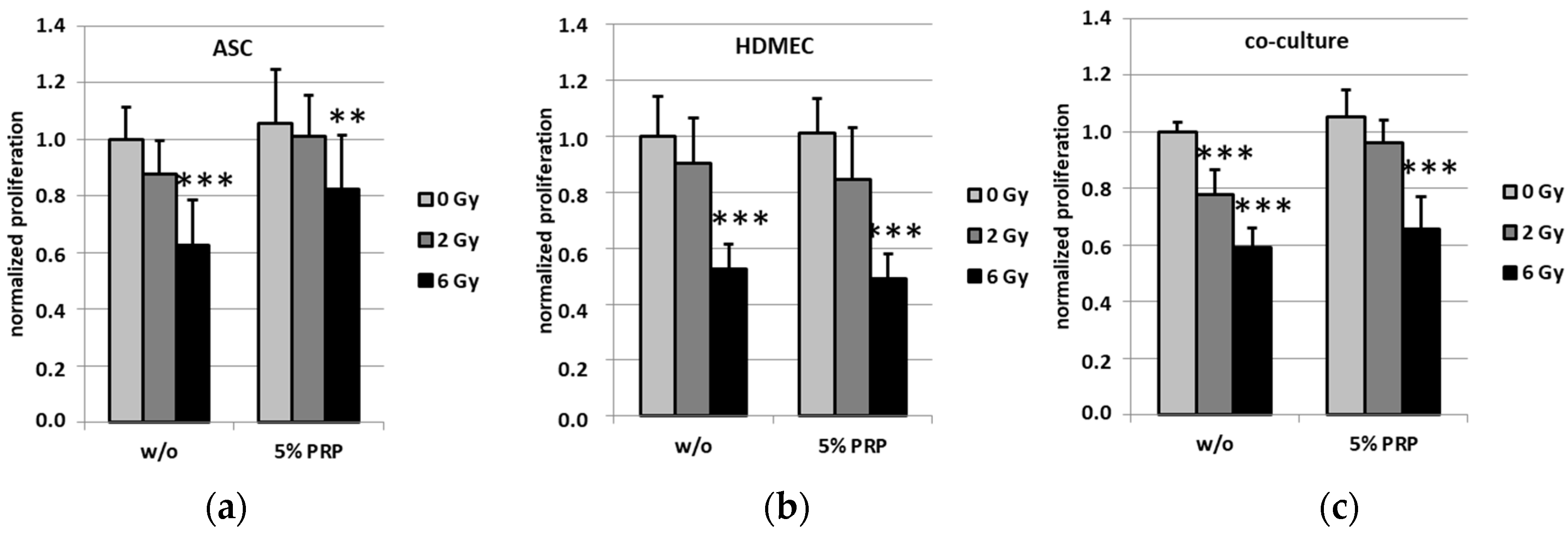
2.3. Effect of Irradiation on Cell Proliferation
3. Discussion and Conclusions
4. Materials and Methods
4.1. Cell Culture of HDMEC, ASC and the Respective Co-Culture
4.2. PRP Preparation
4.3. External Radiation
4.4. Cell Proliferation Assay and Vitality Assay
4.5. Flow Cytometry
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ASC | Adipose-derived stem cells |
| HDMEC | Human dermal microvascular endothelial cells |
| BrdU | 5-Bromo-2′-Deoxyuridine |
| WST-8 | (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium) |
References
- Haubner, F.; Ohmann, E.; Pohl, F.; Strutz, J.; Gassner, H.G. Wound healing after radiation therapy: Review of the literature. Radiat. Oncol. 2012, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Haubner, F.; Leyh, M.; Ohmann, E.; Pohl, F.; Prantl, L.; Gassner, H.G. Effects of external radiation in a co-culture model of endothelial cells and adipose-derived stem cells. Radiat. Oncol. 2013, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Landthaler, M.; Babilas, P. Wound healing in the 21st century. J. Am. Acad. Dermatol. 2010, 63, 866–881. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.L.; Neumeister, M.W.; Levi, B. Stem cells and tissue engineering: Regeneration of the skin and its contents. Clin. Plast. Surg. 2017, 44, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Haubner, F.; Gassner, H.G. Potential of adipose-derived stem cells concerning the treatment of wound healing complications after radiotherapy. HNO 2015, 63, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Smith, O.J.; Kanapathy, M.; Khajuria, A.; Prokopenko, M.; Hachach-Haram, N.; Mann, H.; Mosahebi, A. Protocol for a systematic review of the efficacy of fat grafting and platelet-rich plasma for wound healing. Syst. Rev. 2017, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Hyldig, K.; Riis, S.; Pennisi, C.P.; Zachar, V.; Fink, T. Implications of extracellular matrix production by adipose tissue-derived stem cells for development of wound healing therapies. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Rigotti, G.; Marchi, A.; Stringhini, P.; Baroni, G.; Galie, M.; Molino, A.M.; Mercanti, A.; Micciolo, R.; Sbarbati, A. Determining the oncological risk of autologous lipoaspirate grafting for post-mastectomy breast reconstruction. Aesthetic. Plast. Surg. 2010, 34, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Haubner, F.; Muschter, D.; Schuster, N.; Pohl, F.; Ahrens, N.; Prantl, L.; Gassner, H.G. Platelet-rich plasma stimulates dermal microvascular endothelial cells and adipose derived stem cells after external radiation. Clin. Hemorheol. Microcirc. 2015, 61, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Amable, P.R.; Carias, R.B.; Teixeira, M.V.; da Cruz Pacheco, I.; Correa do Amaral, R.J.; Granjeiro, J.M.; Borojevic, R. Platelet-rich plasma preparation for regenerative medicine: Optimization and quantification of cytokines and growth factors. Stem Cell. Res. Ther. 2013, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Frykberg, R.G.; Driver, V.R.; Carman, D.; Lucero, B.; Borris-Hale, C.; Fylling, C.P.; Rappl, L.M.; Clausen, P.A. Chronic wounds treated with a physiologically relevant concentration of platelet-rich plasma gel: A prospective case series. Ostomy Wound Manag. 2010, 56, 36–44. [Google Scholar]
- Liao, H.T.; Marra, K.G.; Rubin, J.P. Application of platelet-rich plasma and platelet-rich fibrin in fat grafting: Basic science and literature review. Tissue Eng. Part. B. Rev. 2014, 20, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.N.; Simmons, B.J.; Wolfson, A.H.; Nouri, K. Acute and chronic cutaneous reactions to ionizing radiation therapy. Dermatol. Ther. 2016, 6, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Driver, V.R.; Hanft, J.; Fylling, C.P.; Beriou, J.M. A prospective, randomized, controlled trial of autologous platelet-rich plasma gel for the treatment of diabetic foot ulcers. Ostomy Wound Manag. 2006, 52, 68–70. [Google Scholar]
- Van den Dolder, J.; Mooren, R.; Vloon, A.P.; Stoelinga, P.J.; Jansen, J.A. Platelet-rich plasma: Quantification of growth factor levels and the effect on growth and differentiation of rat bone marrow cells. Tissue Eng. 2006, 12, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Busilacchi, A.; Gigante, A.; Mattioli-Belmonte, M.; Manzotti, S.; Muzzarelli, R.A. Chitosan stabilizes platelet growth factors and modulates stem cell differentiation toward tissue regeneration. Carbohydr. Polym. 2013, 98, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Frechette, J.P.; Martineau, I.; Gagnon, G. Platelet-rich plasmas: Growth factor content and roles in wound healing. J. Dent. Res. 2005, 84, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Adeghate, J.; Nurulain, S.; Tekes, K.; Feher, E.; Kalasz, H.; Adeghate, E. Novel biological therapies for the treatment of diabetic foot ulcers. Expert Opin. Biol. Ther. 2017, 17, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Wettstein, R.; Senaldi, C.; Kalbermatten, D.F.; Konerding, M.A.; Raffoul, W.; Erba, P. Impact of platelet rich plasma and adipose stem cells on lymphangiogenesis in a murine tail lymphedema model. Microvasc. Res. 2015, 102, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Prantl, L.; Muehlberg, F.; Navone, N.M.; Song, Y.H.; Vykoukal, J.; Logothetis, C.J.; Alt, E.U. Adipose tissue-derived stem cells promote prostate tumor growth. Prostate 2010, 70, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Tajima, S.; Tobita, M.; Orbay, H.; Hyakusoku, H.; Mizuno, H. Direct and indirect effects of a combination of adipose-derived stem cells and platelet-rich plasma on bone regeneration. Tissue Eng. Part. A 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Seyhan, N.; Alhan, D.; Ural, A.U.; Gunal, A.; Avunduk, M.C.; Savaci, N. The effect of combined use of platelet-rich plasma and adipose-derived stem cells on fat graft survival. Ann. Plast. Surg. 2015, 74, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Caruana, G.; Bertozzi, N.; Boschi, E.; Pio Grieco, M.; Grignaffini, E.; Raposio, E. Role of adipose-derived stem cells in chronic cutaneous wound healing. Ann. Ital. Chir. 2015, 86, 1–4. [Google Scholar] [PubMed]
- Rigotti, G.; Charles-de-Sa, L.; Gontijo-de-Amorim, N.F.; Takiya, C.M.; Amable, P.R.; Borojevic, R.; Benati, D.; Bernardi, P.; Sbarbati, A. Expanded stem cells, stromal-vascular fraction, and platelet-rich plasma enriched fat: Comparing results of different facial rejuvenation approaches in a clinical trial. Aesthet. Surg. J. 2016, 36, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, J.; Dong, Q.; Liu, Y.; Mao, T.; Chen, F. Platelet-rich plasma—a promising cell carrier for micro-invasive articular cartilage repair. Med. Hypotheses 2009, 72, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Anitua, E.; Sanchez, M.; Nurden, A.T.; Nurden, P.; Orive, G.; Andia, I. New insights into and novel applications for platelet-rich fibrin therapies. Trends Biotechnol. 2006, 24, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Pohl, F.; Hassel, S.; Nohe, A.; Flentje, M.; Knaus, P.; Sebald, W.; Koelbl, O. Radiation-induced suppression of the Bmp2 signal transduction pathway in the pluripotent mesenchymal cell line C2C12: An in vitro model for prevention of heterotopic ossification by radiotherapy. Radiat. Res. 2003, 159, 345–350. [Google Scholar] [CrossRef]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinders, Y.; Felthaus, O.; Brockhoff, G.; Pohl, F.; Ahrens, N.; Prantl, L.; Haubner, F. Impact of Platelet-Rich Plasma on Viability and Proliferation in Wound Healing Processes after External Radiation. Int. J. Mol. Sci. 2017, 18, 1819. https://doi.org/10.3390/ijms18081819
Reinders Y, Felthaus O, Brockhoff G, Pohl F, Ahrens N, Prantl L, Haubner F. Impact of Platelet-Rich Plasma on Viability and Proliferation in Wound Healing Processes after External Radiation. International Journal of Molecular Sciences. 2017; 18(8):1819. https://doi.org/10.3390/ijms18081819
Chicago/Turabian StyleReinders, Yvonne, Oliver Felthaus, Gero Brockhoff, Fabian Pohl, Norbert Ahrens, Lukas Prantl, and Frank Haubner. 2017. "Impact of Platelet-Rich Plasma on Viability and Proliferation in Wound Healing Processes after External Radiation" International Journal of Molecular Sciences 18, no. 8: 1819. https://doi.org/10.3390/ijms18081819
APA StyleReinders, Y., Felthaus, O., Brockhoff, G., Pohl, F., Ahrens, N., Prantl, L., & Haubner, F. (2017). Impact of Platelet-Rich Plasma on Viability and Proliferation in Wound Healing Processes after External Radiation. International Journal of Molecular Sciences, 18(8), 1819. https://doi.org/10.3390/ijms18081819