Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease

 , and
, and
Abstract
1. Introduction
2. The γ-Aminobutyric Acid (GABA) Signaling System
2.1. GABA Synthesis
2.2. GABA Metabolism and Homeostasis
2.3. Mechanisms of GABA Transport and Synaptic Uptake
2.4. GABA Receptors
3. GABA System Changes in Alzheimer’s Disease (AD)
3.1. Alterations in GABA Levels and Glutamic Acid Decarboxylase (GAD) Enzyme Activity
3.2. GABAergic Neurons and Synaptic Dysfunction in AD
3.3. Alterations in GABA Receptor Distribution and Subunit Composition in AD
3.4. GABA Transporter Alterations
3.5. Tau Pathology and GABA Signaling
4. Disruption of the Excitatory/Inhibitory (E/I) Balance in AD
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Vinters, H.V. Emerging concepts in Alzheimer’s disease. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.; Wu, Y.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Burns, A.; Iliffe, S. Dementia. BMJ 2009, 338, b75. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Kukull, W.A.; Higdon, R.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Schellenberg, G.D.; van Belle, G.; Jolley, L.; Larson, E.B. Dementia and Alzheimer disease incidence: A prospective cohort study. Arch. Neurol. 2002, 59, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T. Glutamatergic systems in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S15–S21. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A. GABA as an inhibitory neurotransmitter in human cerebral cortex. J. Neurophysiol. 1989, 62, 1018–1027. [Google Scholar] [PubMed]
- Faingold, C.L.; Gehlbach, G.; Caspary, D.M. On the role of GABA as an inhibitory neurotransmitter in inferior colliculus neurons: Iontophoretic studies. Brain Res. 1989, 500, 302–312. [Google Scholar] [CrossRef]
- Cobb, S.R.; Buhl, E.H.; Halasy, K.; Paulsen, O.; Somogyi, P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 1995, 378, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Tamás, G.; Buhl, E.H.; Lorincz, A.; Somogyi, P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nat. Neurosci. 2000, 3, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Szabadics, J.; Lorincz, A.; Tamas, G. Beta and gamma frequency synchronization by dendritic gabaergic synapses and gap junctions in a network of cortical interneurons. J. Neurosci. 2001, 21, 5824–5831. [Google Scholar] [PubMed]
- Möhler, H. Role of GABAA receptors in cognition. Biochem. Soc. Trans. 2009, 37, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.H.; Kelly, P.H.; Bettler, B.; Kaupmann, K.; Cryan, J.F. Specific roles of GABAB(1) receptor isoforms in cognition. Behav. Brain Res. 2007, 181, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Chapouthier, G.; Venault, P. GABA-A receptor complex and memory processes. Curr. Top. Med. Chem. 2002, 2, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, I.; Medina, J.H. GABAA receptor modulation of memory: The role of endogenous benzodiazepines. Trends Pharmacol. Sci. 1991, 12, 260–265. [Google Scholar] [CrossRef]
- Heaney, C.F.; Kinney, J.W. Role of GABAB receptors in learning and memory and neurological disorders. Neurosci. Biobehav. Rev. 2016, 63, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, O.; Moser, E. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci. 1998, 21, 273–278. [Google Scholar] [CrossRef]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.D.; Stanford, I.M.; Yamawaki, N.; McAllister, C.J.; Ronnqvist, K.C.; Woodhall, G.L.; Furlong, P.L. The role of GABAergic modulation in motor function related neuronal network activity. NeuroImage 2011, 56, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Stagg, C.J.; Bachtiar, V.; Johansen-Berg, H. The role of GABA in human motor learning. Curr. Biol. 2011, 21, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Castel, M.; Gainer, H.; Yarom, Y. GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature 1997, 387, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y.; Speh, J.C. GABA is the principal neurotransmitter of the circadian system. Neurosci. Lett. 1993, 150, 112–116. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory actions of GABA during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Hensch, T.K.; Fagiolini, M.; Mataga, N.; Stryker, M.P.; Baekkeskov, S.; Kash, S.F. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 1998, 282, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Luján, R.; Shigemoto, R.; López-Bendito, G. Glutamate and GABA receptor signalling in the developing brain. Neuroscience 2005, 130, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Represa, A.; Ben-Ari, Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005, 28, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Pradhan, D.A.; Ming, G.L.; Song, H. GABA sets the tempo for activity-dependent adult neurogenesis. Trends Neurosci. 2007, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pallotto, M.; Deprez, F. Regulation of adult neurogenesis by GABAergic transmission: Signaling beyond GABAA-receptors. Front. Cell. Neurosci. 2014, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Vicini, S. The role of GABA and glutamate on adult neurogenesis. J. Physiol. 2008, 586, 3737–3738. [Google Scholar] [CrossRef] [PubMed]
- Pontes, A.; Zhang, Y.; Hu, W. Novel functions of GABA signaling in adult neurogenesis. Front. Biol. 2013, 8, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Mitsushima, D.; Hei, D.L.; Terasawa, E. γ-Aminobutyric acid is an inhibitory neurotransmitter restricting the release of luteinizing hormone-releasing hormone before the onset of puberty. Proc. Natl. Acad. Sci. USA 1994, 91, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-K.; Abraham, I.M.; Herbison, A.E. Effect of GABA on GnRH neurons switches from depolarization to hyperpolarization at puberty in the female mouse. Endocrinology 2002, 143, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.; Herbison, A.E. Development of GABA and glutamate signaling at the GnRH neuron in relation to puberty. Mol. Cell. Endocrinol. 2006, 254–255, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, E.; Nyberg, C.L.; Mogi, K.; Terasawa, E. A role of γ-amino butyric acid (GABA) and glutamate in control of puberty in female rhesus monkeys: Effect of an antisense oligodeoxynucleotide for GAD67 messenger ribonucleic acid and MK801 on luteinizing hormone-releasing hormone release. Endocrinology 1999, 140, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Cossart, R.; Bernard, C.; Ben-Ari, Y. Multiple facets of GABAergic neurons and synapses: Multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005, 28, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Maglóczky, Z.; Freund, T.F. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005, 28, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Kang, J.Q.; Gallagher, M.J. Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 2010, 588, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; De Blas, A.L.; Armstrong, D.M. GABAA receptors in aging and Alzheimer’s disease. J. Neurochem. 2007, 103, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Marczynski, T.J. GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease revisited. Brain Res. Bull. 1998, 45, 341–379. [Google Scholar] [CrossRef]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Möhler, H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology 2012, 62, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B. The role of GABA in the pathophysiology and treatment of anxiety disorders. Psychopharmacol. Bull. 2003, 37, 133–146. [Google Scholar] [PubMed]
- Durant, C.; Christmas, D.; Nutt, D. The pharmacology of anxiety. Curr. Top. Behav. Neurosci. 2010, 2, 303–330. [Google Scholar] [PubMed]
- Pizzarelli, R.; Cherubini, E. Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011, 297153. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, S.; Horder, J.; Inkster, B.; Mendez, M.A.; Murphy, D.G.; Nutt, D.J. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci. Biobehav. Rev. 2012, 36, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Auta, J.; Davis, J.M.; Dong, E.; Grayson, D.R.; Veldic, M.; Zhang, X.; Costa, E. GABAergic dysfunction in schizophrenia: New treatment strategies on the horizon. Psychopharmacology 2005, 180, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Auta, J.; Chen, Y.; Davis, J.M.; Dong, E.; Gavin, D.P.; Grayson, D.R.; Matrisciano, F.; Pinna, G.; Satta, R.; et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 2011, 60, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.; Frankel, S. γ-Aminobutyric acid in brain: Its formation from glutamic acid. J. Biol. Chem. 1950, 187, 55–63. [Google Scholar] [PubMed]
- Roberts, E.; Eidelberg, E. Metabolic and neurophysiological roles of γ-aminobutyric acid. Int. Rev. Neurobiol. 1960, 2, 279–332. [Google Scholar] [PubMed]
- Wu, J.-Y.; Matsuda, T.; Roberts, E. Purification and characterization of glutamate decarboxylase from mouse brain. J. Biol. Chem. 1973, 248, 3029–3034. [Google Scholar] [PubMed]
- Martin, D.L.; Martin, S.; Wu, S.; Espina, N. Regulatory properties of brain glutamate decarboxylase (GAD): The apoenzyme of GAD is present principally as the smaller of two molecular forms of GAD in brain. J. Neurosci. 1991, 11, 2725–2731. [Google Scholar] [PubMed]
- Sheikh, S.; Martin, S.; Martin, D. Regional distribution and relative amounts of glutamate decarboxylase isoforms in rat and mouse brain. Neurochem. Int. 1999, 35, 73–80. [Google Scholar] [CrossRef]
- Kaufman, D.L.; Houser, C.R.; Tobin, A.J. Two forms of the γ-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J. Neurochem. 1991, 56, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.L.; Rimvall, K. Regulation of γ-aminobutyric acid synthesis in the brain. J. Neurochem. 1993, 60, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Esclapez, M.; Tillakaratne, N.; Kaufman, D.L.; Tobin, A.; Houser, C. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J. Neurosci. 1994, 14, 1834–1855. [Google Scholar] [PubMed]
- Fukuda, T.; Aika, Y.; Heizmann, C.W.; Kosaka, T. GABAergic axon terminals at perisomatic and dendritic inhibitory sites show different immunoreactivities against two GAD isoforms, GAD67 and GAD65, in the mouse hippocampus: A digitized quantitative analysis. J. Comp. Neurol. 1998, 395, 177–194. [Google Scholar] [CrossRef]
- Mackie, M.; Hughes, D.; Maxwell, D.; Tillakaratne, N.; Todd, A. Distribution and colocalisation of glutamate decarboxylase isoforms in the rat spinal cord. Neuroscience 2003, 119, 461–472. [Google Scholar] [CrossRef]
- Castañeda, M.T.; Sanabria, E.R.G.; Hernandez, S.; Ayala, A.; Reyna, T.A.; Wu, J.-Y.; Colom, L.V. Glutamic acid decarboxylase isoforms are differentially distributed in the septal region of the rat. Neurosci. Res. 2005, 52, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Belhage, B.; Hansen, G.H.; Schousboe, A. Depolarization by K+ and glutamate activates different neurotransmitter release mechanisms in GABAergic neurons: Vesicular versus non-vesicular release of GABA. Neuroscience 1993, 54, 1019–1034. [Google Scholar] [CrossRef]
- Soghomonian, J.J.; Martin, D.L. Two isoforms of glutamate decarboxylase: Why? Trends Pharmacol. Sci. 1998, 19, 500–505. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Thomas, C.; Chen, W.; Davis, K.M.; Foos, T.; Chen, J.L.; Wu, E.; Floor, E.; Schloss, J.V.; Wu, J.-Y. Role of synaptic vesicle proton gradient and protein phosphorylation on ATP-mediated activation of membrane-associated brain glutamate decarboxylase. J. Biol. Chem. 1999, 274, 24366–24371. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wu, H.; Osterhaus, G.; Wei, J.; Davis, K.; Sha, D.; Floor, E.; Hsu, C.-C.; Kopke, R.D.; Wu, J.-Y. Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. USA 2003, 100, 4293–4298. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Schikorski, T.; Stevens, C. Quantitative fine-structural analysis of olfactory cortical synapses. Proc. Natl. Acad. Sci. USA 1999, 96, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.R.; Park, Y.D.; Lee, M.R.; Sharma, S.; Harris, K.M.; Kirov, S.A. Three-dimensional relationships between perisynaptic astroglia and human hippocampal synapses. Glia 2010, 58, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Hertz, L.; Svenneby, G. Uptake and metabolism of GABA in astrocytes cultured from dissociated mouse brain hemispheres. Neurochem. Res. 1977, 2, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hertz, L.; Huang, R.; Sonnewald, U.; Petersen, S.B.; Westergaard, N.; Larsson, O.; Schousboe, A. Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev. Neurosci. 1993, 15, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Schousboe, A. Glutamate transport and metabolism in astrocytes. Glia 1997, 21, 56–63. [Google Scholar] [CrossRef]
- Patel, A.; Balazs, R.; Richter, D. Contribution of the GABA bypath to glucose oxidation, and the development of compartmentation in the brain. Nature 1970, 226, 1160–1161. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Reubi, J.-C.; van Der Berg, C.; Cuenod, M. Glutamine as precursor for the GABA and glutamate trasmitter pools. Neurosci. Lett. 1978, 10, 171–174. [Google Scholar] [CrossRef]
- Yu, A.; Fisher, T.; Hertz, E.; Tildon, J.; Schousboe, A.; Hertz, L. Metabolic fate of [14C]-glutamine in mouse cerebral neurons in primary cultures. J. Neurosci. Res. 1984, 11, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, E.; Torgner, I.A.; Roberg, B. Kinetics and localization of brain phosphate activated glutaminase. J. Neurosci. Res. 2001, 66, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Svenneby, G. Time and temperature dependent activation of pig brain glutaminase. J. Neurochem. 1972, 19, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Weil-Malherbe, H. Modulators of glutaminase activity. J. Neurochem. 1972, 19, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, E.; Torgner, I.A. The effect of acetyl-coenzyme A on phosphate-activated glutaminase from pig kidney and brain. Biochem. J. 1974, 137, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci. 2014, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.L.; Neal, M.J. The uptake of [3H]GABA by slices of rat cerebral cortex. J. Neurochem. 1968, 15, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Henn, F.A.; Hamberger, A. Glial cell function: Uptake of transmitter substances. Proc. Natl. Acad. Sci. USA 1971, 68, 2686–2690. [Google Scholar] [CrossRef] [PubMed]
- McIntire, S.L.; Reimer, R.J.; Schuske, K.; Edwards, R.H.; Jorgensen, E.M. Identification and characterization of the vesicular GABA transporter. Nature 1997, 389, 870–876. [Google Scholar] [PubMed]
- Dingledine, R.; Korn, S.J. γ-aminobutyric acid uptake and the termination of inhibitory synaptic potentials in the rat hippocampal slice. J. Physiol. 1985, 366, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, A.; Lambert, J.D. Factors contributing to the decay of the stimulus-evoked IPSC in rat hippocampal CA1 neurons. J. Neurophysiol. 1994, 72, 2911–2926. [Google Scholar] [PubMed]
- Roepstorff, A.; Lambert, J.D. Comparison of the effect of the GABA uptake blockers, tiagabine and nipecotic acid, on inhibitory synaptic efficacy in hippocampal CA1 neurones. Neurosci. Lett. 1992, 146, 131–134. [Google Scholar] [CrossRef]
- Puia, G.; Costa, E.; Vicini, S. Functional diversity of GABA-activated Cl− currents in Purkinje versus granule neurons in rat cerebellar slices. Neuron 1994, 12, 117–126. [Google Scholar] [CrossRef]
- Engel, D.; Schmitz, D.; Gloveli, T.; Frahm, C.; Heinemann, U.; Draguhn, A. Laminar difference in GABA uptake and GAT-1 expression in rat CA1. J. Physiol. 1998, 512, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Semyanov, A.; Walker, M.C.; Kullmann, D.M. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat. Neurosci. 2003, 6, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, W.; Richerson, G.B. Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J. Neurophysiol. 2003, 89, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.R.; Lopez-Corcuera, B.; Mandiyan, S.; Nelson, H.; Nelson, N. Molecular characterization of four pharmacologically distinct γ-aminobutyric acid transporters in mouse brain. J. Biol. Chem. 1993, 268, 2106–2112. [Google Scholar] [PubMed]
- Dalby, N.O. GABA-level increasing and anticonvulsant effects of three different GABA uptake inhibitors. Neuropharmacology 2000, 39, 2399–2407. [Google Scholar] [CrossRef]
- Grossman, T.R.; Nelson, N. Effect of sodium lithium and proton concentrations on the electrophysiological properties of the four mouse GABA transporters expressed in Xenopus oocytes. Neurochem. Int. 2003, 43, 431–443. [Google Scholar] [CrossRef]
- Radian, R.; Ottersen, O.P.; Storm-Mathisen, J.; Castel, M.; Kanner, B.I. Immunocytochemical localization of the GABA transporter in rat brain. J. Neurosci. 1990, 10, 1319–1330. [Google Scholar] [PubMed]
- Rattray, M.; Priestley, J.V. Differential expression of GABA transporter-1 messenger RNA in subpopulations of GABA neurones. Neurosci. Lett. 1993, 156, 163–166. [Google Scholar] [CrossRef]
- Minelli, A.; Brecha, N.C.; Karschin, C.; DeBiasi, S.; Conti, F. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci. 1995, 15, 7734–7746. [Google Scholar] [PubMed]
- Itouji, A.; Sakai, N.; Tanaka, C.; Saito, N. Neuronal and glial localization of two GABA transporters (GAT1 and GAT3) in the rat cerebellum. Mol. Brain Res. 1996, 37, 309–316. [Google Scholar] [CrossRef]
- Nishimura, M.; Sato, K.; Mizuno, M.; Yoshiya, I.; Shimada, S.; Saito, N.; Tohyama, M. Differential expression patterns of GABA transporters (GAT1–3) in the rat olfactory bulb. Mol. Brain Res. 1997, 45, 268–274. [Google Scholar] [CrossRef]
- Conti, F.; Melone, M.; De Biasi, S.; Minelli, A.; Brecha, N.C.; Ducati, A. Neuronal and glial localization of GAT-1, a high-affinity γ-aminobutyric acid plasma membrane transporter, in human cerebral cortex: With a note on its distribution in monkey cortex. J. Comp. Neurol. 1998, 396, 51–63. [Google Scholar] [CrossRef]
- Kinney, G.A.; Spain, W.J. Synaptically evoked GABA transporter currents in neocortical glia. J. Neurophysiol. 2002, 88, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão-Ferreira, S.; Navarro, G.; Brugarolas, M.; Pérez-Capote, K.; Vaz, S.H.; Fattorini, G.; Conti, F.; Lluis, C.; Ribeiro, J.A.; McCormick, P.J.; et al. A1R-A2AR heteromers coupled to Gs and Gi/o proteins modulate GABA transport into astrocytes. Purinerg. Signal. 2013, 9, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.; Ciappelloni, S.; Conti, F. A quantitative analysis of cellular and synaptic localization of GAT-1 and GAT-3 in rat neocortex. Brain Struct. Funct. 2015, 220, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Minelli, A.; Melone, M. GABA transporters in the mammalian cerebral cortex: Localization, development and pathological implications. Brain Res. Rev. 2004, 45, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Borden, L.A.; Smith, K.E.; Hartig, P.R.; Branchek, T.A.; Weinshank, R.L. Molecular heterogeneity of the g-aminobutyric acid (GABA) transport system. Cloning of two novel high affinity GABA transporters from rat brain. J. Biol. Chem. 1992, 267, 21098–21104. [Google Scholar] [PubMed]
- Ikegaki, N.; Saito, N.; Hashima, M.; Tanaka, C. Production of specific antibodies against GABA transporter subtypes (GAT1, GAT2, GAT3) and their application to immunocytochemistry. Mol. Brain Res. 1994, 26, 47–54. [Google Scholar] [CrossRef]
- Minelli, A.; DeBiasi, S.; Brecha, N.C.; Vitellaro Zuccarello, L.; Conti, F. GAT-3, a high-affinity GABA plasma membrane transporter, Is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci. 1996, 16, 6255–6264. [Google Scholar] [PubMed]
- Nayeem, N.; Green, T.P.; Martin, I.L.; Barnard, E.A. Quaternary structure of the native GABAA receptor determined by electron microscopic image analysis. J. Neurochem. 1994, 62, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Ehya, N.; Fuchs, K.; Sieghart, W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J. Neurosci. 1997, 17, 2728–2737. [Google Scholar] [PubMed]
- Schofield, P.R.; Darlison, M.G.; Fujita, N.; Burt, D.R.; Stephenson, F.A.; Rodriguez, H.; Rhee, L.M.; Ramachandran, J.; Reale, V.; Glencorse, T.A.; et al. Sequence and functional expression of the GABAA receptor shows a ligand-gated receptor super-family. Nature 1987, 328, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Barnard, E.A.; Skolnick, P.; Olsen, R.W.; Mohler, H.; Sieghart, W.; Biggio, G.; Braestrup, C.; Bateson, A.N.; Langer, S.Z. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: Classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 1998, 50, 291–313. [Google Scholar] [PubMed]
- Bonnert, T.P.; McKernan, R.M.; Farrar, S.; le Bourdellès, B.; Heavens, R.P.; Smith, D.W.; Hewson, L.; Rigby, M.R.; Sirinathsinghji, D.J.S.; Brown, N.; et al. θ, a novel γ-aminobutyric acid type A receptor subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 9891–9896. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Olsen, R.W. GABAA receptor channels. Annu. Rev. Neurosci. 1994, 17, 569–602. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Wakimoto, H.; Fujita, N.; Lalande, M.; Barnard, E.A. Analysis of the set of GABAA receptor genes in the human genome. J. Biol. Chem. 2004, 279, 41422–41435. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.; McKernan, R.M.; Iversen, L.L. Another mechanism for creating diversity in γ-aminobutyrate type A receptors: RNA splicing directs expression of two forms of γ2 phosphorylation site. Proc. Natl. Acad. Sci. USA 1990, 87, 9966–9970. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Wang, J.B.; Moss, S.J.; Huganir, R.L.; Burt, D.R. Generation of two forms of the γ-aminobutyric acidA receptor γ2-subunit in mice by alternative splicing. J. Neurochem. 1991, 56, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Farrar, S.J.; Whiting, P.J.; Bonnert, T.P.; McKernan, R.M. Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J. Biol. Chem. 1999, 274, 10100–10104. [Google Scholar] [CrossRef] [PubMed]
- Jechlinger, M.; Pelz, R.; Tretter, V.; Klausberger, T.; Sieghart, W. Subunit composition and quantitative importance of hetero-oligomeric receptors: GABAA receptors containing α6 subunits. J. Neurosci. 1998, 18, 2449–2457. [Google Scholar] [PubMed]
- Li, M.; De Blas, A.L. Coexistence of two b subunit isoforms in the same γ-aminobutyric acid type A receptor. J. Biol. Chem. 1997, 272, 16564–16569. [Google Scholar] [CrossRef] [PubMed]
- Möhler, H.; Fritschy, J.M.; Rudolph, U. A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 2002, 300, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Möhler, H. GABAA receptor diversity and pharmacology. Cell Tissue Res. 2006, 326, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Sperk, G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr. Top. Med. Chem. 2002, 2, 795–816. [Google Scholar] [CrossRef] [PubMed]
- Quirk, K.; Gillard, N.P.; Ragan, C.I.; Whiting, P.J.; McKernan, R.M. Model of subunit composition of γ-aminobutyric acid A receptor subtypes expressed in rat cerebellum with respect to their α and γ/δ subunits. J. Biol. Chem. 1994, 269, 16020–16028. [Google Scholar] [PubMed]
- Nusser, Z.; Ahmad, Z.; Tretter, V.; Fuchs, K.; Wisden, W.; Sieghart, W.; Somogyi, P. Alterations in the expression of GABAA receptor subunits in cerebellar granule cells after the disruption of the α6 subunit gene. Eur. J. Neurosci. 1999, 11, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Benke, D.; Fritschy, J.M.; Trzeciak, A.; Bannwarth, W.; Mohler, H. Distribution, prevalence, and drug binding profile of γ-aminobutyric acid type A receptor subtypes differing in the β-subunit variant. J. Biol. Chem. 1994, 269, 27100–27107. [Google Scholar] [PubMed]
- Benke, D.; Mertens, S.; Trzeciak, A.; Gillessen, D.; Mohler, H. GABAA receptors display association of γ2-subunit with α1- and β2/3-subunits. J. Biol. Chem. 1991, 266, 4478–4483. [Google Scholar] [PubMed]
- Quirk, K.; Gillard, N.P.; Ragan, C.I.; Whiting, P.J.; McKernan, R.M. γ-Aminobutyric acid type A receptors in the rat brain can contain both γ2 and γ3 subunits, but γ1 does not exist in combination with another γ subunit. Mol. Pharmacol. 1994, 45, 1061–1070. [Google Scholar] [PubMed]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Verdoorn, T.A.; Draguhn, A.; Ymer, S.; Seeburg, P.H.; Sakmann, B. Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron 1990, 4, 919–928. [Google Scholar] [CrossRef]
- Rudolph, U.; Mohler, H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 475–498. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.B.; Olsen, R.W. Functional domains of GABAA receptors. Trends Pharmacol. Sci. 1995, 16, 162–168. [Google Scholar] [CrossRef]
- Ebert, B.; Thompson, S.A.; Saounatsou, K.; McKernan, R.; Krogsgaard-Larsen, P.; Wafford, K.A. Differences in agonist/antagonist binding affinity and receptor transduction using recombinant human γ-aminobutyric acid type A receptors. Mol. Pharmacol. 1997, 52, 1150–1156. [Google Scholar] [PubMed]
- Gunther, U.; Benson, J.; Benke, D.; Fritschy, J.M.; Reyes, G.; Knoflach, F.; Crestani, F.; Aguzzi, A.; Arigoni, M.; Lang, Y.; et al. Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of γ-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 7749–7753. [Google Scholar] [CrossRef] [PubMed]
- Hosie, A.M.; Dunne, E.L.; Harvey, R.J.; Smart, T.G. Zinc-mediated inhibition of GABAA receptors: Discrete binding sites underlie subtype specificity. Nat. Neurosci. 2003, 6, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Perrot, C.; Feltz, P.; Poulter, M.O. Recombinant GABAA receptor desensitization: The role of the γ2 subunit and its physiological significance. J. Physiol. 1996, 497, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, A.M.; Tingey, J.J.; Harrison, N.L.; Pritchett, D.B.; Twyman, R.E. Activation and deactivation rates of recombinant GABAA receptor channels are dependent on α-subunit isoform. Biophys. J. 1997, 73, 2518–2526. [Google Scholar] [CrossRef]
- Brickley, S.G.; Mody, I. Extrasynaptic GABAA receptors: Their function in the CNS and implications for disease. Neuron 2012, 73, 23–34. [Google Scholar]
- Sieghart, W. Structure, Pharmacology, and function of GABAA receptor subtypes. Adv. Pharmacol. 2006, 54, 231–263. [Google Scholar] [PubMed]
- Fritschy, J.M.; Mohler, H. GABAA-receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 1995, 359, 154–194. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; Mobley, W.C. Implications for treatment: GABAA receptors in aging, Down syndrome and Alzheimer’s disease. J. Neurochem. 2011, 117, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Loup, F.; Wieser, H.-G.; Yonekawa, Y.; Aguzzi, A.; Fritschy, J.-M. Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. J. Neurosci. 2000, 20, 5401–5419. [Google Scholar] [PubMed]
- Allen, K.L.; Waldvogel, H.J.; Glass, M.; Faull, R.L.M. Cannabinoid (CB1), GABAA and GABAB receptor subunit changes in the globus pallidus in Huntington’s disease. J. Chem. Neuroanat. 2009, 37, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Galvez, T.; Duthey, B.; Kniazeff, J.; Blahos, J.; Rovelli, G.; Bettler, B.; Prezeau, L.; Pin, J.P. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO J. 2001, 20, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Kaupmann, K.; Huggel, K.; Heid, J.; Flor, P.J.; Bischoff, S.; Mickel, S.J.; McMaster, G.; Angst, C.; Bittiger, H.; Froestl, W.; et al. Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature 1997, 386, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Emson, P.C. GABAB receptors: Structure and function. Prog. Brain Res. 2007, 160, 43–57. [Google Scholar] [PubMed]
- Marshall, F.H.; Jones, K.A.; Kaupmann, K.; Bettler, B. GABAB receptors—The first 7TM heterodimers. Trends Pharmacol. Sci. 1999, 20, 396–399. [Google Scholar] [CrossRef]
- Padgett, C.L.; Slesinger, P.A. GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. 2010, 58, 123–147. [Google Scholar] [PubMed]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABAB receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Fam, S.R.; Hall, R.A. GABAB receptor association with the PDZ scaffold Mupp1 alters receptor stability and function. J. Biol. Chem. 2007, 282, 4162–4171. [Google Scholar] [CrossRef] [PubMed]
- Galvez, T.; Parmentier, M.L.; Joly, C.; Malitschek, B.; Kaupmann, K.; Kuhn, R.; Bittiger, H.; Froestl, W.; Bettler, B.; Pin, J.P. Mutagenesis and modeling of the GABAB receptor extracellular domain support a venus flytrap mechanism for ligand binding. J. Biol. Chem. 1999, 274, 13362–13369. [Google Scholar] [CrossRef] [PubMed]
- Kniazeff, J.; Galvez, T.; Labesse, G.; Pin, J.P. No ligand binding in the GB2 subunit of the GABAB receptor is required for activation and allosteric interaction between the subunits. J. Neurosci. 2002, 22, 7352–7361. [Google Scholar] [PubMed]
- Margeta-Mitrovic, M.; Jan, Y.N.; Jan, L.Y. A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 2000, 27, 97–106. [Google Scholar] [CrossRef]
- Robbins, M.J.; Calver, A.R.; Filippov, A.K.; Hirst, W.D.; Russell, R.B.; Wood, M.D.; Nasir, S.; Couve, A.; Brown, D.A.; Moss, S.J.; et al. GABAB2 is essential for G-protein coupling of the GABAB receptor heterodimer. J. Neurosci. 2001, 21, 8043–8052. [Google Scholar] [PubMed]
- Duthey, B.; Caudron, S.; Perroy, J.; Bettler, B.; Fagni, L.; Pin, J.P.; Prezeau, L. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABAB receptor. J. Biol. Chem. 2002, 277, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.Y.; Clark, J.; Coulombe, N.; Ethier, N.; Hebert, T.E.; Sullivan, R.; Kargman, S.; Chateauneuf, A.; Tsukamoto, N.; McDonald, T.; et al. Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J. Biol. Chem. 1999, 274, 7607–7610. [Google Scholar] [CrossRef] [PubMed]
- Baloucoune, G.A.; Chun, L.; Zhang, W.; Xu, C.; Huang, S.; Sun, Q.; Wang, Y.; Tu, H.; Liu, J. GABAB receptor subunit GB1 at the cell surface independently activates ERK1/2 through IGF-1R transactivation. PLoS ONE 2012, 7, e39698. [Google Scholar] [CrossRef] [PubMed]
- Möhler, H.; Benke, D.; Fritschy, J.M. GABAB-receptor isoforms: Molecular architecture and distribution. Life Sci. 2001, 68, 2297–2300. [Google Scholar] [CrossRef]
- Fritschy, J.-M.; Meskenaite, V.; Weinmann, O.; Honer, M.; Benke, D.; Mohler, H. GABAB-receptor splice variants GB1a and GB1b in rat brain: Developmental regulation, cellular distribution and extrasynaptic localization. Eur. J. Neurosci. 1999, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Tiao, J.Y.; Bradaia, A.; Biermann, B.; Kaupmann, K.; Metz, M.; Haller, C.; Rolink, A.G.; Pless, E.; Barlow, P.N.; Gassmann, M.; et al. The sushi domains of secreted GABAB1 isoforms selectively impair GABAB heteroreceptor function. J. Biol. Chem. 2008, 283, 31005–31011. [Google Scholar] [CrossRef] [PubMed]
- Biermann, B.; Ivankova-Susankova, K.; Bradaia, A.; Abdel Aziz, S.; Besseyrias, V.; Kapfhammer, J.P.; Missler, M.; Gassmann, M.; Bettler, B. The Sushi domains of GABAB receptors function as axonal targeting signals. J. Neurosci. 2010, 30, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Garci, E.; Gassmann, M.; Bettler, B.; Larkum, M.E. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron 2006, 50, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Mohanakrishnan, P.; Fowler, A.H.; Vonsattel, J.P.; Husain, M.M.; Jolles, P.R.; Liem, P.; Komoroski, R.A. An in vitro 1H nuclear magnetic resonance study of the temporoparietal cortex of Alzheimer brains. Exp. Brain Res. 1995, 102, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.L.; Francis, P.T.; Procter, A.W.; Palmer, A.M.; Davison, A.N.; Bowen, D.M. Gamma-aminobutyric acid concentration in brain tissue at two stages of Alzheimer’s disease. Brain 1988, 111, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Kobayashi, K.; Ichimiya, Y.; Kosaka, K.; Iizuka, R. A preliminary study of free amino acids in the postmortem temporal cortex from Alzheimer-type dementia patients. Neurobiol. Aging 1984, 5, 319–321. [Google Scholar] [CrossRef]
- Rossor, M.N.; Iversen, L.L.; Reynolds, G.P.; Mountjoy, C.Q.; Roth, M. Neurochemical characteristics of early and late onset types of Alzheimer’s disease. Br. Med. J. 1984, 288, 961–964. [Google Scholar] [CrossRef]
- Ellison, D.W.; Beal, M.F.; Mazurek, M.F.; Bird, E.D.; Martin, J.B. A postmortem study of amino acid neurotransmitters in Alzheimer’s disease. Ann. Neurol. 1986, 20, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Rossor, M.N.; Garrett, N.J.; Johnson, A.L.; Mountjoy, C.Q.; Roth, M.; Iversen, L.L. A post-mortem study of the cholinergic and GABA systems in senile dementia. Brain 1982, 105, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Seidl, R.; Cairns, N.; Singewald, N.; Kaehler, S.T.; Lubec, G. Differences between GABA levels in Alzheimer’s disease and Down syndrome with Alzheimer-like neuropathology. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 363, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gueli, M.C.; Taibi, G. Alzheimer’s disease: Amino acid levels and brain metabolic status. Neurol. Sci. 2013, 34, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W.; Bergeron, C.; Hansen, S.; Jones, K. Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1987, 21, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Muramoto, O.; Kanazawa, I.; Arai, H.; Kosaka, K.; Iizuka, R. Regional distribution of amino acid transmitters in postmortem brains of presenile and senile dementia of Alzheimer type. Ann. Neurol. 1986, 19, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Yew, D.T.; Li, W.P.; Webb, S.E.; Lai, H.W.; Zhang, L. Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and Alzheimer patients: A comparison. Exp. Gerontol. 1999, 34, 117–133. [Google Scholar] [CrossRef]
- Tarbit, I.; Perry, E.K.; Perry, R.H.; Blessed, G.; Tomlinson, B.E. Hippocampal free amino acids in Alzheimer’s disease. J. Neurochem. 1980, 35, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [PubMed]
- Mitew, S.; Kirkcaldie, M.T.K.; Dickson, T.C.; Vickers, J.C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 2013, 34, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Lanctôt, K.L.; Herrmann, N.; Mazzotta, P.; Khan, L.R.; Ingber, N. GABAergic function in Alzheimer’s disease: Evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can. J. Psychiatry 2004, 49, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Vawter, M.P.; Walsh, D.M.; Evans, S.J.; Choudary, P.V.; Li, J.; Overman, K.M.; Atz, M.E.; Myers, R.M.; Jones, E.G.; et al. Effect of agonal and postmortem factors on gene expression profile: Quality control in microarray analyses of postmortem human brain. Biol. Psychiatry 2004, 55, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, J.; Tennant, M.C.; Yates, C.M. Brain enzymes in agonal state and dementia. Biochem. Soc. Trans. 1989, 17, 208–209. [Google Scholar] [CrossRef]
- Butterworth, J. Changes in nine enzyme markers for neurons, glia, and endothelial cells in agonal state and Huntington’s disease caudate nucleus. J. Neurochem. 1986, 47, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Heath, P.R.; Eastwood, S.L.; Burnet, P.W.J.; McDonald, B.; Pearson, R.C.A. The relative Importance of premortem acidosis and postmortem interval for human brain gene expression studies: Selective mRNA vulnerability and comparison with their encoded proteins. Neurosci. Lett. 1995, 200, 151–154. [Google Scholar] [CrossRef]
- Spokes, E.G.S.; Garrett, N.J.; Iversen, L.L. Differential effects of agonal status on measurements of GABA and glutamate decarboxylase in human post-mortem brain tissue from control and Huntington’s chorea subjects. J. Neurochem. 1979, 33, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Lewohl, J.M.; Scott, H.L.; Dodd, P.R. Biochemical and molecular studies using human autopsy brain tissue. J. Neurochem. 2003, 85, 543–562. [Google Scholar] [CrossRef] [PubMed]
- Kammoun, S.; Gold, G.; Bouras, C.; Giannakopoulos, P.; McGee, W.; Herrmann, F.; Michel, J.P. Immediate causes of death of demented and non-demented elderly. Acta Neurol. Scand. 2000, 176, 96–99. [Google Scholar] [CrossRef]
- Erdő, S.L. Postmortem increase of GABA levels in peripheral rat tissues: Prevention by 3-mercapto-propionic acid. J. Neural Transm. 1984, 60, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Hansen, S.; Gandham, S.S. Postmortem changes of amino compounds in human and rat brain. J. Neurochem. 1981, 36, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Molina, J.A.; Gómez, P.; Vargas, C.; de Bustos, F.; Benito-León, J.; Tallón-Barranco, A.; Ortí-Pareja, M.; Gasalla, T.; Arenas, J. Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. J. Neural Transm. 1998, 105, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Mochizuki, Y.; Yoshihashi, H.; Takasu, T.; Nakano, E. Laboratory examinations correlated with severity of dementia. Ann. Clin. Lab. Sci. 1996, 26, 340–345. [Google Scholar] [PubMed]
- Weiner, M.F.; Speciale, S.G.; Risser, R.C.; Kramer, G.L.; Petty, F. Cerebrospinal fluid and plasma gamma-aminobutyric acid in Alzheimer’s disease. Biol. Psychiatry 1996, 40, 933–934. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Takahashi, S.; Kimura, M. A selective reduction of excitatory amino acids in cerebrospinal fluid of patients with Alzheimer type dementia compared with vascular dementia of the Binswanger type. Neurosci. Lett. 1992, 141, 5–8. [Google Scholar] [CrossRef]
- Tosca, P.; Canevari, L.; Di Paolo, E.; Ferrari, R.; Verze, S.; Zerbi, F.; Dagani, F. Glutamate and GABA levels in CSF from patients affected by dementia and olivo-ponto-cerebellar atrophy. Acta Neurol. Scand. 1992, 85, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Pomara, N.; Deptula, D.; Galloway, M.P.; LeWitt, P.A.; Stanley, M. CSF GABA in caregiver spouses of Alzheimer patients. Am. J. Psychiatry 1989, 146, 787–788. [Google Scholar] [PubMed]
- Kuroda, H. Gamma-aminobutyric acid (GABA) in cerebrospinal fluid. Acta Med. Okayama 1983, 37, 167–177. [Google Scholar] [PubMed]
- Bareggi, S.R.; Franceschi, M.; Bonini, L.; Zecca, L.; Smirne, S. Decreased CSF concentrations of homovanillic acid and γ-aminobutyric acid in alzheimer’s disease: Age- or disease-related modifications? Arch. Neurol. 1982, 39, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Mohr, E.; Bruno, G.; Foster, N.; Gillespie, M.; Cox, C.; Hare, T.A.; Tamminga, C.; Fedio, P.; Chase, T.N. GABA-agonist therapy for Alzheimer’s disease. Clin. Neuropharmacol. 1986, 9, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, R.; Teelken, A.W.; Trieling, W.B.; Weber, W.; Weihmayr, T.; Lauter, H. γ-Aminobutyric acid and homovanillic acid concentration in the CSF of patients with senile dementia of Alzheimer’s type. Arch. Neurol. 1984, 41, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Manyam, N.V.; Katz, L.; Hare, T.A.; Gerber, J.C., 3rd; Grossman, M.H. Levels of γ-aminobutyric acid in cerebrospinal fluid in various neurologic disorders. Arch. Neurol. 1980, 37, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Enna, S.J.; Stern, L.Z.; Wastek, G.J.; Yamamura, H.I. Cerebrospinal fluid γ-aminobutyric acid variations in neurological disorders. Arch. Neurol. 1977, 34, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Tomlinson, B.E.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J. 1978, 2, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Smith, C.B.; White, P.; Goodhardt, M.J.; Spillane, J.A.; Flack, R.H.A.; Davison, A.N. Chemical pathology of the organic dementias: Validity of biochemical measurements on human post-mortem brain specimens. Brain 1977, 100, 397–426. [Google Scholar] [CrossRef] [PubMed]
- Spillane, J.A.; White, P.; Goodhardt, M.J.; Flack, R.H.A.; Bowen, D.M.; Davison, A.N. Selective vulnerability of neurones in organic dementia. Nature 1977, 266, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.; Goodhardt, M.; Strong, A.; Smith, C.; White, P.; Branston, N.; Symon, L.; Davison, A. Biochemical indices of brain structure, function and ‘hypoxia’ in cortex from baboons with middle cerebral artery occlusion. Brain Res. 1976, 117, 503–507. [Google Scholar] [CrossRef]
- Bowen, D.M.; Flack, R.H.A.; White, P.; Smith, C.; Davison, A.N. Brain-decarboxylase activities as indices of pathological change in senile dementia. Lancet 1974, 303, 1247–1249. [Google Scholar] [CrossRef]
- Davies, P. Neurotransmitter-related enzymes in senile dementia of the Alzheimer type. Brain Res. 1979, 171, 319–327. [Google Scholar] [CrossRef]
- Reinikainen, K.J.; Paljärvi, L.; Huuskonen, M.; Soininen, H.; Laakso, M.; Riekkinen, P.J. A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer’s disease. J. Neurol. Sci. 1988, 84, 101–116. [Google Scholar] [CrossRef]
- Levy, R.; Ruberg, M.; Herrero, M.T.; Villares, J.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Alterations of GABAergic neurons in the basal ganglia of patients with progressive supranuclear palsy: An in situ hybridization study of GAD67 messenger RNA. Neurology 1995, 45, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Moore, R.Y. Glutamic acid decarboxylase message isoforms in human suprachiasmatic nucleus. J. Biol. Rhythms 1996, 11, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Yu, S.; Wong, W.; McGeer, E.G.; McGeer, P.L. GAD65, GAD67, and GABAT immunostaining in human brain and apparent GAD65 loss in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 1073–1088. [Google Scholar] [PubMed]
- Boissière, F.; Faucheux, B.; Duyckaerts, C.; Hauw, J.-J.; Agid, Y.; Hirsch, E.C. Striatal expression of glutamic acid decarboxylase gene in Alzheimer’s disease. J. Neurochem. 1998, 71, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Cowburn, R.; Barton, A.; Reynolds, G.; Dodd, P.; Wester, P.; O’Carroll, A.-M.; Lofdahl, E.; Winblad, B. A disorder of cortical GABAergic innervation in Alzheimer’s disease. Neurosci. Lett. 1987, 73, 192–196. [Google Scholar] [CrossRef]
- Fuhrer, T.E.; Palpagama, T.H.; Waldvogel, H.J.; Synek, B.J.L.; Turner, C.; Faull, R.L.; Kwakowsky, A. Impaired expression of GABA transporters in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. Neuroscience 2017, 351, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.D.C.; Cross, A.J.; Slater, P.; Deakin, J.F.W. Loss of cortical GABA uptake sites in Alzheimer’s disease. J. Neural Transm. 1988, 71, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.; Ducatenzeiler, A.; Ribeiro-da-Silva, A.; Duff, K.; Bennett, D.A.; Cuello, A.C. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol. Aging 2006, 27, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.S.; de Kort, G.J.L.; Steggerda, S.; Shigemoto, R.; Ribeiro-da-Silva, A.; Cuello, A.C. Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci. Lett. 2003, 353, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Brasnjevic, I.; Rutten, B.P.F.; Van Der Kolk, N.; Perl, D.P.; Bouras, C.; Steinbusch, H.W.M.; Schmitz, C.; Hof, P.R.; Dickstein, D.L. Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer’s disease. Brain Struct. Funct. 2010, 214, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Marin, V.; Blazquez-Llorca, L.; Rodriguez, J.-R.; Boluda, S.; Muntane, G.; Ferrer, I.; DeFelipe, J. Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front. Neuroanat. 2009, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Koliatsos, V.E.; Kecojevic, A.; Troncoso, J.C.; Gastard, M.C.; Bennett, D.A.; Schneider, J.A. Early involvement of small inhibitory cortical interneurons in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Miguel, A.; Hercher, C.; Beasley, C.L.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G. Loss of Munc18-1 long splice variant in GABAergic terminals is associated with cognitive decline and increased risk of dementia in a community sample. Mol. Neurodegener. 2015, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso, A.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Piccione, R.; Ballesteros-Yáñez, I.; DeFelipe, J.; Yuste, R. Cortical area and species differences in dendritic spine morphology. J. Neurocytol. 2002, 31, 337–346. [Google Scholar] [CrossRef] [PubMed]
- DeFelipe, J. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Front. Neuroanat. 2011, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Hof, P.R.; Glezer, I.I.; Nimchinsky, E.A.; Erwin, J.M. Neurochemical and cellular specializations in the mammalian neocortex reflect phylogenetic relationships: Evidence from primates, cetaceans, and artiodactyls. Brain Behav. Evol. 2000, 55, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.C.; Penney, J.B.; Young, A.B. Cortical GABAB and GABAA receptors in Alzheimer’s disease: A quantitative autoradiographic study. Neurology 1987, 37, 1454. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, M.; Mizukami, K.; Ikonomovic, M.D.; Ishikawa, M.; Hidaka, S.; Abrahamson, E.E.; DeKosky, S.T.; Asada, T. Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: Association with Braak staging. Acta Neuropathol. 2005, 109, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Ikonomovic, M.D.; Grayson, D.R.; Sheffield, R.; Armstrong, D.M. Immunohistochemical study of GABAA receptor α1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res. 1998, 799, 148–155. [Google Scholar] [CrossRef]
- Rissman, R.A.; Mishizen-Eberz, A.J.; Carter, T.L.; Wolfe, B.B.; De Blas, A.L.; Miralles, C.P.; Ikonomovic, M.D.; Armstrong, D.M. Biochemical analysis of GABAA receptor subunits α1, α5, β1, β2 in the hippocampus of patients with Alzheimer’s disease neuropathology. Neuroscience 2003, 120, 695–704. [Google Scholar] [CrossRef]
- Rissman, R.A.; Bennett, D.A.; Armstrong, D.M. Subregional analysis of GABAA receptor subunit mRNAs in the hippocampus of older persons with and without cognitive impairment. J. Chem. Neuroanat. 2004, 28, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Howell, O.; Atack, J.R.; Dewar, D.; McKernan, R.M.; Sur, C. Density and pharmacology of α5 subunit-containing GABAA receptors are preserved in hippocampus of Alzheimer’s disease patients. Neuroscience 2000, 98, 669–675. [Google Scholar] [CrossRef]
- Mizukami, K.; Ikonomovic, M.D.; Grayson, D.R.; Rubin, R.T.; Warde, D.; Sheffield, R.; Hamilton, R.L.; Davies, P.; Armstrong, D.M. Immunohistochemical study of GABAA receptor β2/3 subunits in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Exp. Neurol. 1997, 147, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Grayson, D.R.; Ikonomovic, M.D.; Sheffield, R.; Armstrong, D.M. GABAA receptor β2 and β3 subunits mRNA in the hippocampal formation of aged human brain with Alzheimer-related neuropathology. Mol. Brain Res. 1998, 56, 268–272. [Google Scholar] [CrossRef]
- Iwakiri, M.; Mizukami, K.; Ikonomovic, M.D.; Ishikawa, M.; Abrahamson, E.E.; DeKosky, S.T.; Asada, T. An immunohistochemical study of GABAA receptor gamma subunits in Alzheimer’s disease hippocampus: Relationship to neurofibrillary tangle progression. Neuropathology 2009, 29, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, S.; Bossers, K.; Van de Bilt, S.; Agrapart, V.; Morales, R.R.; Frajese, G.V.; Swaab, D.F. Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1964–1976. [Google Scholar] [CrossRef] [PubMed]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of functional GABAA receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef] [PubMed]
- Nägga, K.; Bogdanovic, N.; Marcusson, J. GABA transporters (GAT-1) in Alzheimer’s disease. J. Neural Transm. 1999, 106, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.M.; Sheffield, R.; Mishizen-Eberz, A.J.; Carter, T.L.; Rissman, R.A.; Mizukami, K.; Ikonomovic, M.D. Plasticity of glutamate and GABAA receptors in the hippocampus of patients with Alzheimer’s disease. Cell. Mol. Neurobiol. 2003, 23, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.D.; Penney, J.B., Jr.; Young, A.B. NMDA, AMPA, and benzodiazepine binding site changes in Alzheimer’s disease visual cortex. Neurobiol. Aging 1993, 14, 343–352. [Google Scholar] [CrossRef]
- Shimohama, S.; Taniguchi, T.; Fujiwara, M.; Kameyama, M. Changes in benzodiazepine receptors in Alzheimer-type dementia. Ann. Neurol. 1988, 23, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Crow, T.J.; Ferrier, I.N.; Johnson, J.A. The selectivity of the reduction of serotonin S2 receptors in Alzheimer-type dementia. Neurobiol. Aging 1986, 7, 3–7. [Google Scholar] [CrossRef]
- Owen, F.; Poulter, M.; Waddington, J.L.; Mashal, R.D.; Crow, T.J. [3H]R05-4864 and [3H]flunitrazepam binding in kainate-lesioned rat striatum and in temporal cortex of brains from patients with senile dementia of the Alzheimer type. Brain Res. 1983, 278, 373–375. [Google Scholar] [CrossRef]
- Jansen, K.L.R.; Faull, R.L.M.; Dragunow, M.; Synek, B.L. Alzheimer’s disease: Changes in hippocampal N-methyl-d-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors—An autoradiographic study. Neuroscience 1990, 39, 613–627. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Penney, J.B.; D’Amato, C.J.; Young, A.B. Dementia of the Alzheimer’s type: Changes in hippocampal L-[3H]glutamate binding. J. Neurochem. 1987, 48, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Perry, R.H.; Crossman, A.R. A detailed anatomical analysis of neurotransmitter receptors in the putamen and caudate in Parkinson’s disease and Alzheimer’s disease. Neurosci. Lett. 1994, 169, 68–72. [Google Scholar] [CrossRef]
- Syapin, P.J.; Skolnick, P. Characterization of benzodiazepine binding sites in cultured cells of neural origin. J. Neurochem. 1979, 32, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- McEnery, M.W. The mitochondrial benzodiazepine receptor: Evidence for association with the voltage-dependent anion channel (VDAC). J. Bioenerg. Biomembr. 1992, 24, 63–69. [Google Scholar] [CrossRef] [PubMed]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef] [PubMed]
- Riond, J.; Vita, N.; Fur, G.L.; Ferrara, P. Characterization of a peripheral-type benzodiazepine-binding site in the mitochondria of Chinese hamster ovary cells. FEBS Lett. 1989, 245, 238–244. [Google Scholar] [CrossRef]
- Regan, J.W.; Yamamura, H.I.; Yamada, S.; Roeske, W.R. High affinity renal [3H]flunitrazepam binding: Characterization, localization, and alteration in hypertension. Life Sci. 1981, 28, 991–998. [Google Scholar] [CrossRef]
- Veenman, L.; Leschiner, S.; Spanier, I.; Weisinger, G.; Weizman, A.; Gavish, M. PK 11195 attenuates kainic acid-induced seizures and alterations in peripheral-type benzodiazepine receptor (PBR) protein components in the rat brain. J. Neurochem. 2002, 80, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, G.; Edison, P.; Bertoldo, A.; Roncaroli, F.; Singh, P.; Gerhard, A.; Cobelli, C.; Brooks, D.J.; Turkheimer, F.E. Novel reference region model reveals increased microglial and reduced vascular binding of 11C-(R)-PK11195 in patients with Alzheimer’s disease. J. Nucl. Med. 2008, 49, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Diorio, D.; Welner, S.A.; Butterworth, R.F.; Meaney, M.J.; Suranyi-Cadotte, B.E. Peripheral benzodiazepine binding sites in Alzheimer’s disease frontal and temporal cortex. Neurobiol. Aging 1991, 12, 255–258. [Google Scholar] [CrossRef]
- Ohyama, M.; Senda, M.; Ishiwata, K.; Kitamura, S.; Mishina, M.; Ishii, K.; Toyama, H.; Oda, K.; Katayama, Y. Preserved benzodiazepine receptors in Alzheimer’s disease measured with C-11 flumazenil PET and I-123 iomazenil SPECT in comparison with CBF. Ann. Nucl. Med. 1999, 13, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Koeppe, R.A.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Positron emission tomography measures of benzodiazepine binding in Alzheimer’s disease. Arch. Neurol. 1995, 52, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Wyper, D.; Kelly, C.; Patterson, J. Single photon emission computed tomography in Alzheimer’s disease: Cerebral perfusion and acetylcholine muscarinic receptor imaging and a novel study of the GABA/benzodiazepine system. Int. J. Geriatr. Psychopharmacol. 1998, 1, 126–133. [Google Scholar]
- Fukuchi, K.; Hashikawa, K.; Seike, Y.; Moriwaki, H.; Oku, N.; Ishida, M.; Fujita, M.; Uehara, T.; Tanabe, H.; Kusuoka, H.; et al. Comparison of iodine-123-iomazenil SPECT and technetium-99m-HMPAO-SPECT in Alzheimer’s disease. J. Nucl. Med. 1997, 38, 467–470. [Google Scholar] [PubMed]
- Birdsill, A.C.; Walker, D.G.; Lue, L.; Sue, L.I.; Beach, T.G. Postmortem interval effect on RNA and gene expression in human brain tissue. Cell Tissue Bank. 2011, 12, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Barrett-Jolley, R. Nipecotic acid directly activates GABAA-like ion channels. Br. J. Pharmacol. 2001, 133, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Pasantes-Morales, H.; Schousboe, A. Role of taurine in osmoregulation in brain cells: Mechanisms and functional implications. Amino Acids 1997, 12, 281–292. [Google Scholar] [CrossRef]
- Zhu, X.M.; Ong, W.Y. Changes in GABA transporters in the rat hippocampus after kainate-induced neuronal injury: Decrease in GAT-1 and GAT-3 but upregulation of betaine/GABA transporter BGT-1. J. Neurosci. Res. 2004, 77, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Lehre, A.C.; Rowley, N.M.; Zhou, Y.; Holmseth, S.; Guo, C.; Holen, T.; Hua, R.; Laake, P.; Olofsson, A.M.; Poblete-Naredo, I.; et al. Deletion of the betaine–GABA transporter (BGT1; slc6a12) gene does not affect seizure thresholds of adult mice. Epilepsy Res. 2011, 95, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.E.; Frostholm, A.; Rotter, A. Embryonic and postnatal expression of four gamma-aminobutyric acid transporter mRNAs in the mouse brain and leptomeninges. J. Comp. Neurol. 1996, 376, 431–446. [Google Scholar] [CrossRef]
- Zhou, Y.; Holmseth, S.; Hua, R.; Lehre, A.C.; Olofsson, A.M.; Poblete-Naredo, I.; Kempson, S.A.; Danbolt, N.C. The betaine-GABA transporter (BGT1, slc6a12) is predominantly expressed in the liver and at lower levels in the kidneys and at the brain surface. Am. J. Physiol. Ren. Physiol. 2012, 302, F316–F328. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Corcuera, B.; Liu, Q.-R.; Mandiyan, S.; Nelson, H.; Nelson, N. Expression of a mouse brain cDNA encoding novel γ-aminobutyric acid transporter. J. Biol. Chem. 1992, 267, 17491–17493. [Google Scholar] [PubMed]
- Madsen, K.K.; Ebert, B.; Clausen, R.P.; Krogsgaard-Larsen, P.; Schousboe, A.; White, H.S. Selective GABA transporter inhibitors tiagabine and EF1502 exhibit mechanistic differences in their ability to modulate the ataxia and anticonvulsant action of the extrasynaptic GABAA receptor agonist gaboxadol. J. Pharmacol. Exp. Ther. 2011, 338, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Kempson, S.A.; Zhou, Y.; Danbolt, N.C. The betaine/GABA transporter and betaine: Roles in brain, kidney, and liver. Front. Physiol. 2014, 5, 159. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Tolnay, M.; Probst, A. REVIEW: Tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol. Appl. Neurobiol. 1999, 25, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Loreth, D.; Ozmen, L.; Revel, F.G.; Knoflach, F.; Wetzel, P.; Frotscher, M.; Metzger, F.; Kretz, O. Selective degeneration of septal and hippocampal GABAergic neurons in a mouse model of amyloidosis and tauopathy. Neurobiol. Dis. 2012, 47, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Levenga, J.; Krishnamurthy, P.; Rajamohamedsait, H.; Wong, H.; Franke, T.F.; Cain, P.; Sigurdsson, E.M.; Hoeffer, C.A. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol. Commun. 2013, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, L.H.; Rae, C.; Ittner, L.M.; Gotz, J.; Sonnewald, U. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J. Cereb. Blood Flow Metab. 2013, 33, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Nykänen, N.P.; Kysenius, K.; Sakha, P.; Tammela, P.; Huttunen, H.J. γ-Aminobutyric acid type A (GABAA) receptor activation modulates tau phosphorylation. J. Biol. Chem. 2012, 287, 6743–6752. [Google Scholar] [CrossRef] [PubMed]
- Hapfelmeier, G.; Schneck, H.; Kochs, E. Sevoflurane potentiates and blocks GABA-induced currents through recombinant a1b2g2 GABAA receptors: Implications for an enhanced GABAergic transmission. Eur. J. Anaesthesiol. 2001, 18, 377–383. [Google Scholar] [PubMed]
- Le Freche, H.; Brouillette, J.; Fernandez-Gomez, F.-J.; Patin, P.; Caillierez, R.; Zommer, N.; Sergeant, N.; Buée-Scherrer, V.; Lebuffe, G.; Blum, D.; et al. Tau phosphorylation and sevoflurane anesthesia: An association to postoperative cognitive impairment. Anesthesiology 2012, 116, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.A.; Virág, L.; Marcouiller, F.; Papon, M.-A.; Khoury, N.B.E.; Julien, C.; Morin, F.; Emala, C.W.; Planel, E. Propofol directly increases tau phosphorylation. PLoS ONE 2011, 6, e16648. [Google Scholar] [CrossRef] [PubMed]
- Planel, E.; Krishnamurthy, P.; Miyasaka, T.; Liu, L.; Herman, M.; Kumar, A.; Bretteville, A.; Figueroa, H.Y.; Yu, W.H.; Whittington, R.A.; et al. Anesthesia-induced hyperphosphorylation detaches 3-repeat tau from microtubules without affecting their stability in vivo. J. Neurosci. 2008, 28, 12798–12807. [Google Scholar] [CrossRef] [PubMed]
- Run, X.; Liang, Z.; Zhang, L.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Anesthesia induces phosphorylation of tau. J. Alzheimers Dis. 2009, 16, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Ramos Bernardes da Silva Filho, S.; Oliveira Barbosa, J.H.; Rondinoni, C.; Dos Santos, A.C.; Garrido Salmon, C.E.; da Costa Lima, N.K.; Ferriolli, E.; Moriguti, J.C. Neuro-degeneration profile of Alzheimer’s patients: A brain morphometry study. Neuroimage Clin. 2017, 15, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.-Q.; Kreitzer, A.; et al. Aberrant ecitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Hazra, A.; Corbett, B.F.; You, J.C.; Aschmies, S.; Zhao, L.; Li, K.; Lepore, A.C.; Marsh, E.D.; Chin, J. Corticothalamic network dysfunction and behavioral deficits in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.L.; Pow, D.V.; Tannenberg, A.E.; Dodd, P.R. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer’s disease. J. Neurosci. 2002, 22, RC206. [Google Scholar] [PubMed]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 2013, 70, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, I.; Medina, J.H. Memory formation: The sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol. Learn. Mem. 1997, 68, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Okumura, S.; Mizuo, H.; Iwata, H. Inhibition of diazepam and gamma-aminobutyric acid of depolarization-induced release of [14C]cysteine sulfinate and [3H]glutamate in rat hippocampal slices. J. Neurochem. 1983, 40, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Minc-Golomb, D.; Eimerl, S.; Schramm, M. Cysteine sulfinic acid-induced release of D-[3H]aspartate and [14C]GABA in hippocampus slices: The role of sodium channels and cAMP. Brain Res. 1989, 490, 205–211. [Google Scholar] [CrossRef]
- Erdő, S.; Michler, A.; Wolff, J.R. GABA accelerates excitotoxic cell death in cortical cultures: Protection by blockers of GABA-gated chloride channels. Brain Res. 1991, 542, 254–258. [Google Scholar] [CrossRef]
- Sims, N.R.; Bowen, D.M.; Allen, S.J.; Smith, C.C.; Neary, D.; Thomas, D.J.; Davison, A.N. Presynaptic cholinergic dysfunction in patients with dementia. J. Neurochem. 1983, 40, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Nyakas, C.; Granic, I.; Halmy, L.G.; Banerjee, P.; Luiten, P.G. The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-β42 with memantine. Behav. Brain Res. 2011, 221, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.I.; Kitt, C.A.; Simonds, W.F.; Price, D.L.; Brann, M.R. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 1991, 11, 3218–3226. [Google Scholar] [PubMed]
- Levey, A.I.; Edmunds, S.M.; Koliatsos, V.; Wiley, R.G.; Heilman, C.J. Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J. Neurosci. 1995, 15, 4077–4092. [Google Scholar] [PubMed]
- Li, G.; Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Ring, K.; Halabisky, B.; Deng, C.; Mahley, R.W.; Huang, Y. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 2009, 5, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [PubMed]
- Cowburn, R.F.; Wiehager, B.; Trief, E.; Li-Li, M.; Sundstrom, E. Effects of β-amyloid-(25–35) peptides on radioligand binding to excitatory amino acid receptors and voltage-dependent calcium channels: Evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochem. Res. 1997, 22, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Roselli, F.; Tirard, M.; Lu, J.; Hutzler, P.; Lamberti, P.; Livrea, P.; Morabito, M.; Almeida, O.F.X. Soluble β-amyloid1–40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 2005, 25, 11061–11070. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tomé, P.; Brera, B.; Arévalo, M.-A.; de Ceballos, M.L. β-Amyloid25–35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Neurobiol. Dis. 2004, 15, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Watson, J.B.; Xie, C.-W. Amyloid β prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J. Neurophysiol. 2004, 92, 2853–2858. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-Methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hou, L.; Gao, X.; Guo, F.; Jing, W.; Qi, J.; Qiao, J. Amyloid β-protein differentially affects NMDA receptor- and GABAA receptor-mediated currents in rat hippocampal CA1 neurons. Prog. Nat. Sci. 2009, 19, 963–972. [Google Scholar] [CrossRef]
- Lei, M.; Xu, H.; Li, Z.; Wang, Z.; O’Malley, T.T.; Zhang, D.; Walsh, D.M.; Xu, P.; Selkoe, D.J.; Li, S. Soluble Abeta oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016, 85, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, D. Amyloid-β impairs synaptic inhibition via GABAA receptor endocytosis. J. Neurosci. 2015, 35, 9205–9210. [Google Scholar] [CrossRef] [PubMed]
- Nava-Mesa, M.; Jimenez-Diaz, L.; Yajeya, J.; Navarro-Lopez, J. Amyloid-β induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria-CA3 hippocampal synapse. Front. Cell. Neurosci. 2013, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Slesinger, P.A. Emerging concepts for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Massone, S.; Vassallo, I.; Fiorino, G.; Castelnuovo, M.; Barbieri, F.; Borghi, R.; Tabaton, M.; Robello, M.; Gatta, E.; Russo, C.; et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 2011, 41, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, P.; Klausberger, T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J. Physiol. 2005, 562, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.; Li, Y.; Cai, G.Q.; Niu, R.F.; Fang, Q.; Wu, K.; Chen, Z.; Lin, L.N.; Xu, L.; Fei, J.; et al. GABA transporter-1 activity modulates hippocampal theta oscillation and theta burst stimulation-induced long-term potentiation. J. Neurosci. 2009, 29, 15836–15845. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Laviolette, P.S.; O’Keefe, K.; O’Brien, J.; Rentz, D.M.; Pihlajamaki, M.; Marshall, G.; Hyman, B.T.; Selkoe, D.J.; Hedden, T.; et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009, 63, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Epilepsy and cognitive impairments in alzheimer disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Vignes, M. Regulation of spontaneous inhibitory synaptic transmission by endogenous glutamate via non-NMDA receptors in cultured rat hippocampal neurons. Neuropharmacology 2001, 40, 737–748. [Google Scholar] [CrossRef]
- Sawada, M.; Ichinose, M. Amyloid β proteins reduce the GABA-induced Cl− current in identified Aplysia neurons. Neurosci. Lett. 1996, 213, 213–215. [Google Scholar] [CrossRef]
- Pakaski, M.; Farkas, Z.; Kasa, P., Jr.; Forgon, M.; Papp, H.; Zarandi, M.; Penke, B.; Kasa, P., Sr. Vulnerability of small GABAergic neurons to human β-amyloid pentapeptide. Brain Res. 1998, 796, 239–246. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Louzada, P.R.; De Mello, F.G.; Ferreira, S.T. Neuroprotection against Aβ and glutamate toxicity by melatonin: Are GABA receptors involved? Neurotox. Res. 2003, 5, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Louzada, P.R.; Paula Lima, A.C.; Mendonca-Silva, D.L.; Noel, F.; De Mello, F.G.; Ferreira, S.T. Taurine prevents the neurotoxicity of β-amyloid and glutamate receptor agonists: Activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 2004, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Ban, J.Y.; Seong, Y.H. Chronic stimulation of GABAA receptor with muscimol reduces amyloid β protein (25–35)-induced neurotoxicity in cultured rat cortical cells. Neurosci. Res. 2005, 52, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Kater, S.B. Excitatory and inhibitory neurotransmitters in the generation and degeneration of hippocampal neuroarchitecture. Brain Res. 1989, 478, 337–348. [Google Scholar] [CrossRef]
- Alagiakrishnan, K. Melatonin based therapies for delirium and dementia. Discov. Med. 2016, 21, 363–371. [Google Scholar] [PubMed]
- Shao, H.; Zhang, Y.; Dong, Y.; Yu, B.; Xia, W.; Xie, Z. Chronic treatment with anesthetic propofol improves cognitive function and attenuates caspase activation in both aged and Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2014, 41, 499–513. [Google Scholar] [PubMed]
- Zhang, Y.; Shao, H.; Dong, Y.; Swain, C.A.; Yu, B.; Xia, W.; Xie, Z. Chronic treatment with anesthetic propofol attenuates β-amyloid protein levels in brain tissues of aged mice. Transl. Neurodegener. 2014, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic neurotransmission in Alzheimer’s disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Atack, J.R. Preclinical and clinical pharmacology of the GABAA receptor α5 subtype-selective inverse agonist α5IA. Pharmacol. Ther. 2010, 125, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Atack, J.R. GABAA receptor subtype-selective modulators. II. a5-selective inverse agonists for cognition enhancement. Curr. Top. Med. Chem. 2011, 11, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials.gov [Internet]. Identifier NCT00880412, A Study to Determine the Clinical Safety/Tolerability and Exploratory Efficacy of EHT 0202 as Adjunctive Therapy to Acetylcholinesterase Inhibitor in Mild to Moderate Azheimer’s Disease (EHT0202/002). National Library of Medicine (US): Bethesda, MD, USA, 2009. Available online: https://clinicaltrials.gov/ct2/show/NCT00880412 (accessed on 13 July 2017).
- Drott, J.; Desire, L.; Drouin, D.; Pando, M.; Haun, F. Etazolate improves performance in a foraging and homing task in aged rats. Eur. J. Pharmacol. 2010, 634, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Siopi, E.; Llufriu-Dabén, G.; Cho, A.H.; Vidal-Lletjós, S.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Etazolate, an α-secretase activator, reduces neuroinflammation and offers persistent neuroprotection following traumatic brain injury in mice. Neuropharmacology 2013, 67, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Sol, O.; Snyder, P.J.; Ousset, P.J.; Haddad, R.; Maurin, M.; Lemarie, J.C.; Desire, L.; Pando, M.P. EHT0202 in Alzheimer’s disease: A 3-month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 2011, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- De Gage, S.B.; Moride, Y.; Ducruet, T.; Kurth, T.; Verdoux, H.; Tournier, M.; Pariente, A.; Bégaud, B. Benzodiazepine use and risk of Alzheimer’s disease: Case-control study. BMJ 2014, 349, g5205. [Google Scholar] [CrossRef] [PubMed]
- Fastbom, J.; Forsell, Y.; Winblad, B. Benzodiazepines may have protective effects against Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1998, 12, 14–17. [Google Scholar] [CrossRef] [PubMed]


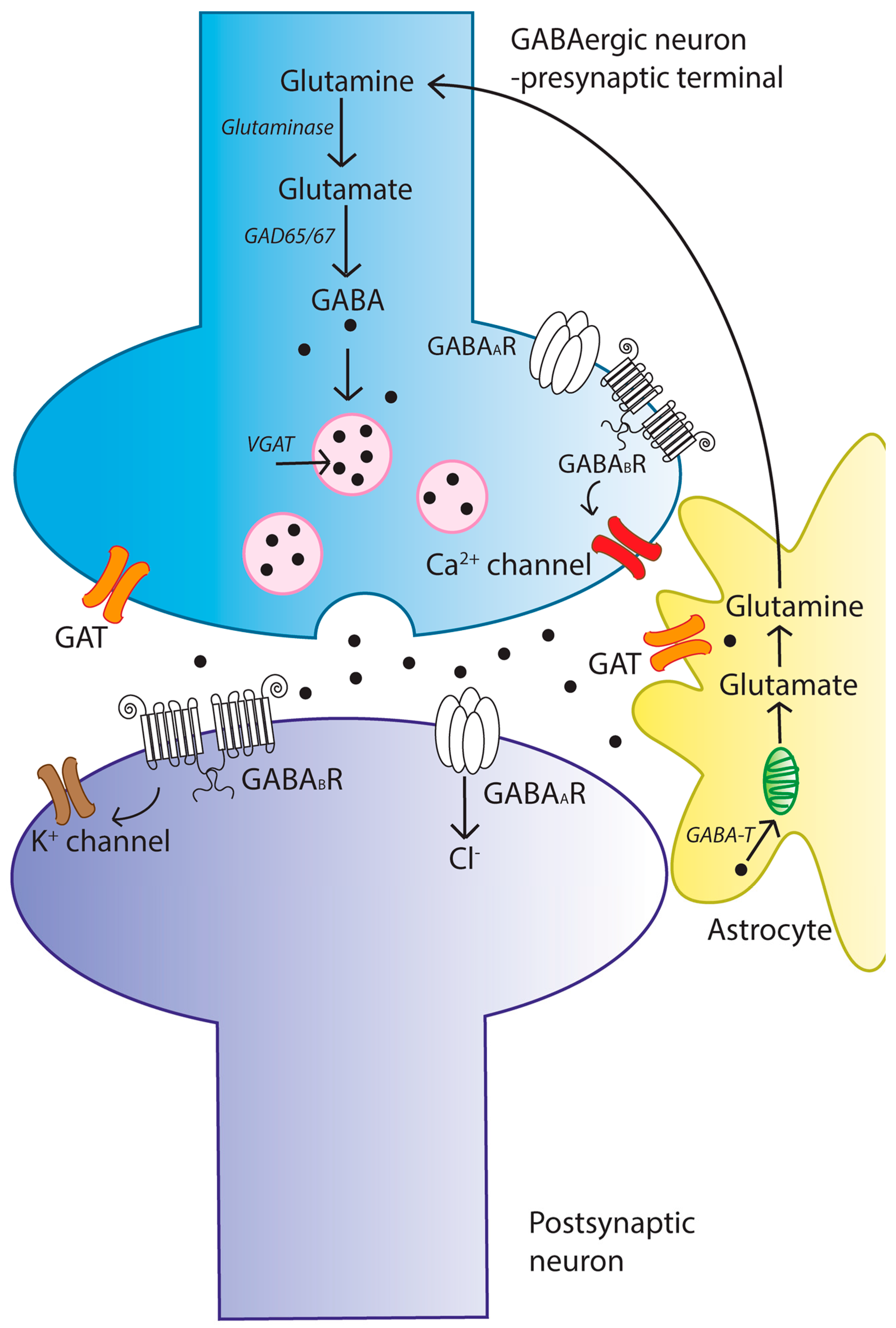{kind=link}
| Region | Change | References |
|---|---|---|
| Hippocampus | ↓ | [172,174,177] |
| ↔ | [173,178,180] | |
| Subiculum | ↔ | [174] |
| Thalamus (Whole/Subregion Unspecified) | ↔ | [175] |
| Thalamus (Dorsolateral Nucleus) | ↔ | [178] |
| Thalamus (Dorsomedial Nucleus) | ↓ | [173] |
| Thalamus (Ventrolateral Nucleus) | ↓ | [178] |
| ↔ | [173,174] | |
| Thalamus (Anterior Nucleus) | ↔ | [173,174] |
| Subthalamic Nucleus | ↔ | [174] |
| Cingulate Cortex | ↓ | [173,178] |
| ↔ | [170,174] | |
| Amygdala | ↓ | [172,178] |
| ↔ | [174] | |
| Caudate Nucleus | ↔ | [173,174,175] |
| ↑ | [178] | |
| Putamen | ↔ | [173,174,178] |
| Substantia Nigra | ↔ | [174,177,178] |
| Substantia Innominata | ↔ | [174,177] |
| Globus Pallidus (Whole) | ↔ | [174] |
| Globus Pallidus (Interna) | ↔ | [178] |
| Globus Pallidus (Externa) | ↔ | [178] |
| Nucleus Accumbens | ↔ | [173,174,178] |
| Septal Nuclei/Medial Olfactory Area | ↔ | [174] |
| Frontal Cortex (Subregion Unspecified) | ↓ | [177] |
| Frontal Cortex (Prefrontal Cortex) | ↓ | [173,174] |
| ↔ | [172,179] | |
| ↑↓ | [170] | |
| Frontal Cortex (Superior) | ↔ | [178] |
| Frontal Cortex (Orbitofrontal Cortex) | ↓ | [178] |
| ↔ | [174] | |
| Frontal Cortex (Premotor Cortex) | ↓ | [173] |
| ↔ | [174] | |
| Frontal Cortex (Primary Motor Cortex) | ↓ | [174] |
| Insular Cortex | ↔ | [178] |
| Temporal Cortex (Subregion Unspecified) | ↓ | [176] |
| Temporal Cortex (Superior) | ↓ | [170,174,175] |
| Temporal Cortex (Middle) | ↓ | [170,172,173,174] |
| Temporal Cortex (Inferior) | ↓ | [171,174] |
| ↔ | [173,178] | |
| Temporal Cortex (Temporal Pole) | ↓ | [173] |
| ↔ | [174] | |
| Temporal Cortex (Entorhinal Cortex) | ↓ | [173] |
| Temporoparietal Cortex (Posterior) | ↓ | [169] |
| Parietal Cortex (Superior) | ↓ | [170] |
| Parietal Cortex (Primary Somatosensory Cortex) | ↓ | [173] |
| Parietal Cortex (Somatosensory Association Cortex) | ↔ | [172,174] |
| Parietal Cortex (Angular Cortex) | ↔ | [178] |
| Occipital Cortex | ↓ | [177,178] |
| Occipital Cortex (Visual Cortex) | ↓ | [173,175] |
| ↔ | [172] | |
| Cerebellum | ↓ | [175] |
| ↔ | [174] | |
| Hypothalamus | ↔ | [174,178] |
| Hypothalamus (Mammillary Body) | ↔ | [178] |
| Nucleus Rubor | ↔ | [174] |
| Periaqueductal Gray | ↔ | [174] |
| Raphe Nucleus | ↔ | [178] |
| Pons (Basilar) | ↔ | [174] |
| Medulla Oblongata (Olivary Body) | ↔ | [174] |
| Component | Methodology | Subregion | Change | Comments | References |
|---|---|---|---|---|---|
| Hippocampal Formation | |||||
| GABAAR α1 | IHC | CA1, CA2, prosubiculum | ↓ | [235] | |
| DG, subiculum, presubiculum | ↔ | ||||
| WB | CA1, CA2, CA3, DG, subiculum | ↔ | [236] | ||
| ISH | CA1/subiculum, CA2, CA3, CA4, DG | ↓ | [237] | ||
| GABAAR α5 | WB | CA1, CA2, CA3 | ↓ | Significant change from mild/moderate to severe AD | [236] |
| DG, subiculum | ↔ | ||||
| ISH | CA1/subiculum, CA2, CA3, CA4, DG | ↓ | [237] | ||
| Autoradiography | CA1 | ↓ | [238] | ||
| CA2, CA3, DG, subiculum, presubiculum, parasubiculum | ↔ | ||||
| GABAAR β1 | WB | CA1, CA2, CA3, DG, subiculum | ↔ | [236] | |
| GABAAR β2Subunit | WB | CA1, CA2, CA3, DG, subiculum | ↔ | [236] | |
| ISH | CA1, CA2, CA3, CA4, DG | ↔ | [240] | ||
| GABAAR β3 Subunit | ISH | CA1, CA2, CA3, DG | ↓ | [240] | |
| CA4 | ↔ | ||||
| GABAAR β2/3 Subunit | IHC | CA1, CA2, CA3, subiculum | ↔ | Antibody specific for both β2 and β3 | [239] |
| GABAAR γ2 Subunit | IHC | CA1, CA2/3, CA4, DG | ↔ | Qualitative study | [241] |
| GABAAR γ1/3 Subunit | IHC | CA1, CA2/3, CA4/DG | ↑ | Antibody specific for both γ1 and γ3, Qualitative study | [241] |
| GABABR R1 Subunit | IHC | DG, CA2/3, CA4 | ↑↓ | Antibody primarily recognizes R1a isoform, increase observed from control to moderate AD, but no change from control to severe AD—transient upregulation | [234] |
| CA1, DG, subiculum | ↔ | Non-significant but trend as in other regions | |||
| GAD65/2 | IHC | DG | ↓ | [215] | |
| GAT1 | [3H]tiagabine radioligand binding | DG | ↔ | [244] | |
| IHC | CA1, CA2, CA3, DG, subiculum | ↔ | [218] | ||
| GAT3 | IHC | DG | ↑ | Significant increase in reactive astrocytes | [181] |
| CA1, CA3 | ↓ | Observed in stratum pyramidale and also in astrocytes specifically | [218] | ||
| Subiculum | Observed across region and in astrocytes specifically | ||||
| CA2, DG | ↔ | Large, non-significant downregulation observed | |||
| BGT1 | IHC | CA2, CA3 | ↑ | Observed in stratum radiatum and also in CA3 astrocytes specifically | [218] |
| DG | Observed in all layers | ||||
| CA1, subiculum | ↔ | Increase observed in astrocytes specifically | |||
| Cerebral Cortex | |||||
| GABAAR | Autoradiography [3H] GABA radioligand binding | Superior frontal gyrus | ↔ | GABABR blocked with baclofen | [233] |
| GABAAR α1 Subunit | qPCR | Prefrontal cortex | ↓ | [242] | |
| Temporal cortex | [243] | ||||
| WB | Temporal cortex | ↓ | [243] | ||
| GABAAR α2 Subunit | qPCR | Prefrontal cortex | ↓ | No change when patients classified by Aβ plaque load | [242] |
| Temporal cortex | [243] | ||||
| GABAAR α4 Subunit | qPCR | Prefrontal cortex | ↓ | [242] | |
| GABAAR α5 Subunit | Autoradiography | Entorhinal cortex, perirhinal cortex | ↓ | [238] | |
| Temporal cortex | ↔ | ||||
| qPCR | Temporal cortex | ↓ | [243] | ||
| GABAAR β1 Subunit | qPCR | Temporal cortex | ↔ | [243] | |
| WB | |||||
| GABAAR β2 Subunit | qPCR | Temporal cortex | ↓ | [243] | |
| GABAAR β3 Subunit | qPCR | Temporal cortex | ↓ | [243] | |
| GABAAR γ1 Subunit | qPCR | Temporal cortex | ↔ | [243] | |
| WB | |||||
| GABAAR γ2 Subunit | qPCR | Prefrontal cortex | ↔ | [242] | |
| Temporal cortex | ↓ | [243] | |||
| WB | Temporal cortex | ↓ | [243] | ||
| GABAAR δ Subunit | qPCR | Prefrontal cortex | ↓ | [242] | |
| Temporal cortex | [243] | ||||
| GABAAR ε Subunit | qPCR | Prefrontal cortex | ↔ | [242] | |
| GABAAR θ Subunit | qPCR | Prefrontal cortex | ↔ | [242] | |
| GABABR R1 Subunit | qPCR | Prefrontal cortex | ↔ | [242] | |
| GABABR R2 Subunit | qPCR | Prefrontal cortex | ↓ | Patients classified by Aβ plaque load | [242] |
| GABABR | Autoradiography [3H]GABA radioligand binding | Superior frontal gyrus | ↓ | GABAAR blocked with isoguvacine | [233] |
| GAD67/1 | qPCR | Prefrontal cortex | ↓ | Patients classified by Aβ plaque load | [242] |
| WB | Middle temporal gyrus | ↔ | Only 2 AD and 2 control cases | [215] | |
| GAD65/2 | IHC | Middle temporal gyrus | ↓ | Layers II/III and IV | [215] |
| Primary visual cortex | ↔ | Trend towards reduction | |||
| WB | Middle temporal gyrus | ↓ | Only 2 AD and 2 control cases | [215] | |
| GATs | [3H]nipecotic acid radioligand binding | Temporal cortex | ↓ | Nipecotic acid is a GABA reuptake inhibitor | [219] |
| Frontal cortex | ↔ | ||||
| GAT1 | [3H]tiagabine radioligand binding | Frontal cortex | ↔ | [244] | |
| Temporal cortex | |||||
| IHC | Superior temporal gyrus | ↓ | Observed across both regions, and also in astrocytes specifically | [218] | |
| Entorhinal cortex | |||||
| GAT3 | IHC | Superior temporal gyrus | ↔ | Large, non-significant downregulation observed | [218] |
| Entorhinal cortex | ↓ | Observed across region, and also in astrocytes specifically | |||
| BGT1 | IHC | Superior temporal gyrus | ↑ | [218] | |
| Entorhinal cortex | ↔ | Increase observed in astrocytes specifically | |||
| Subcortical Structures | |||||
| GAD67/1 | ISH | Hypothalamus (superchiasmatic nucleus and retrochiasmatic area) | ↔ | [214] | |
| ISH | Caudate nucleus, putamen | ↑ | Attributed to increase in neuron number, but no increase per neuron | [216] | |
| Ventral striatum | ↔ | ||||
| GAD65/2 | ISH | Hypothalamus (superchiasmatic nucleus and retrochiasmatic area) | ↔ | [214] | |
| IHC | Putamen | ↓ | [215] | ||
| Globus pallidus | ↔ | ||||
| GATs | [3H]nipecotic acid radioligand binding | Caudate nucleus, putamen, globus pallidus | ↔ | Nipecotic acid is a GABA reuptake inhibitor | [219] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govindpani, K.; Calvo-Flores Guzmán, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1813. https://doi.org/10.3390/ijms18081813
Govindpani K, Calvo-Flores Guzmán B, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A. Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. International Journal of Molecular Sciences. 2017; 18(8):1813. https://doi.org/10.3390/ijms18081813
Chicago/Turabian StyleGovindpani, Karan, Beatriz Calvo-Flores Guzmán, Chitra Vinnakota, Henry J. Waldvogel, Richard L. Faull, and Andrea Kwakowsky. 2017. "Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease" International Journal of Molecular Sciences 18, no. 8: 1813. https://doi.org/10.3390/ijms18081813
APA StyleGovindpani, K., Calvo-Flores Guzmán, B., Vinnakota, C., Waldvogel, H. J., Faull, R. L., & Kwakowsky, A. (2017). Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. International Journal of Molecular Sciences, 18(8), 1813. https://doi.org/10.3390/ijms18081813