Protocatechuic Acid from Pear Inhibits Melanogenesis in Melanoma Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
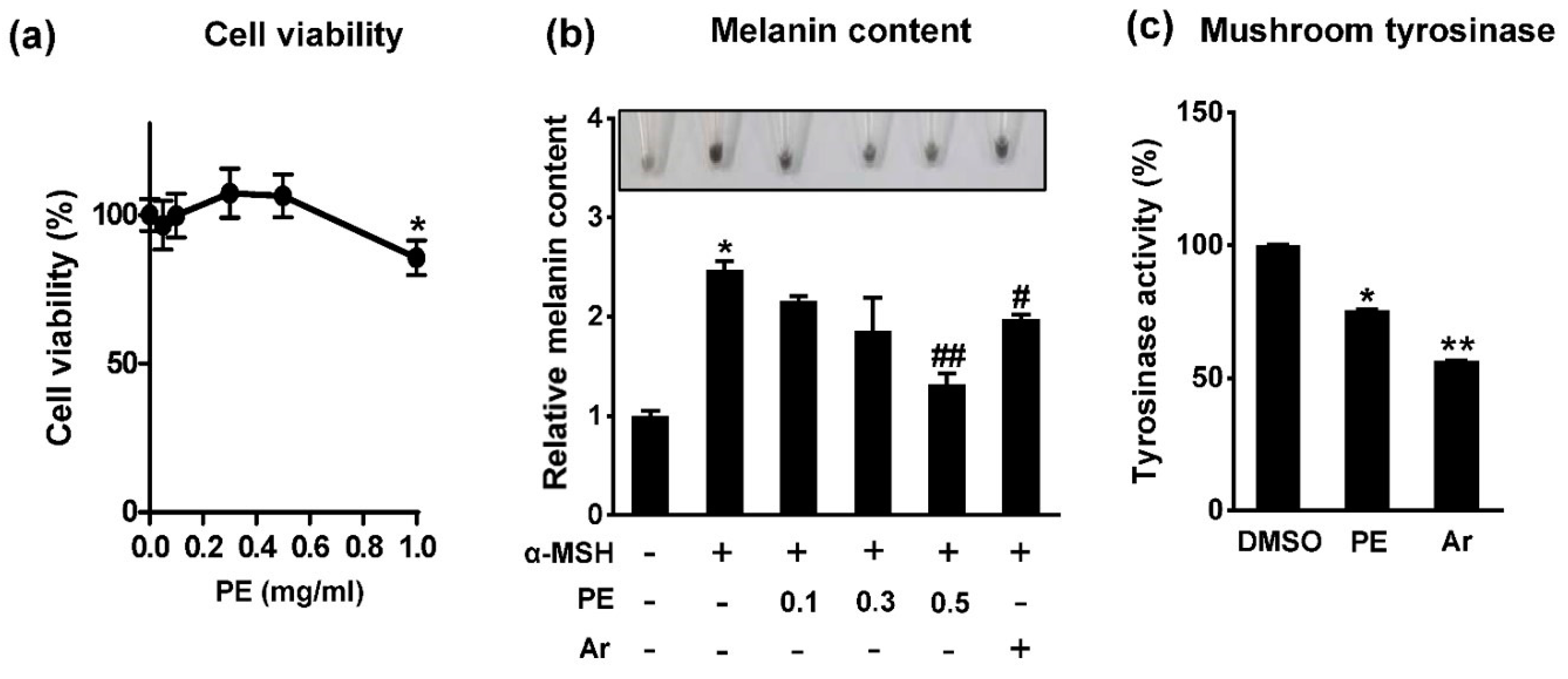
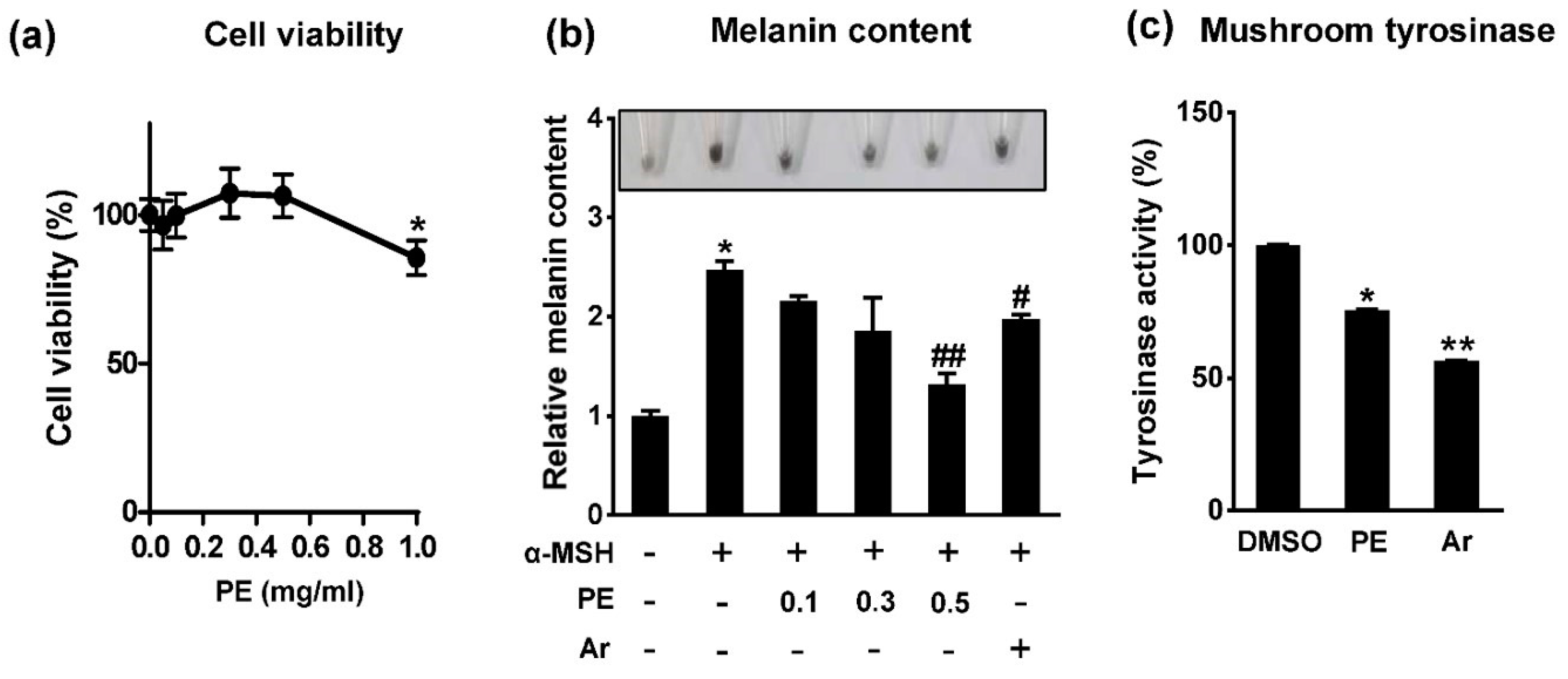
2.1. Pear Extract (PE) Inhibits Melanin Synthesis in B16F10 Cells
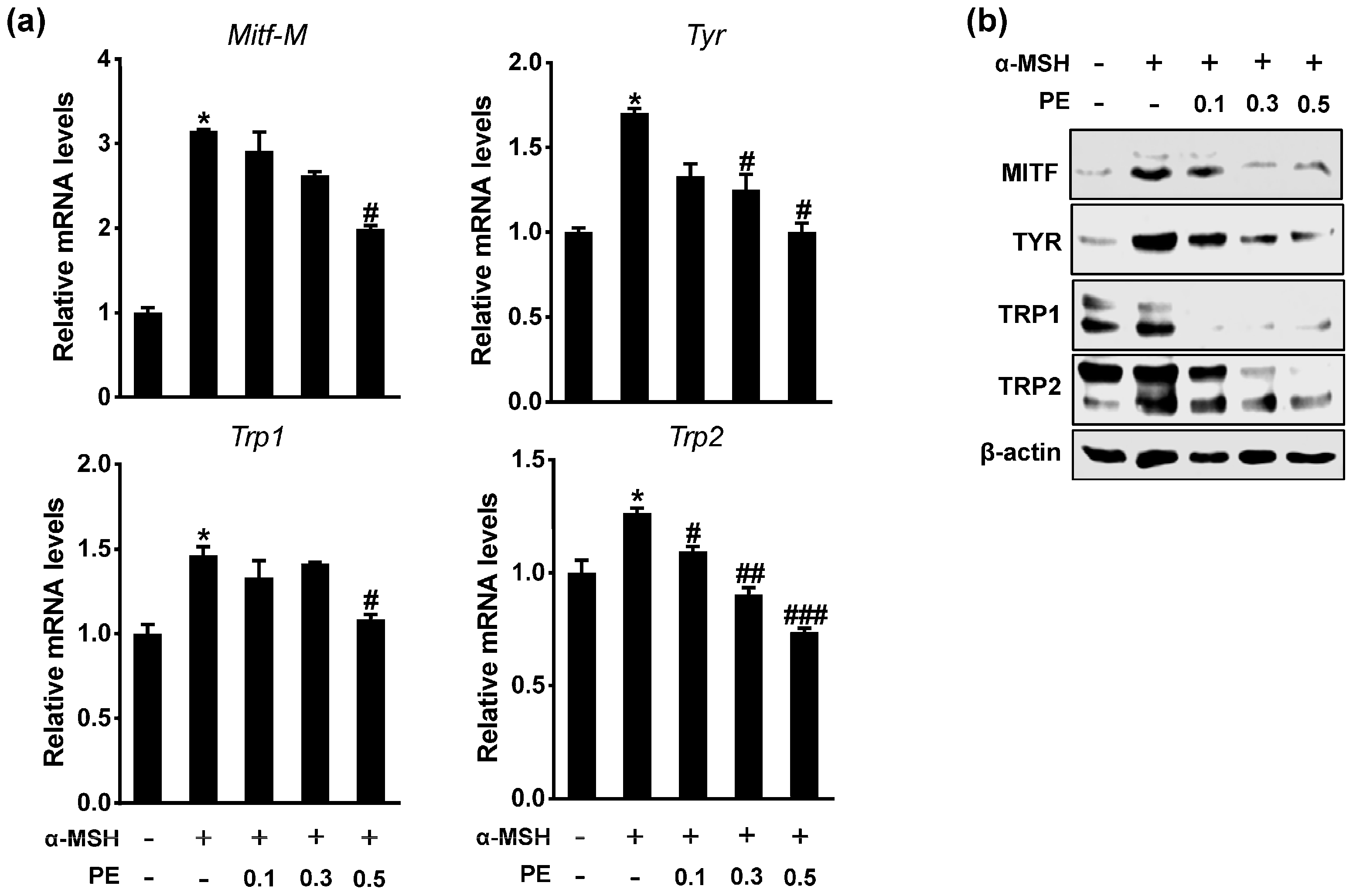
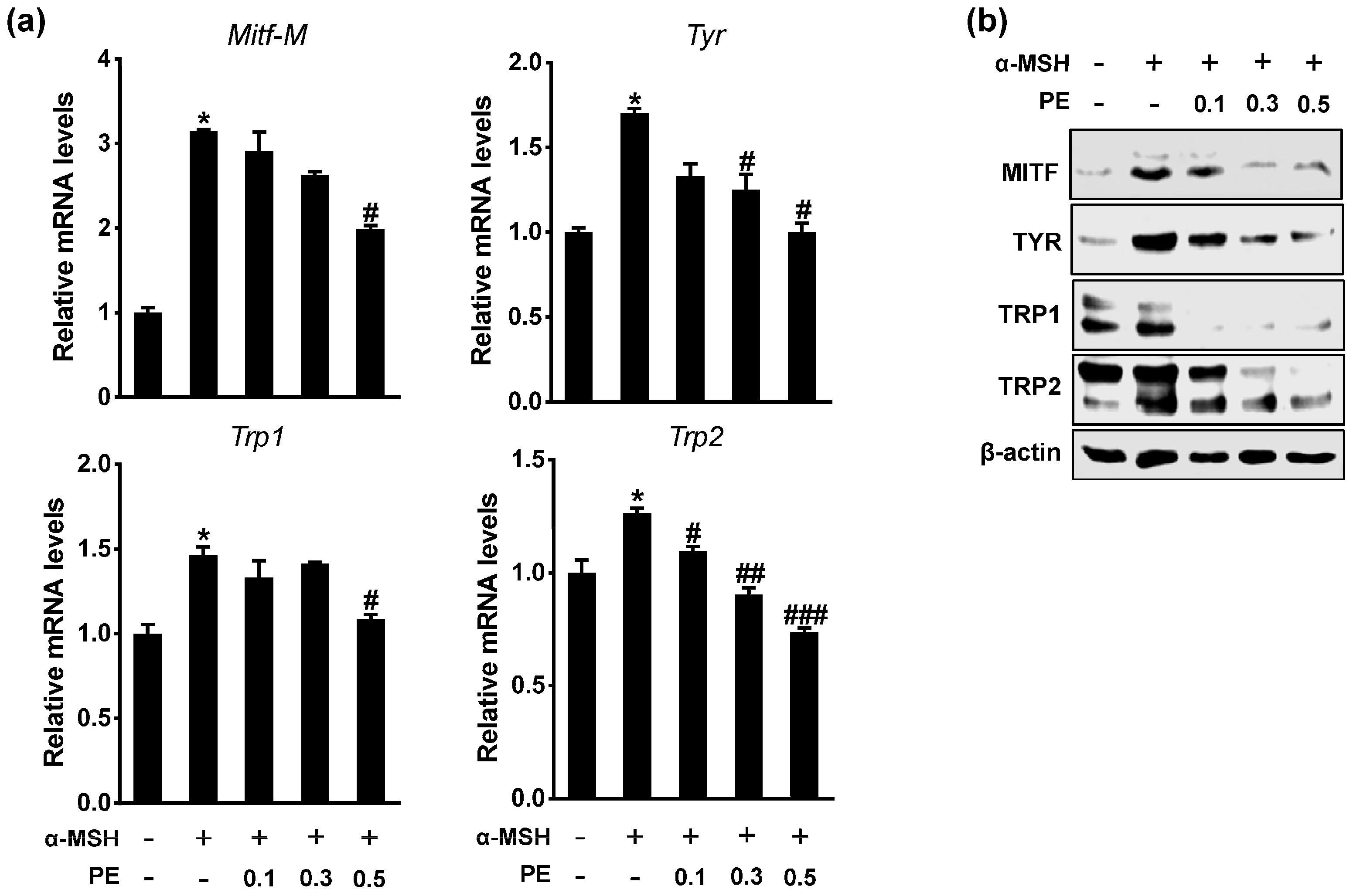
2.2. PE Inhibits Melanogenic Gene Expression
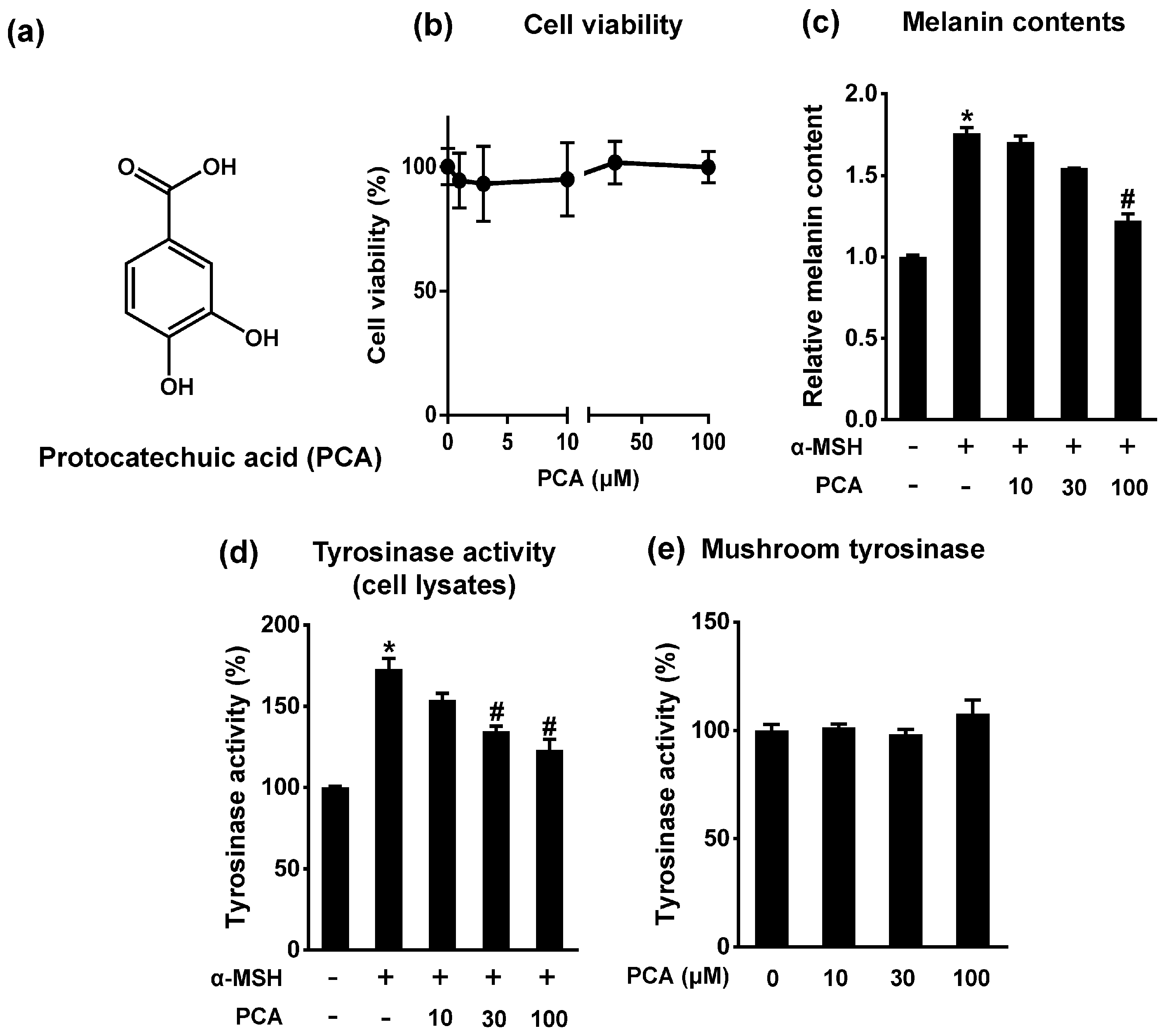
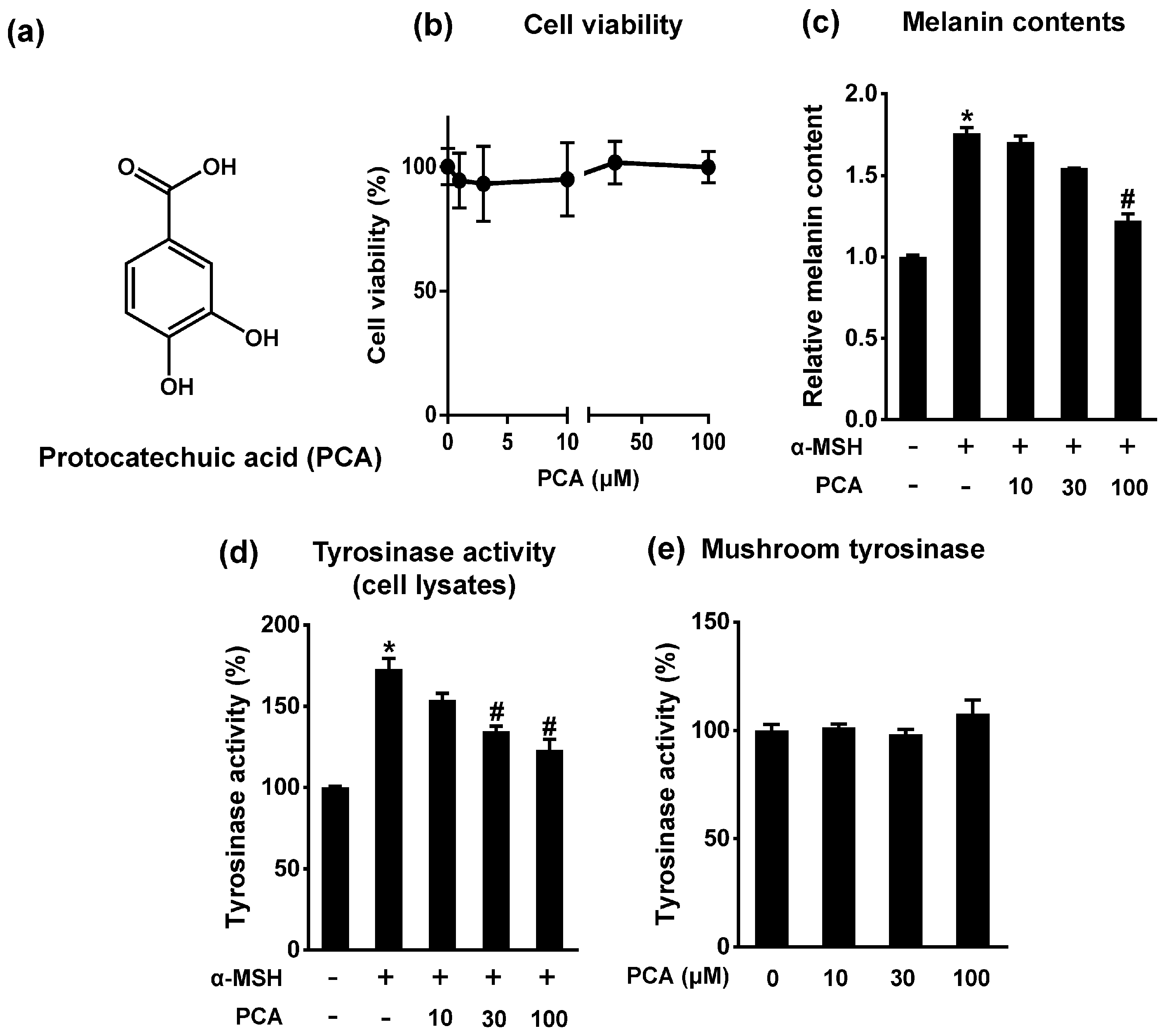
2.3. Protocatechuic Acid Inhibits Melanogenesis
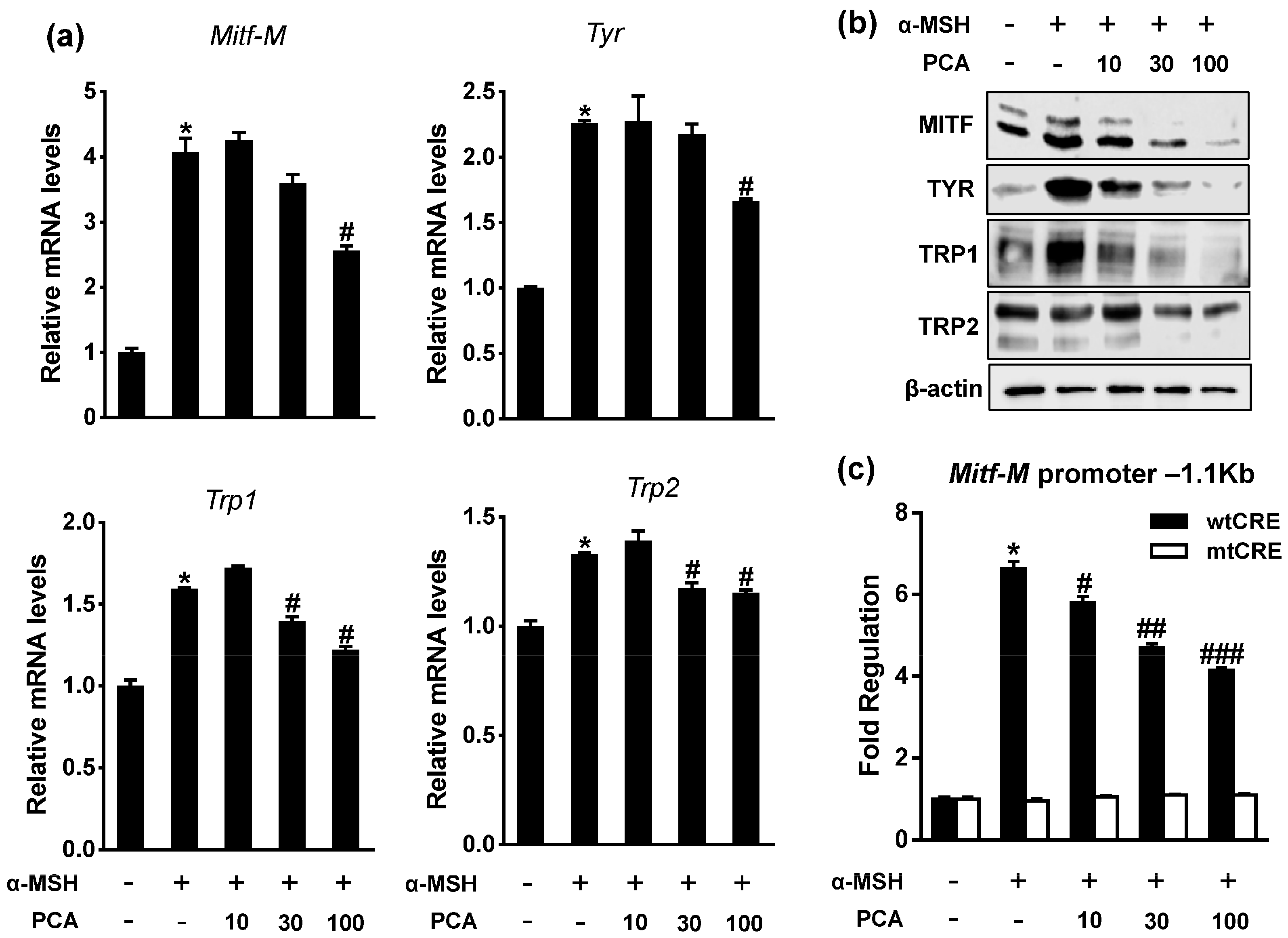
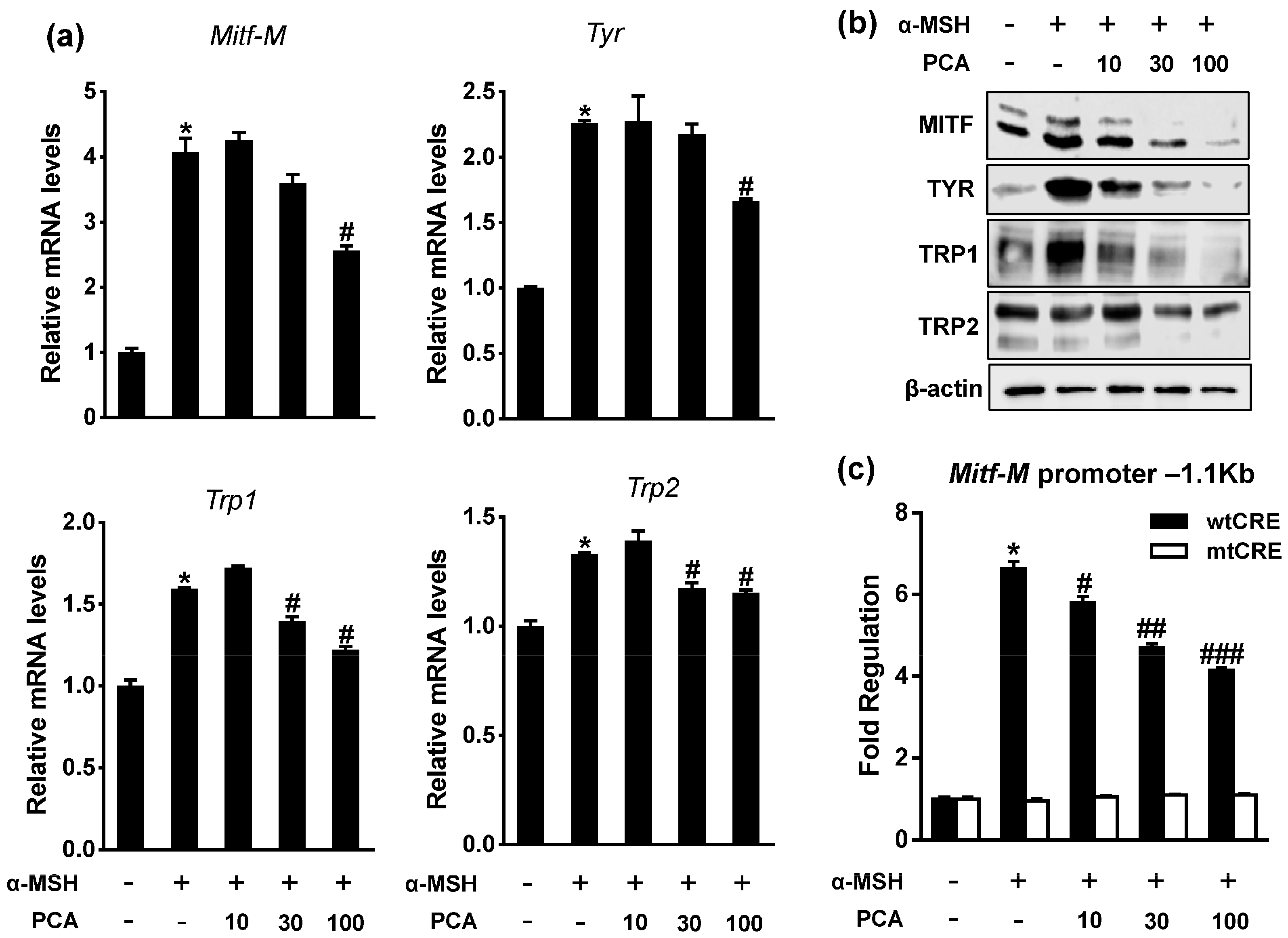
2.4. Protocatechuic Acid (PCA) Inhibits Melanogenic Genes through the Downregulation of Microphthalmia-Associated Transcription Factor (MITF) Transcription
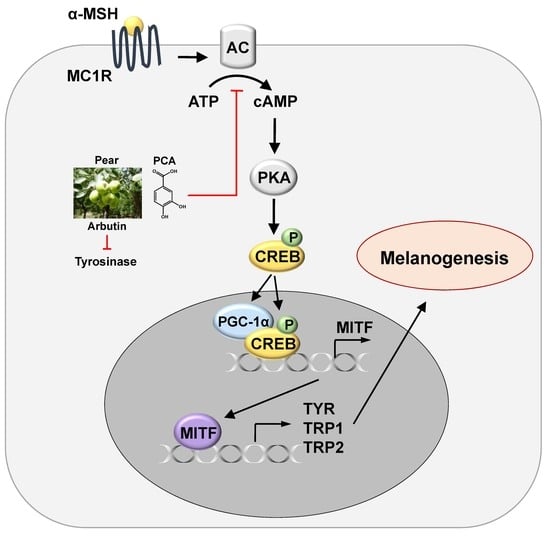
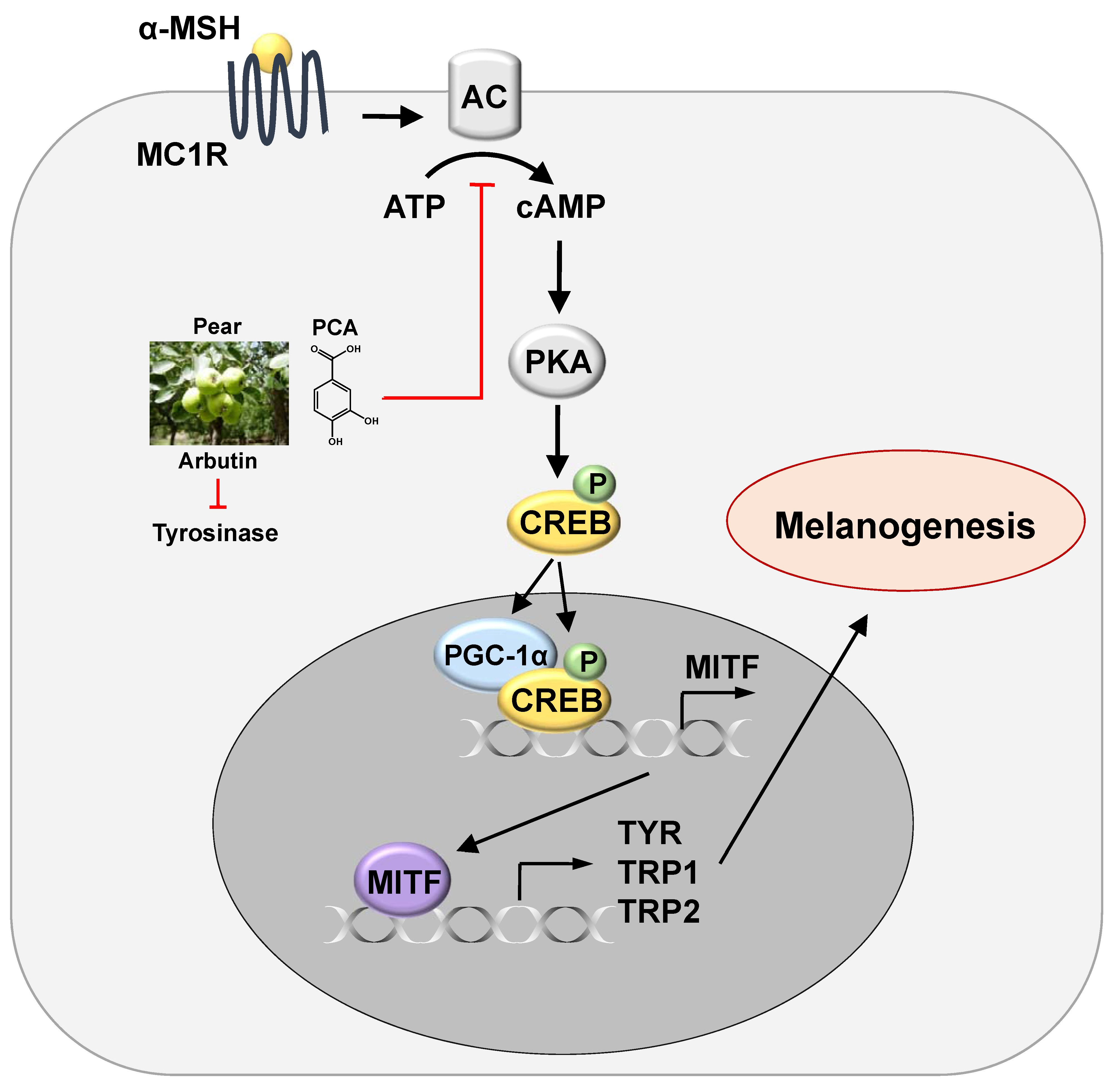
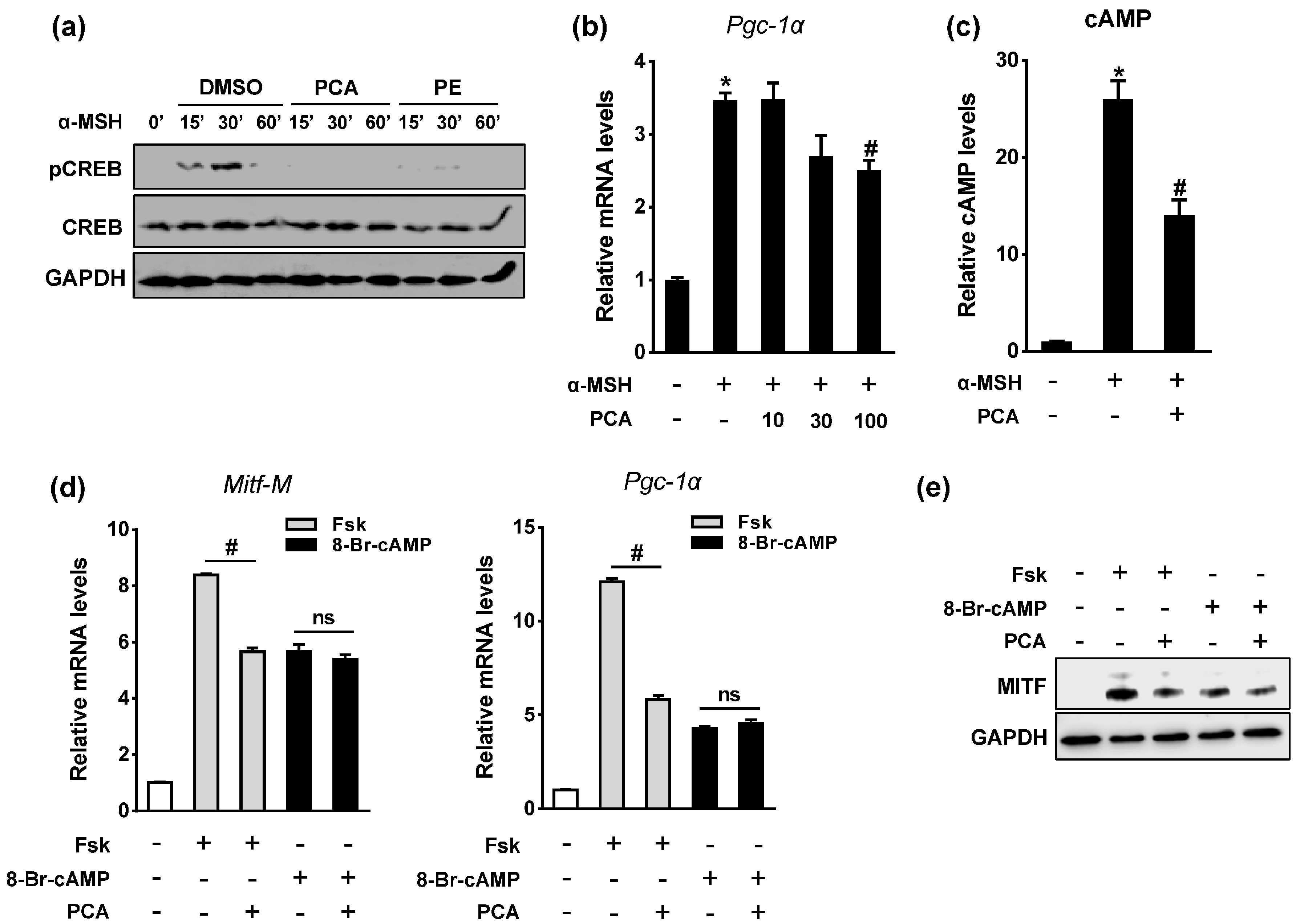
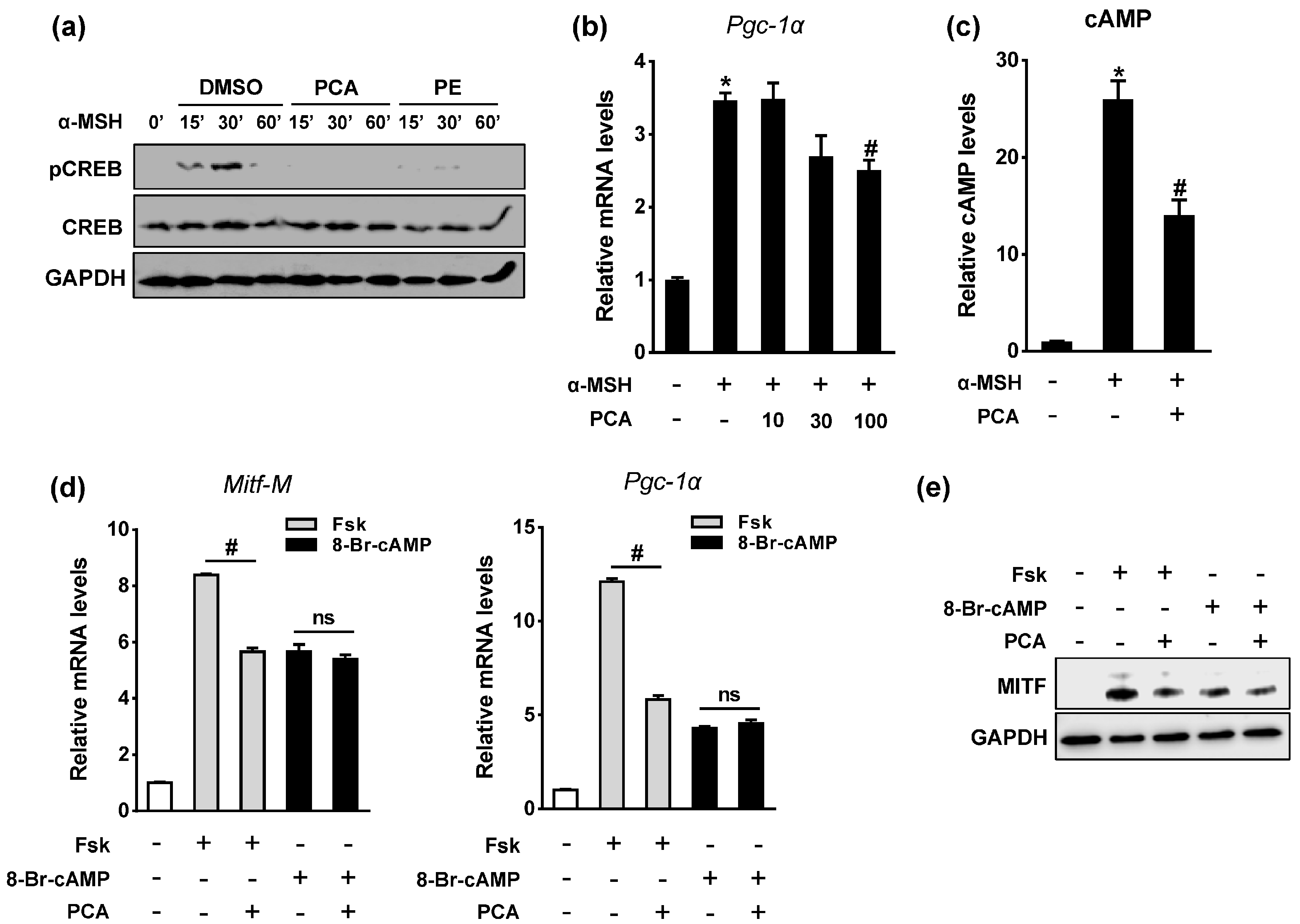
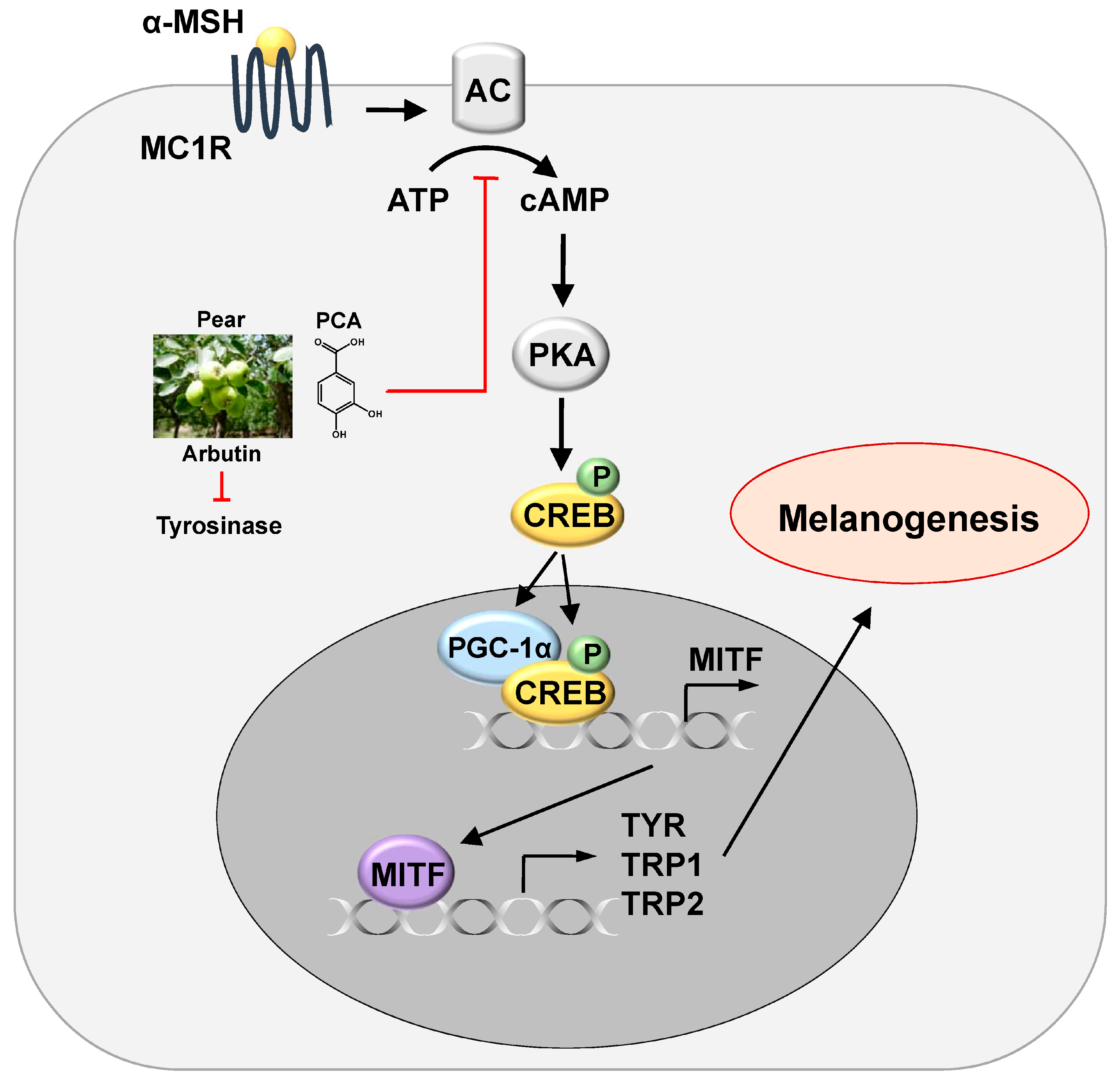
2.5. PCA Downregulates Cyclic Adenosine Monophosphate (cAMP)/cAMP-Responsive Element Binding Protein (CREB) Signaling Pathway
3. Materials and Methods
3.1. Materials and Cell Culture
3.2. Sample Preparation
3.3. Determination of Arbutin and PCA Contents in Immature Pear Fruit
3.4. Cell Viability
3.5. Determination of Melanin Content
3.6. Measurement of Tyrosinase Activity
3.7. RNA Analysis
3.8. Immunoblotting
3.9. Luciferase Reporter Assay
3.10. cAMP Determination
3.11. Statistical Analysis
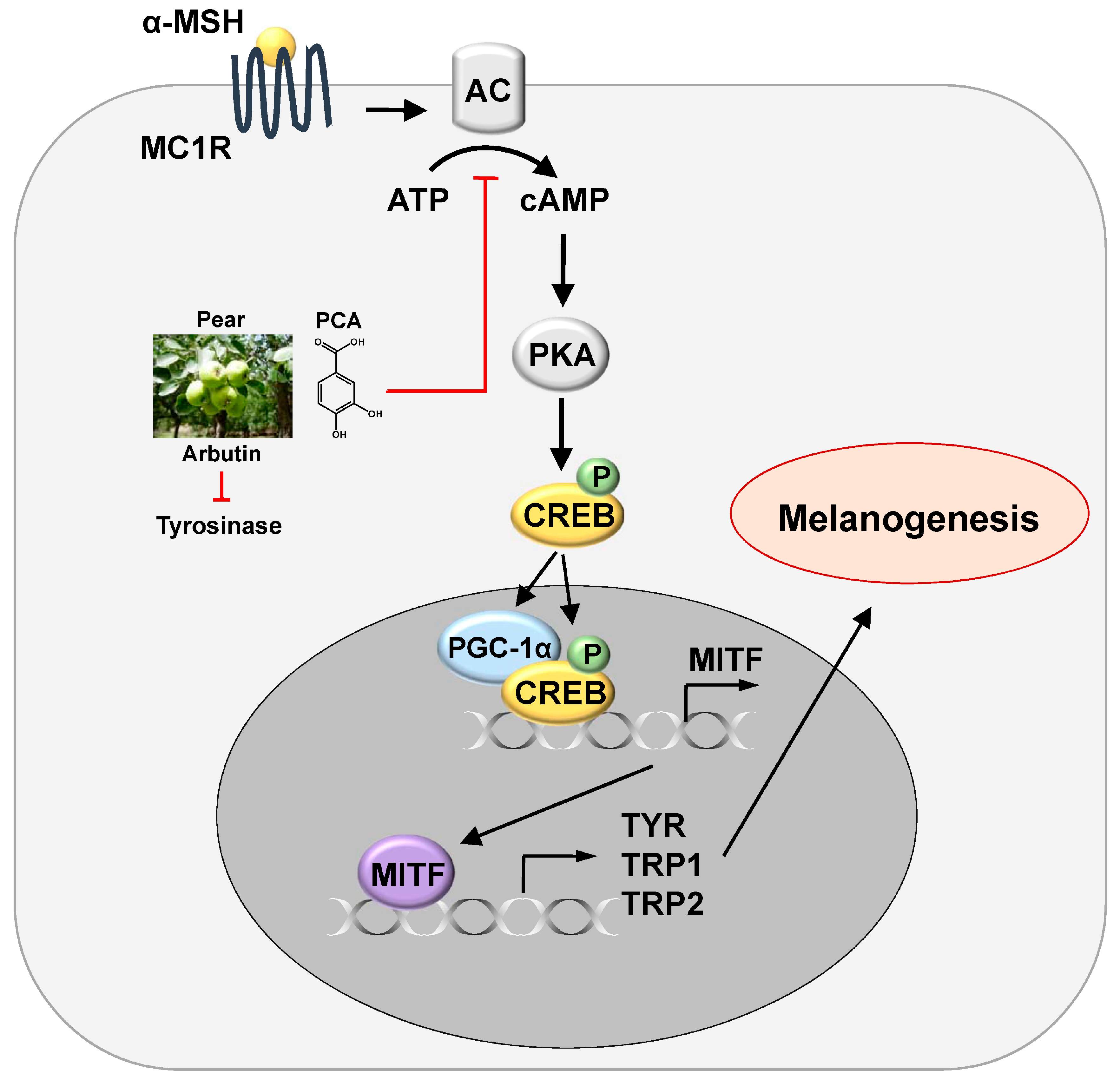
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jablonski, N.G. The evolution of human skin and skin color. Annu. Rev. Anthropol. 2004, 33, 585–623. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Leyden, J.J.; Shergill, B.; Micali, G.; Downie, J.; Wallo, W. Natural options for the management of hyperpigmentation. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling pathways in melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed]
- Solano, F.; Briganti, S.; Picardo, M.; Ghanem, G. Hypopigmenting agents: An updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006, 19, 550–571. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. Biofactors 2009, 35, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Toriyama, M. Depigmenting effect of catechins. Molecules 2009, 14, 4425–4432. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Nakamura, K.; Ma, L.; Li, J.Z.; Kayahara, H. Analyses of arbutin and chlorogenic acid, the major phenolic constituents in Oriental pear. J. Agric. Food Chem. 2005, 53, 3882–3887. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cho, J.Y.; Lee, H.J.; Park, K.Y.; Ma, Y.K.; Lee, S.H.; Cho, J.A.; Kim, W.S.; Park, K.H.; Moon, J.H. Isolation and identification of phenolic compounds from an Asian pear (Pyrus pyrifolia Nakai) fruit peel. Food Sci. Biotechnol. 2011, 20, 1539–1545. [Google Scholar] [CrossRef]
- Lee, K.H.; Cho, J.Y.; Lee, H.J.; Ma, Y.K.; Kwon, J.; Park, S.H.; Lee, S.H.; Cho, J.A.; Kim, W.S.; Park, K.H.; et al. Hydroxycinnamoylmalic Acids and Their Methyl Esters from Pear (Pyrus pyrifolia Nakai) Fruit Peel. J. Agric. Food Chem. 2011, 59, 10124–10128. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.G.; Cho, J.Y.; Kim, C.M.; Lee, S.H.; Kim, W.S.; Jeon, T.I.; Park, K.H.; Moon, J.H. Coumaroyl quinic acid derivatives and flavonoids from immature pear (Pyrus pyrifolia Nakai) fruit. Food Sci. Biotechnol. 2013, 22, 803–810. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Park, K.Y.; Lee, K.H.; Lee, H.J.; Lee, S.H.; Cho, J.A.; Kim, W.S.; Shin, S.C.; Park, K.H.; Moon, J.H. Recovery of arbutin in high purity from fruit peels of pear (Pyrus pyrifolia Nakai). Food Sci. Biotechnol. 2011, 20, 801–807. [Google Scholar] [CrossRef]
- Widlund, H.R.; Fisher, D.E. Microphthalamia-associated transcription factor: A critical regulator of pigment cell development and survival. Oncogene 2003, 22, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, H.K.; Lee, K.J.; Jeon, H.W.; Cui, S.; Lee, Y.M.; Moon, B.J.; Kim, Y.H.; Lee, Y.S. Inhibitory effect of glyceollin isolated from soybean against melanogenesis in B16 melanoma cells. BMB Rep. 2010, 43, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Jo, Y.K.; Park, S.J.; Chang, H.; Shin, J.H.; Choi, E.S.; Kim, J.B.; Seok, S.H.; Kim, J.S.; Oh, J.S.; et al. ARP101 inhibits α-MSH-stimulated melanogenesis by regulation of autophagy in melanocytes. FEBS Lett. 2013, 587, 3955–3960. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, C.M.; Kim, Y.C. Anti-melanogenic effect of oenothera laciniata methanol extract in melan-a cells. Toxicol Res. 2017, 33, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Ding, H.Y.; Lin, R.J.; Liang, J.Y.; Liang, C.H. Inhibition of melanogenesis and oxidation by protocatechuic acid from Origanum vulgare (oregano). J. Nat. Prod. 2010, 73, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J. Cell Biol. 1998, 142, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Haq, R.; Zhang, M.; Liu, L.; Rowe, G.C.; Jiang, A.; Koulisis, N.; Farrel, C.; Amos, C.I.; Wei, Q.; et al. PGC-1 coactivators regulate MITF and the tanning response. Mol. Cell 2013, 49, 145–157. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truong, X.T.; Park, S.-H.; Lee, Y.-G.; Jeong, H.Y.; Moon, J.-H.; Jeon, T.-I. Protocatechuic Acid from Pear Inhibits Melanogenesis in Melanoma Cells. Int. J. Mol. Sci. 2017, 18, 1809. https://doi.org/10.3390/ijms18081809
Truong XT, Park S-H, Lee Y-G, Jeong HY, Moon J-H, Jeon T-I. Protocatechuic Acid from Pear Inhibits Melanogenesis in Melanoma Cells. International Journal of Molecular Sciences. 2017; 18(8):1809. https://doi.org/10.3390/ijms18081809
Chicago/Turabian StyleTruong, Xuan T., Seo-Hee Park, Yu-Geon Lee, Hang Yeon Jeong, Jae-Hak Moon, and Tae-Il Jeon. 2017. "Protocatechuic Acid from Pear Inhibits Melanogenesis in Melanoma Cells" International Journal of Molecular Sciences 18, no. 8: 1809. https://doi.org/10.3390/ijms18081809
APA StyleTruong, X. T., Park, S.-H., Lee, Y.-G., Jeong, H. Y., Moon, J.-H., & Jeon, T.-I. (2017). Protocatechuic Acid from Pear Inhibits Melanogenesis in Melanoma Cells. International Journal of Molecular Sciences, 18(8), 1809. https://doi.org/10.3390/ijms18081809