A Critical Review on the Effect of Docosahexaenoic Acid (DHA) on Cancer Cell Cycle Progression


Abstract
1. Introduction
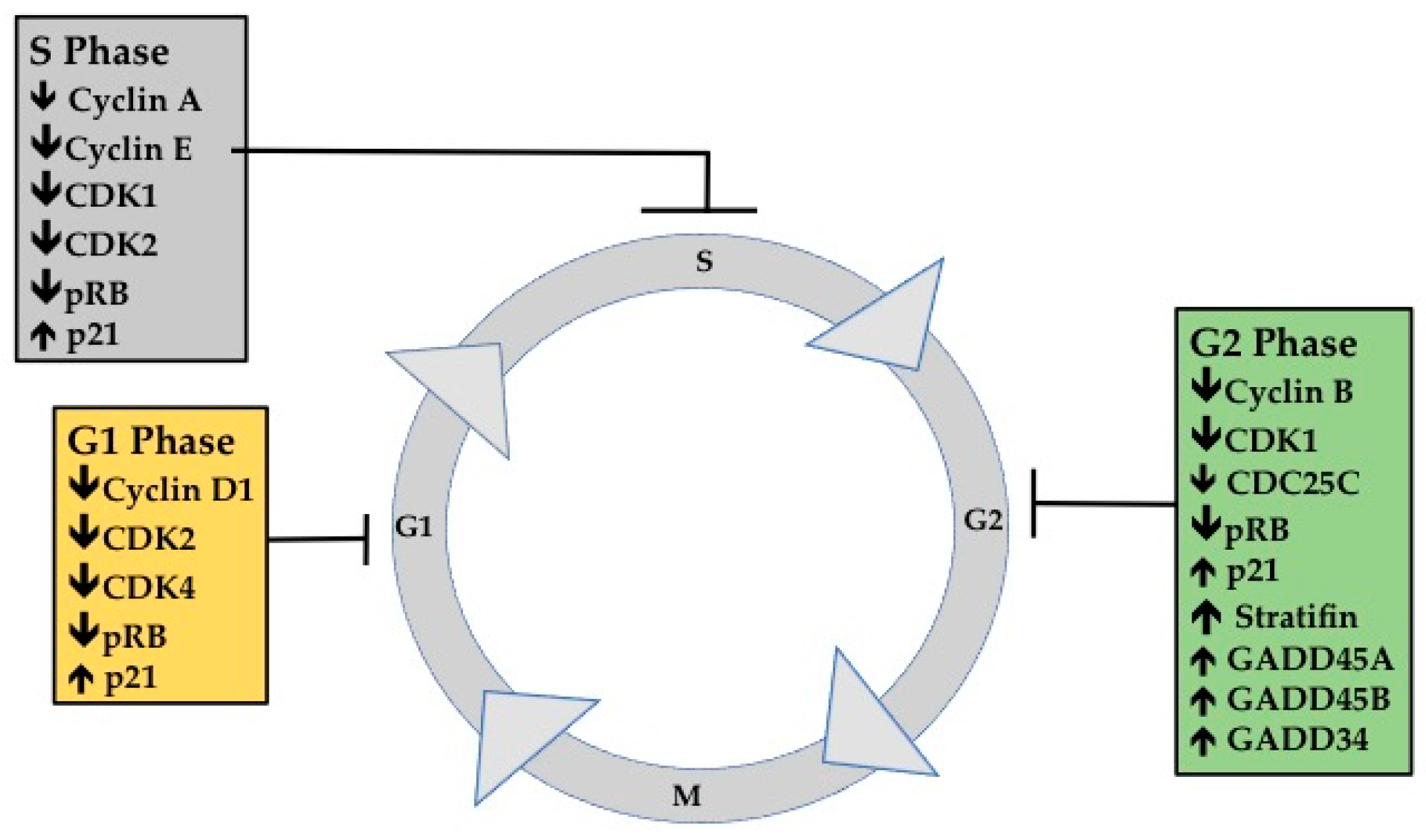
2. Cell Cycle Progression in Cancer
3. Effect of Docosahexaenoic Acid on Cell Cycle Progression
3.1. G1 Phase Analysis
3.2. G1 Phase Cellular Markers
3.3. S Phase Analysis
3.4. S Phase Cellular Markers
3.5. G2M Phase Analysis
3.6. G2M Phase Cellular Markers
3.7. Multi Cell Cycle Phase Analysis
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil |
| AA | Arachidonic acid |
| ALA | Alpha linolenic acid |
| AURK | Aurora kinase |
| BHT | Butylated hydroxyanisole |
| BSA | Bovine serum albumin |
| CDHA | Conjugated DHA |
| CDK | Cyclin dependent kinase |
| DHA | Docosahexaenoic acid |
| DOX | Doxorubicin |
| ER | Estrogen-receptor |
| EPA | Eicosapentaenoic acid |
| FO | Fish oil |
| G1 | Gap 1 |
| G2 | Gap 2 |
| HER2 | Human epidermal growth factor receptor 2/erbB-2 receptor tyrosine-protein kinase ERBB2 |
| LA | Linoleic acid |
| LCPUFA | Long chain polyunsaturated fatty acid |
| METC | Mitochondrial electron transfer chain |
| MMP | Mitochondrial membrane potential |
| mRNA | Messenger RNA |
| PLK1 | Polo-like kinase 1/Serine/threonine-protein kinase |
| PPAR | Peroxisome proliferator-activated receptors |
| PR | Progesterone-receptor |
| pRB | Retinoblastoma protein |
| RTPCR | Real time quantitative PCR |
| TXT | Docetaxel |
References
- World Health Organization: Cancer. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 25 July 2017).
- Berquin, I.; Edwards, I.; Chen, Y. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008, 269, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Docasahexaenoic acid. Ann. Nutr. Metab. 2016, 69, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, D.W. The role of n-3 polyunsaturated fatty acids in the prevention and treatment of breast cancer. Nutrients 2014, 6, 5184–5223. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W.S.; Shaikh, S.R.; Zerouga, M.; Siddiqui, R.; Wassall, S.R. Docosahexaenoic acid affects cell signalling by altering lipid rafts. Reprod. Nutr. Dev. 2005, 45, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.J.; Schemmel, R.A.; Dugan, L.J.; Gray, J.I.; Welsch, C.W. Dietary fish oil inhibits human breast carcinoma growth: A function of increased lipid peroxidation. Lipids 1993, 28, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.; Chajes, V.; Cognault, S.; Lhuillery, C.; Bougnoux, P. Enhancement of doxorubicin cytotoxicity by polyunsaturated fatty acids in the human breast tumor cell line MDA-MB-231: Relationship to lipid peroxidation. Int. J. Cancer 1998, 75, 578–583. [Google Scholar] [CrossRef]
- D’Eliseo, D.; Velotti, F. Omega-3 fatty acids and cancer cell cytotoxicity: Implications for mult-targeted cancer therapy. J. Clin. Med. 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Yamagami, T.; Porada, C.D.; Pardini, R.S.; Zanjani, E.D.; Almeida-Porada, G. Docosahexaenoic acid induces dose dependent cell death in an early undifferentiated subtype of acute myeloid leukemia cell line. Cancer Biol. Ther. 2009, 8, 331–337. [Google Scholar] [CrossRef] [PubMed]
- So, W.W.; Liu, W.N.; Leung, K.N. Omega-3 polyunsaturated fatty acids trigger cell cycle arrest and induce apoptosis in human neuroblastoma LA-N-1 cells. Nutrients 2015, 7, 6956–6973. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Istfan, N.W. Docosahexaenoic acid, a major constituent of fish oil diets, prevents activation of cyclin-dependent kinases and S-phase entry by serum stimulation in HT-29 cells. Prostaglandins Leukot. Essent. Fat. Acids 2001, 64, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Wang, Q.; Zhao, J.; Dong, L.; Ge, Y.; Hou, L.; Liu, Y.; Zheng, Z. Docosahexaenoic acid inhibited the Wnt/β-catenin pathway and suppressed breast cancer cells in vitro and in vivo. J. Nut. Biochem. 2014, 25, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhu, S.; Wu, Y.; Song, C.; Wang, W.; Zhang, Y.; Chen, Y.-L.; He, Z. Omega-3 free fatty acids and all-trans retinoic acid synergistically induce growth inhibition of three subtypes of breast cancer cell lines. Sci. Rep. 2017, 7, 2929. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Nishimura, K.; Aires, V.; Yamashita, T.; Oaxaca-Castillo, D.; Kashiwagi, K.; Igarashi, K. Docosahexaenoic acid inhibits cancer cell growth via p27kip1, CDK2, ERK1/ERK2, and retinoblastoma phosphorylation. J. Lipid Res. 2006, 47, 2306–2313. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, T.; Capasso, A.; Tecce, M.F. Effect of docosahexaenoic acid on cell cycle pathways in breast cell lines with different transformation degree. J. Cell. Physiol. 2016, 231, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Liang, Q.; Zhao, Z.H.; Li, Y.F.; Wang, S.F. Synergistic anticancer properties of docosahexaenoic acid and 5-fluorouracil through interference with energy metabolism and cell cycle arrest in human gastric cancer cell line AGS cells. World J. Gastroenterol. 2016, 22, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.A.; Jenski, L.J.; Harvey, K.A.; Wiesehan, J.D.; Stillwell, W.; Zaloga, G.P. Cell-cycle arrest in jurkat leukaemic cells: A possible role for docosahexaenoic acid. Biochem. J. 2003, 371, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Albino, A.P.; Juan, G.; Traganos, F.; Reinhart, L.; Connolly, J.; Rose, D.P.; Darzynkiewicz, Z. Cell cycle arrest and apoptosis of melanoma cells by docosahexaenoic acid: Association with decreased PRB phosphorylation. Cancer Res. 2000, 60, 4139–4145. [Google Scholar] [PubMed]
- Lee, C.Y.; Sit, W.; Fan, S.; Man, K.; Jor, I.W.; Wong, L.L.; Wan, M.L.; Tan-Un, K.C.; Wan, J.M. The cell cycle effects of docosahexaenoic acid on human metastatic hepatocellular carcinoma proliferation. Int. J. Oncol. 2010, 36, 991–998. [Google Scholar] [PubMed]
- Dekoj, T.; Lee, S.; Desai, S.; Trevino, J.; Babcock, T.A.; Helton, W.S.; Espat, N.J. G2/M cell-cycle arrest and apoptosis by n-3 fatty acids in a pancreatic cancer model. J. Surg. Res. 2007, 139, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Barascu, A.; Besson, P.; Le, F.O.; Bougnoux, P.; Jourdan, M.L. Cdk1-cyclin B1 mediates the inhibition of proliferation induced by omega-3 fatty acids in MDA-MB-231 breast cancer cells. Int. J. Biochem. Cell Biol. 2006, 38, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Fahrmann, J.H.; Elaine, W. Omega 3 fatty acids increase the chemo-sensitivity of B-CLL-derived cell lines eheb and MEC-2 and of B-PLL-derived cell line JVM-2 to anti-cancer drugs doxorubicin, vincristine and fludarabine. Lipids Health Dis. 2013, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, I.A.; Brown, I.; Schofield, A.C.; Wahle, K.W.; Heys, S.D. Docosahexaenoic acid enhances the efficacy of docetaxel in prostate cancer cells by modulation of apoptosis: The role of genes associated with the NF-κB pathway. Prostate 2008, 68, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Stein, J. Effect of an omega-3 fatty acid containing lipid emulsion alone and in combination with 5-fluorouracil (5-FU) on growth of the colon cancer cell line Caco-2. Eur. J. Nutr. 2003, 42, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kolenic, N.; Pardini, R.S. Docosahexaenoic acid (DHA), a primary tumor suppressive omega-3 fatty acid, inhibits growth of colorectal cancer independent of p53 mutational status. Nutr. Cancer 2007, 58, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Slagsvold, J.E.; Pettersen, C.H.; Storvold, G.L.; Follestad, T.; Krokan, H.E.; Schonberg, S.A. DHA alters expression of target proteins of cancer therapy in chemotherapy resistant SW620 colon cancer cells. Nutr. Cancer 2010, 62, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Tsujita-Kyutoku, M.; Yuri, T.; Danbara, N.; Senzaki, H.; Kiyozuka, Y.; Uehara, N.; Takada, H.; Hada, T.; Miyazawa, T.; Ogawa, Y.; et al. Conjugated docosahexaenoic acid suppresses KPL-1 human breast cancer cell growth in vitro and in vivo: Potential mechanisms of action. Breast Cancer Res. 2004, 6, R291–R299. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, O.; Shapira, M.; Hershko, D.D. Differential effects of doxorubicin treatment on cell cycle arrest and SKP2 expression in breast cancer cells. Anticancer Drugs 2007, 18, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhu, Z.; McGinley, J.N.; El Bayoumy, K.; Manni, A.; Thompson, H.J. Identification of a molecular signature underlying inhibition of mammary carcinoma growth by dietary n-3 fatty acids. Cancer Res. 2012, 72, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Bieche, I.; Vacher, S.; Lallemand, F.; Tozlu-Kara, S.; Bennani, H.; Beuzelin, M.; Driouch, K.; Rouleau, E.; Lerebours, F.; Ripoche, H.; et al. Expression analysis of mitotic spindle checkpoint genes in breast carcinoma: Role of NDC80/HEC1 in early breast tumorigenicity, and a two-gene signature for aneuploidy. Mol. Cancer 2011, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Weiß, L.; Efferth, T. Polo-like kinase 1 as target for cancer therapy. Exp. Hematol. Oncol. 2012, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.T.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting mitosis in cancer: Emerging strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.; Wong, E.; Ooi, V. Docosahexaenoic acid modulates different genes in cell cycle and apoptosis to control growth of human leukemia HL-60 cells. Int. J. Oncol. 2004, 25, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Crnkovic, S.; Riederer, M.; Lechleitner, M.; Hallstrom, S.; Malli, R.; Graier, W.F.; Lindenmann, J.; Popper, H.; Olschewski, H.; Olschewski, A.; et al. Docosahexaenoic acid-induced unfolded protein response, cell cycle arrest, and apoptosis in vascular smooth muscle cells are triggered by Ca2+-dependent induction of oxidative stress. Free Radic. Biol. Med. 2012, 52, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E.; Daly, R.J.; Sutherland, R.L.; Musgrove, E.A. Cell cycle control in breast cancer cells. J. Cell. Biochem. 2006, 97, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Terano, T.; Tanaka, T.; Tamura, Y.; Kitagawa, M.; Higashi, H.; Saito, Y.; Hirai, A. Eicosapentaenoic acid and docosahexaenoic acid inhibit vascular smooth muscle cell proliferation by inhibiting phosphorylation of CDK2-cycline complex. Biochem. Biophys. Res. Commun. 1999, 254, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 mutation status in human cancer cell lines: A reassessment. Human Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.-H.; Byun, D.-S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the EGFR inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Cancer, COSMIC. Available online: http://cancer.Sanger.Ac.Uk/cell_lines (accessed on 25 July 2017).
- Tronstad, K.J.; Berge, K.; Flindt, E.N.; Kristiansen, K.; Berge, R.K. Optimization of methods and treatment conditions for studying effects of fatty acids on cell growth. Lipids 2001, 36, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Berquin, I.M.; Owens, R.T.; O’Flaherty, J.T.; Edwards, I.J. Peroxisome proliferator-activated receptor γ-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008, 68, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Blethrow, J.D.; Glavy, J.S.; Morgan, D.O.; Shokat, K.M. Covalent capture of kinase-specific phosphopeptides reveals CDK1-Cyclin B substrates. Proc. Nat. Acad. Sci. USA 2008, 105, 1442–1447. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell Cycle | Cancer Model | Cancer Cell Line | Treat | Cell Cycle Markers | Other Markers | Reference |
|---|---|---|---|---|---|---|
| G1 | Leukemia | KG1A | 150 μM | ↑ cells in G1 and ↓ in G2M | ↑ apoptosis; ↑ DNA fragmentation; NC BCL2, ↑ Bax expression | [9] |
| G1 | Neuroblastoma | LA-N-1; (HEK-293; WRL-68-control) | 0–70 μM | ↑ in cells in G1; ↓ expression of CDK2 and Cyclin E | ↑ apoptosis; ↑ PS extern.; ↓ MMP; ↓ BCL-XL and ↑ Bax, Casp-3 and -9; Casp 8 NC | [10] |
| G1 | Colorectal | HT-29 | 150 μM | ↑ in cells in G1; ↓ Cyclin D1, E and A activation; ↓ expression of Cyclin A and pRb; ↓ E2F-1 DNA binding activity | NA | [11] |
| G1 | Breast | 4T1 (Mouse); MCF-7 (Human) | 25–100 μM | ↑ cells in G1; ↓ β-catenin, c-myc, Cyclin D1 in 4T1 cells | ↑ apoptotic in 4T1 and MCF-7 cells | [12] |
| G1 | Breast | MDA-MB-231, MCF-7, SK-BR-3, HCC1806 | 80 μM | ↑ cells in G1; ↓ in p21 in MCF-7 and SK-BR-3, ↑ in HCC1086, NC in MDA-MB-231; NC in p27 or Cyclin D1 | ↑ apoptosis | [13] |
| G1 | Breast | FM3A (Mouse) | 10 μM | ↑ cells in G1; ↑ p27; ↓ MAPK expression; NC p27 mRNA; ↓ Cyclin E, pCDK2 expression; NC Cyclin D; ↓ pRB | NA | [14] |
| G1 | Breast | MCF-7, ZR-75-1, SK-BR-3, MCF-10A | 100 or 300 μM | ↑ in cells in G1; ↑ in sub G1; ↑ p21 (mRNA and protein) in MCF-10A; NC in G1; ↓ sub G1; ↓ p21 (mRNA) in MCF-7; ↑ in G2M; ↑ in G1; ↓ p21 (mRNA) SKBR3 | ↓ p-ERK ½ and STAT3 in SKBR3 and MCF-7 cells; ↑ p-ERK ½, STAT3; ↑ p53 all cell lines | [15] |
| G1 | Gastric | AGS | 7.5–45 μg/mL DHA; 1.5625–50 μg/mL 5-FU | ↑ cells in G1 with DHA or 5-FU alone; ↑ cells in G1 more in combination; ↓ in S-phase | ↓ in METC I, II, V expression | [16] |
| S | Leukemia | E6-1 | 0–30 μM | ↑ cells in S; ↓ Cdk2, pRb and Cyclin A expression; ↑ p21 | 4-fold ↑ ceramide formation; ↓ Casp-3 expression | [17] |
| S | Melanoma | SK-Mel-110 and SK-Mel-29 (control) | 0.5–5 μg/mL | Two fold ↑ SK-Mel-110 cells in S; ↓ pRb in SK-Mel-110; NC Cyclin D, E, p21, p27 | ↑ apoptotis in SK-Mel-110 | [18] |
| S | Liver | MHCC97L | 0–200 μM | ↑ in cells in sub G1; prolonged S phase; ↓ in Cyclin A, E and CDK2 | ↓ COX-2 mRNA; NC protein expression; ↓ Hsp27, GRP78, N-myc protein; ↑ SOD2 | [19] |
| G2M | Pancreatic | MIA PaCa-2 | 10–100 μM n-3 emulsion | ↑ in cells in G2; ↓ in G1, 13% ↑ in S-phase; large sub G1; ↓ Cdc2 (Cdk1) expression | ↑ in apoptotic cells; ↓ BCL-2 expression; ↑ PARP cleavage product | [20] |
| G2M | Breast | MDA-MB-231 | 30–100 μM DHA | ↑ cells in G2M; ↓ CDK1, Cyclin B1, Cyclin A, CDC25C, Cyclin B1p-Ser126 and NC Cyclin E | ↑ apoptotis with ↑ concentrations DHA | [21] |
| G2M | Leukemia | EHEB, JVM-2 and MEC-2 | 50 μM; 0.75 μM Dox | ↑ in cells in G2M with DHA alone; ↑ in G2M with DHA + Dox (EHEB, JVM-2 and MEC-2); ↑ in G2/M DHA + vincristine (JVM-2 and EPA) ↑ in G2/M DHA + fludarabine (EHEB) | ↑ cell death from Dox in EHEB, JVM-2 and MEC-2; ↑ cell death from vincristine in JVM-2 and MEC-2 and fludarabine in EHEB | [22] |
| G2M | Prostate | LNCaP, DU145, PC3 | 25 μM; 0.6 nM TXT | ↑ sub G1 cells; no diff between DHA, TXT, and combo; ↑ in G2M in LNCaP cells; >DHA + TXT than other treatments alone | ↑ MMP collapse in DHA + TXT; ↑ MAP2K4, TNFRSF11A, RIPK1; ↓ FADD, AKT1, MAX (microarray); RT-PCR opposite values | [23] |
| G2M | Colorectal | CaCo2 | FO (10–50 uM EPA 2:1 EPA:DHA); 0.25–1.0 μmol/L 5-FU | ↑ cells in G2M with FO, ↑ in S with 5-FU and ↑ cells in S and ↓ in G2M with 5-FU and FO combined | ↑ in apoptotic cells | [24] |
| G1 and G2M | Colorectal | COLO205 (wt p53) and WiDr (mutated p53) | 125 μM | ↑ in G1 in WiDr; ↑ G2M in COLO205 | ↓ proliferation in WiDr (NC in COLO205), ↑ apoptosis in COLO205, NC in WiDr | [25] |
| G1 and G2M | Colorectal | SW620 (chemotherapy resistant) | 70 μM | ↓ Cyclin D1, D3, A2, B2, F, CDK1, CDK2, CDK4, PCNA, CDC25B, CDC25C; ↑ p21, 14-3-3; ↓ mRNA transcript: G1/S: CCND1, CCND3, CCNG2, CDC42, CDC45L, CDC7, CDK2, CDK2AP, CDK4, CIP1/P21, CDKN1A, E2F1, PCNA, UNG, G2M: AURKA, AURKB, BIRC5, BUB1, CCNA2, CCNA2, CCNB2, CNF, CDC2/CDK1, CDC20, CDC25B, CDC25C, CENPE, FOXO3A, PLK1; ↑ p21, 14-3-3 protein | ↑ Gadd-45A, Gadd45B and Gadd34, Casp-4, 7, TNFRSF10B mRNA; ↓ NFκB, p38-P, α, β-livin, ↑ t-livin (protein); NC total p38 or Survivin (protein) | [26] |
| G1 and G2M | Breast | KLP-1 | 97 (CDHA) 270 (DHA) μmol/L | ↑ cells in G2 with DHA; ↑ cells in G1 with CDHA; Cyclin D1; ↑ p21 expression | ↑ apoptosis; ↑ p53; ↓ BCL-2; NC Bax | [27] |
| G1 and G2M | Breast | MDA-MB-231 MCF-7 | 0–100 nmol/L Dox | ↑ cells in G1 and G2M in MCF-7; ↑ G2M in MDA-MB-231; ↓ expression SKP2, p21, p27, Cyclin B, p53 in MCF-7; ↑ protein expression SKP2, Cyclin B, p53 and ↓ p21 MDA-MB-231 | NA | [28] |
| Animal Model | Tumour Model | Treatment/Diet | Results | Reference |
|---|---|---|---|---|
| BALB/c mice | KLP-1 | 0, 0.2%, 1.0% CDHA | NC body weight; ↓ in tumour volume and ↓ in metastases in 1.0% CDHA, but NC in tumour weight | [27] |
| Rats | mammary tumours induced with 1M1N | high n-3 diet (3:1 EPA:DHA, 45 g/kg diet) | ↓ in Cyclin D1, pRB ↑ p21, ↑ p27 protein expression; ↑ apoptotic markers | [29] |
| BALB/c mice | 4T1; mammary fat pad | 5% fish oil | ↓ tumour weight; ↓ in Cyclin D1, c-myc, B-catenin ↑ TUNEL + cells | [12] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newell, M.; Baker, K.; Postovit, L.M.; Field, C.J. A Critical Review on the Effect of Docosahexaenoic Acid (DHA) on Cancer Cell Cycle Progression. Int. J. Mol. Sci. 2017, 18, 1784. https://doi.org/10.3390/ijms18081784
Newell M, Baker K, Postovit LM, Field CJ. A Critical Review on the Effect of Docosahexaenoic Acid (DHA) on Cancer Cell Cycle Progression. International Journal of Molecular Sciences. 2017; 18(8):1784. https://doi.org/10.3390/ijms18081784
Chicago/Turabian StyleNewell, Marnie, Kristi Baker, Lynne M. Postovit, and Catherine J. Field. 2017. "A Critical Review on the Effect of Docosahexaenoic Acid (DHA) on Cancer Cell Cycle Progression" International Journal of Molecular Sciences 18, no. 8: 1784. https://doi.org/10.3390/ijms18081784
APA StyleNewell, M., Baker, K., Postovit, L. M., & Field, C. J. (2017). A Critical Review on the Effect of Docosahexaenoic Acid (DHA) on Cancer Cell Cycle Progression. International Journal of Molecular Sciences, 18(8), 1784. https://doi.org/10.3390/ijms18081784