FANCD2 and DNA Damage



{kind=link}
Abstract
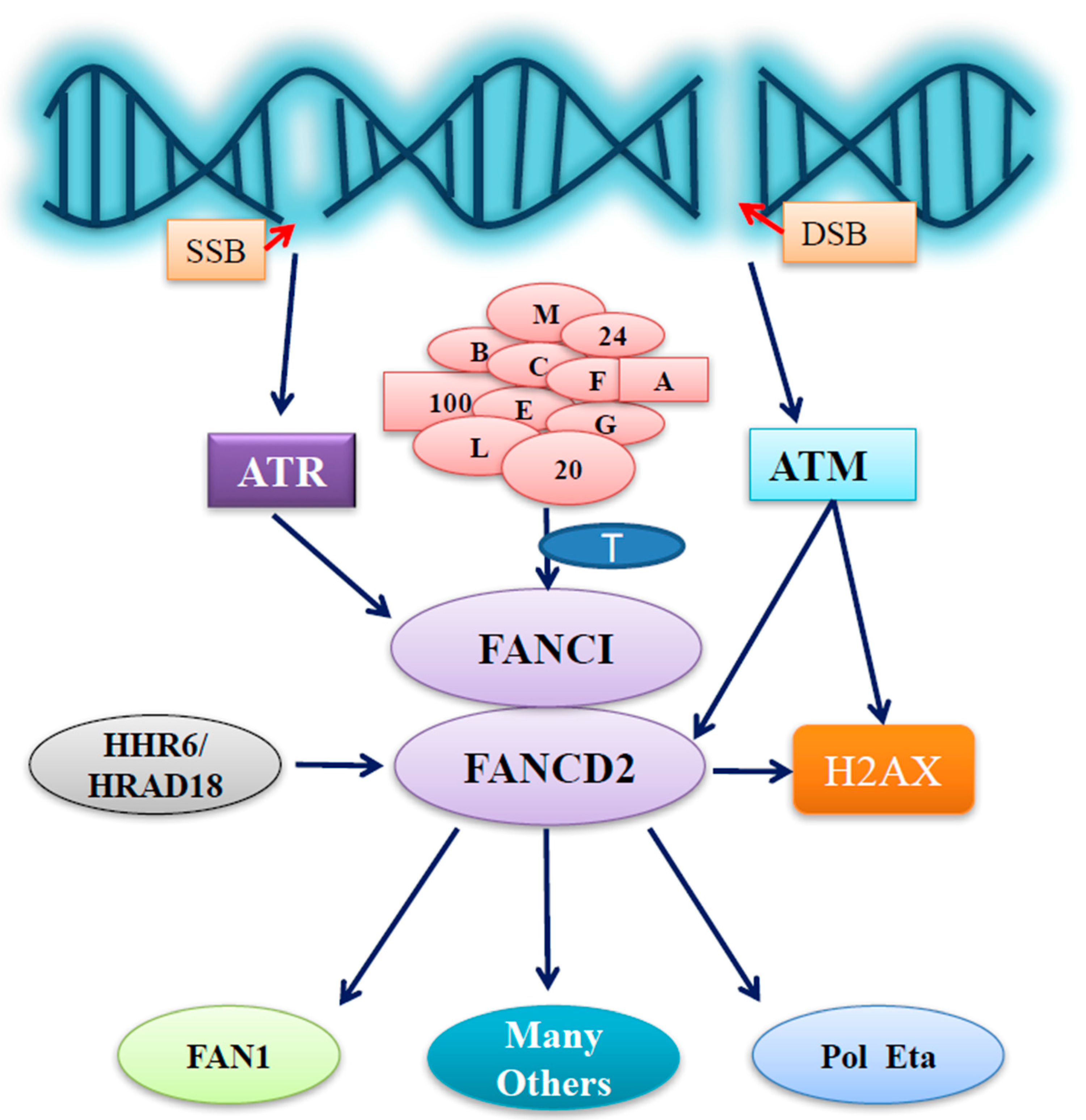
1. Introduction of the Fanconi Anemia Pathway
2. The Importance of Fanconi Anemia Group D2 Protein (FANCD2)
3. FANCD2 under Stressed Conditions
3.1. Cross Talking with Human Homologs of Yeast Rad6 (HHR6) Signaling
3.2. Coupling with Ataxia Telangiectasia and Rad3-Related Protein (ATR)/Ataxia Telangiectasia Mutated (ATM) Signaling
3.3. Cooperating with Other Signaling Pathways
4. FANCD2 in Non-Stressed Condition
5. Conclusive Remarks
Acknowledgments
Conflicts of Interest
References
- Alter, B.P. Cancer in Fanconi anemia, 1927–2001. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. The Fanconi road to cancer. Genes Dev. 2003, 17, 1933–1936. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G.C., Jr. Genetic basis of Fanconi anemia. Curr. Opin. Hematol. 2003, 10, 68–76. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Greenhalf, B.; Ellis, I.; Sina-Frey, M.; Rieder, H.; Korte, B.; Gerdes, B.; Kress, R.; Ziegler, A.; Raeburn, J.A.; et al. BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst. 2003, 95, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; D’Andrea, A.D. Molecular pathogenesis of Fanconi anemia: Recent progress. Blood 2006, 107, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.S.D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Reund, M.; Wurm, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2006, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Roest, H.P.; Baarends, W.M.; de Wit, J.; van Klaveren, J.W.; Wassenaar, E.; Hoogerbrugge, J.W.; van Cappellen, W.A.; Hoeijmakers, J.H.; Grootegoed, J.A. The ubiquitin-conjugating DNA repair enzyme HR6A is a maternal factor essential for early embryonic development in mice. Mol. Cell. Biol. 2004, 24, 5485–5495. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2006, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi aneamia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Genet. 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.; Zhang, J.; Panneerselvam, J.; Fei, P. Advances in the understanding of Fanconi anemia tumor suppressor pathway. Cancer Biol. Ther. 2013, 14, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surralles, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Knies, K.; Inano, S.; Ramirez, M.J.; Ishiai, M.; Surralles, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Sala-Trepat, M.; Rouillard, D.; Escarceller, M.; Laquerbe, A.; Moustacchi, E.; Papadopoulo, D. Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp. Cell Res. 2000, 260, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M. FANCL, as in ligase. Nat. Genet. 2003, 35, 113–114. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; de Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.S.S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2006, 39, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; de Winter, J.P.; Medhurst, A.L.; Wallisch, M.; Waisfisz, Q.; van de Vrugt, H.J.; Oostra, A.B.; Yan, Z.; Ling, C.; Bishop, C.E.; et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003, 35, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Dudimah, F.D.; Zhang, J.; Pickering, A.; Paneerselvam, J.; Palrasu, M.; Wang, H.; Fei, P. Recruitment of DNA polymerase eta by FANCD2 in the early response to DNA damage. Cell Cycle 2013, 12, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; Yin, J.; Wang, W. New advances in the DNA damage response network of Fanconi anemia and BRCA proteins. FAAP95 replaces BRCA2 as the true FANCB protein. Cell Cycle 2005, 4, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Masson, J.Y.; Shah, R.; O’Regan, P.; West, S.C. RAD51C is required for Holliday junction processing in mammalian cells. Science 2004, 303, 243–246. [Google Scholar] [CrossRef] [PubMed]
- McHugh, P.J.; Ward, T.A.; Chovanec, M. A prototypical Fanconi anemia pathway in lower eukaryotes? Cell Cycle 2012, 11, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Alpi, A.F.; Patel, K.J. Monoubiquitylation in the Fanconi anemia DNA damage response pathway. DNA Repair (Amst.) 2009, 8, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; Levitus, M.; Xue, Y.; Medhurst, A.L.; Zwaan, M.; Ling, C.; Rooimans, M.A.; Bier, P.; Hoatlin, M.; Pals, G.; et al. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 2004, 36, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Ameziane, N.; May, P.; Haitjema, A.; van de Vrugt, H.J.; van Rossum-Fikkert, S.E.; Ristic, D.; Williams, G.J.; Balk, J.; Rockx, D.; Li, H.; et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat. Commun. 2015, 6, 8829. [Google Scholar] [CrossRef] [PubMed]
- Jayabal, P.; Ma, C.; Nepal, M.; Shen, Y.; Che, R.; Turkson, J.; Fei, P. Involvement of FANCD2 in Energy Metabolism via ATP5alpha. Sci. Rep. 2017, 7, 4921. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Lui, V.W.; Grandis, J.R.; Wells, S.I. The Fanconi anemia pathway: Repairing the link between DNA damage and squamous cell carcinoma. Mutat. Res. 2013, 743, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Alpi, A.F.; Pace, P.E.; Babu, M.M.; Patel, K.J. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 2008, 32, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M.; D’Andrea, A. Fanconi anemia and DNA repair. Hum. Mol. Genet. 2001, 10, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Murina, O.; Von Aesch, C.; Karakus, U.; Ferretti, L.P.; Bolck, H.A.; Hanggi, K.; Sartori, A.A. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014, 7, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated FANCD2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 351, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Borel, V.; Adelman, C.A.; Schindler, D.; Boulton, S.J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015, 29, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Daschkey, S.; Bienemann, K.; Schuster, V.; Kreth, H.W.; Linka, R.M.; Honscheid, A.; Fritz, G.; Johannes, C.; Fleckenstein, B.; Kempkes, B.; et al. Fatal Lymphoproliferative Disease in Two Siblings Lacking Functional FAAP24. J. Clin. Immunol. 2016, 36, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, X.; Akhter, S.; Georgescu, M.M.; Legerski, R.J. FANCI is a negative regulator of Akt activation. Cell Cycle 2016, 15, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, D.; Wang, H.; Lin, C.J.; Fei, P. FANCD2 monoubiquitination provides a link between the HHR6 and FA-BRCA pathways. Cell Cycle 2008, 7, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Wang, H.; Zhang, J.; Datta, S.; Fei, P. Convergence of Rad6/Rad18 and Fanconi anemia tumor suppressor pathways upon DNA damage. PLoS ONE 2010, 5, e13313. [Google Scholar] [CrossRef] [PubMed]
- Song, I.Y.; Palle, K.; Gurkar, A.; Tateishi, S.; Kupfer, G.M.; Vaziri, C. Rad18-mediated trans-lesion synthesis of bulky DNA adducts is coupled to activation of the fanconi anemia DNA repair pathway. J. Biol. Chem. 2010, 285, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Huntoon, C.J.; Karnitz, L.M. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol. 2010, 191, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Pickering, A.; Panneerselvam, J.; Zhang, J.; Zheng, J.; Zhang, Y.; Fei, P. In vitro FANCD2 monoubiquitination by HHR6 and hRad18. Cell Cycle 2013, 12, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.; Xu, G.; Persky, N.S.; Smogorzewska, A.; Rudge, D.G.; Buzovetsky, O.; Elledge, S.J.; Pavletich, N.P. Structure of the FANCI-FANCD2 complex: Insights into the Fanconi anemia DNA repair pathway. Science 2011, 333, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Taniguchi, T.; Ranganathan, V.; New, H.V.; Moreau, L.A.; Stotsky, M.; Mathew, C.G.; Kastan, M.B.; Weaver, D.T.; D’Andrea, A.D. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat. Cell Biol. 2002, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., 3rd; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Li, Z.; Lopez-Martinez, D.; Nicholson, W.V.; Venien-Bryan, C.; Cohn, M.A. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat. Commun. 2016, 7, 12124. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Wang, H.; Zhang, J.; Che, R.; Yu, H.; Fei, P. BLM promotes the activation of Fanconi Anemia signaling pathway. Oncotarget 2016, 7, 32351–32361. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Su, F.; Mukherjee, S.; Mori, E.; Hu, B.; Asaithamby, A. FANCD2 influences replication fork processes and genome stability in response to clustered DSBs. Cell Cycle 2015, 14, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiacovo, M.P.; et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lachaud, C.; Slean, M.; Marchesi, F.; Lock, C.; Odell, E.; Castor, D.; Toth, R.; Rouse, J. Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev. 2016, 30, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Pizzolato, J.; Mukherjee, S.; Scharer, O.D.; Jiricny, J. FANCD2-associated nuclease 1, but not exonuclease 1 or flap endonuclease 1, is able to unhook DNA interstrand cross-links in vitro. J. Biol. Chem. 2015, 290, 22602–22611. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ghosal, G.; Yuan, J.; Chen, J.; Huang, J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010, 329, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 2013, 73, 3393–3401. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Gunawardena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Wells, S.I. Coordinate regulation of Fanconi anemia gene expression occurs through the Rb/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Houghton, P.J. Targeting FANCD2 for therapy sensitization. Oncotarget 2014, 5, 3426–3427. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Pickering, A.; Han, B.; Li, L.; Zheng, J.; Zhang, J.; Zhang, Y.; Fei, P. Basal level of FANCD2 monoubiquitination is required for the maintenance of a sufficient number of licensed-replication origins to fire at a normal rate. Oncotarget 2014, 5, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Huang, J.; Yan, Z.; Li, Y.; Ohzeki, M.; Ishiai, M.; Xu, D.; Takata, M.; Seidman, M.; Wang, W. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016, 2, 16047. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Xie, G.; Che, R.; Su, M.; Zhang, J.; Jia, W.; Fei, P. Distinct Metabolic Signature of Human Bladder Cancer Cells Carrying an Impaired Fanconi Anemia Tumor-Suppressor Signaling Pathway. J. Proteom. Res. 2016, 15, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Du, W.; Wilson, A.F.; Namekawa, S.H.; Andreassen, P.R.; Meetei, A.R.; Pang, Q. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci. Rep. 2017, 7, 45626. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepal, M.; Che, R.; Ma, C.; Zhang, J.; Fei, P. FANCD2 and DNA Damage. Int. J. Mol. Sci. 2017, 18, 1804. https://doi.org/10.3390/ijms18081804
Nepal M, Che R, Ma C, Zhang J, Fei P. FANCD2 and DNA Damage. International Journal of Molecular Sciences. 2017; 18(8):1804. https://doi.org/10.3390/ijms18081804
Chicago/Turabian StyleNepal, Manoj, Raymond Che, Chi Ma, Jun Zhang, and Peiwen Fei. 2017. "FANCD2 and DNA Damage" International Journal of Molecular Sciences 18, no. 8: 1804. https://doi.org/10.3390/ijms18081804
APA StyleNepal, M., Che, R., Ma, C., Zhang, J., & Fei, P. (2017). FANCD2 and DNA Damage. International Journal of Molecular Sciences, 18(8), 1804. https://doi.org/10.3390/ijms18081804