Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis

Abstract

1. NKG2D/NKG2D Ligand Axis in the Recognition of Damaged Cells
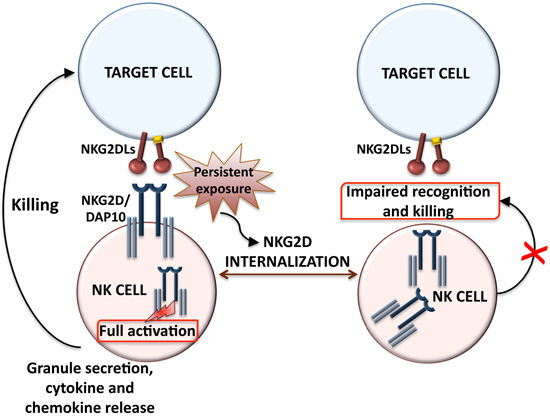
2. Ligand-Dependent NKG2D Down-Modulation: Impact on NK Cell Function
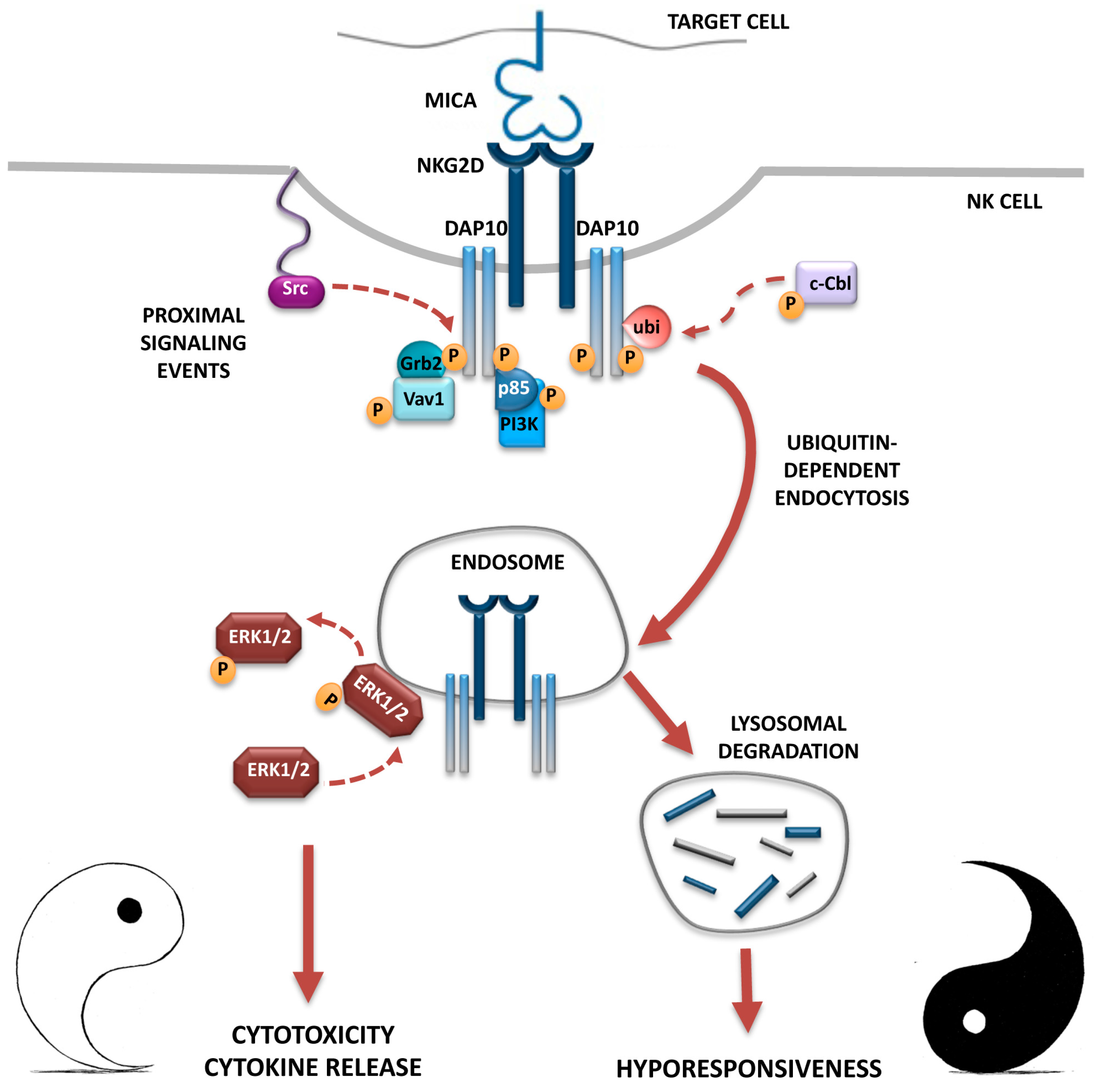
3. NKG2D Endocytosis: Intracellular Receptor Trafficking and Signalling
4. Down-Modulation of Other Activating NK Cell Receptors and Their Impact of NK Cell Function
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.A.; Trowsdale, J. Promiscuity and the single receptor: NKG2D. Nat. Rev. Immunol. 2007, 7, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2013, 2, e26097. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Baron, J.L.; Lanier, L.L. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 11521–11526. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Zafirova, B.; Mandarić, S.; Antulov, R.; Krmpotić, A.; Jonsson, H.; Yokoyama, W.M.; Jonjić, S.; Polić, B. Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-deficient mice. Immunity 2009, 31, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Jelenčić, V.; Lenartić, M.; Wensveen, F.M.; Polić, B. NKG2D: A versatile player in the immune system. Immunol. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; Ljunggren, H.G.; Long, E.O. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 2009, 114, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Das, A.; Gross, C.C.; Bryceson, Y.T.; Long, E.O. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity 2010, 32, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Long, E.O. Complementary phosphorylation sites in the adaptor protein SLP-76 promote synergistic activation of natural killer cells. Sci. Signal. 2012, 5, ra49. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.L.; Leibson, P.J. NKG2D-mediated activation of cytotoxic lymphocytes: Unique signaling pathways and distinct functional outcomes. Semin. Immunol. 2006, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Tomasello, E.; Lucas, M.; Jamieson, A.M.; Hsia, J.K.; Vivier, E.; Raulet, D.H. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 2002, 3, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, S.; Ho, E.L.; Cella, M.; Yokoyama, W.M.; Colonna, M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 2002, 3, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Fionda, C.; Soriani, A.; Zingoni, A.; Doria, M.; Cippitelli, M.; Santoni, A. The DNA Damage Response: A Common Pathway in the Regulation of NKG2D and DNAM-1 Ligand Expression in Normal, Infected, and Cancer Cells. Front. Immunol. 2014, 4, 508. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Iannitto, M.L.; Ricci, B.; Fionda, C.; Malgarini, G.; Morrone, S.; Peruzzi, G.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J. Immunol. 2014, 193, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bahram, S.; Bauer, S.; Herman, A.; Beauchamp, M.; Spies, T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA 1996, 93, 12445–12450. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Zingoni, A.; Cippitelli, M.; Piccoli, M.; Frati, L.; Santoni, A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood 2007, 110, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Davis, Z.; DeHart, J.; Zimmerman, E.; Bosque, A.; Brunetta, E.; Mavilio, D.; Planelles, V.; Barker, E. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 2009, 5, e1000613. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Sindhu, S.; Pham, T.N.; Belzile, J.P.; Cohen, E.A. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 2010, 115, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Pignoloni, B.; Fionda, C.; Dell’Oste, V.; Luganini, A.; Cippitelli, M.; Zingoni, A.; Landolfo, S.; Gribaudo, G.; Santoni, A.; Cerboni, C. Distinct Roles for Human Cytomegalovirus Immediate Early Proteins IE1 and IE2 in the Transcriptional Regulation of MICA and PVR/CD155 Expression. J. Immunol. 2016, 197, 4066–4078. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Vanneman, M.; Munshi, N.C.; Tai, Y.T.; Prabhala, R.H.; Ritz, J.; Neuberg, D.; Anderson, K.C.; Carrasco, D.R.; Dranoff, G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc. Natl. Acad. Sci. USA 2008, 105, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tao, Y.; Hou, J.; Meng, X.; Shi, J. Valproic acid upregulates NKG2D ligand expression through an ERK-dependent mechanism and potentially enhances NK cell-mediated lysis of myeloma. Neoplasia 2012, 14, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef] [PubMed]
- Abruzzese, M.P.; Bilotta, M.T.; Fionda, C.; Zingoni, A.; Soriani, A.; Vulpis, E.; Borrelli, C.; Zitti, B.; Petrucci, M.T.; Ricciardi, M.R.; et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J. Hematol. Oncol. 2016, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Borrelli, C.; Ricci, B.; Molfetta, R.; Zingoni, A.; Fionda, C.; Carnevale, S.; Abruzzese, M.P.; Petrucci, M.T.; Ricciardi, M.R.; et al. p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells. Oncoimmunology 2016, 6, e1264564. [Google Scholar] [CrossRef] [PubMed]
- Welte, S.A.; Sinzger, C.; Lutz, S.Z.; Singh-Jasuja, H.; Sampaio, K.L.; Eknigk, U.; Rammensee, H.G.; Steinle, A. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 2003, 33, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ashiru, O.; Bennett, N.J.; Boyle, L.H.; Thomas, M.; Trowsdale, J.; Wills, M.R. NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J. Virol. 2009, 83, 12345–12354. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.J.; Ashiru, O.; Morgan, F.J.; Pang, Y.; Okecha, G.; Eagle, R.A.; Trowsdale, J.; Sissons, J.G.; Wills, M.R. Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J. Immunol. 2010, 185, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Bacon, L.; Eagle, R.A.; Meyer, M.; Easom, N.; Young, N.T.; Trowsdale, J. Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J. Immunol. 2004, 173, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Xi, X.; Hao, Z.; Li, W.; Kong, Y.; Cui, L.; Ma, C.; Ba, D.; He, W. RAET1E2, a soluble isoform of the UL16-binding protein RAET1E produced by tumor cells, inhibits NKG2D-mediated NK cytotoxicity. J. Biol. Chem. 2007, 282, 18922–18928. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 2006, 66, 2520–2526. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Boutet, P.; Agüera-González, S.; Atkinson, S.; Pennington, C.J.; Edwards, D.R.; Murphy, G.; Reyburn, H.T.; Valés-Gómez, M. Cutting edge: The metalloproteinase ADAM17/TNF-α-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J. Immunol. 2009, 182, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Cecere, F.; Vulpis, E.; Fionda, C.; Molfetta, R.; Soriani, A.; Petrucci, M.T.; Ricciardi, M.R.; Fuerst, D.; Amendola, M.G.; et al. Genotoxic Stress Induces Senescence-Associated ADAM10-Dependent Release of NKG2D MIC Ligands in Multiple Myeloma Cells. J. Immunol. 2015, 195, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Messina, L.; Ashiru, O.; Boutet, P.; Agüera-González, S.; Skepper, J.N.; Reyburn, H.T.; Valés-Gómez, M. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J. Biol. Chem. 2010, 285, 8543–8551. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.S.; Doubrovin, M.M.; Vider, E.; Sisson, R.B.; O’Reilly, R.J.; Dupont, B.; Vyas, Y.M. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J. Immunol. 2003, 171, 6891–6899. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, K.; Hamerman, J.A.; Hsin, H.; Chikuma, S.; Bour-Jordan, H.; Chen, T.; Pertel, T.; Carnaud, C.; Bluestone, J.A.; Lanier, L.L. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity 2003, 18, 41–51. [Google Scholar] [CrossRef]
- Oppenheim, D.E.; Roberts, S.J.; Clarke, S.L.; Filler, R.; Lewis, J.M.; Tigelaar, R.E.; Girardi, M.; Hayday, A.C. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat. Immunol. 2005, 6, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, K.; Mittrücker, H.W.; Feger, U.; Welte, S.A.; Yokoyama, W.M.; Spies, T.; Rammensee, H.G.; Steinle, A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J. Immunol. 2005, 175, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Molfetta, R.; Quatrini, L.; Capuano, C.; Gasparrini, F.; Zitti, B.; Zingoni, A.; Galandrini, R.; Santoni, A.; Paolini, R. c-Cbl regulates MICA- but not ULBP2-induced NKG2D down-modulation in human NK cells. Eur. J. Immunol. 2014, 44, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Molfetta, R.; Quatrini, L.; Zitti, B.; Capuano, C.; Galandrini, R.; Santoni, A.; Paolini, R. Regulation of NKG2D Expression and Signaling by Endocytosis. Trends Immunol. 2016, 37, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Von Lilienfeld-Toal, M.; Frank, S.; Leyendecker, C.; Feyler, S.; Jarmin, S.; Morgan, R.; Glasmacher, A.; Märten, A.; Schmidt-Wolf, I.G.; Brossart, P.; et al. Reduced immune effector cell NKG2D expression and increased levels of soluble NKG2D ligands in multiple myeloma may not be causally linked. Cancer Immunol. Immunother. 2010, 59, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bruhl, A.; El-Gabalawy, H.; Nelson, J.L.; Spies, T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2003, 100, 9452–9457. [Google Scholar] [CrossRef] [PubMed]
- Hüe, S.; Mention, J.J.; Monteiro, R.C.; Zhang, S.; Cellier, C.; Schmitz, J.; Verkarre, V.; Fodil, N.; Bahram, S.; Cerf-Bensussan, N.; et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004, 21, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kim, J.; Cosman, D.; Choi, I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell Immunol. 2006, 239, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Ardolino, M.; Santoni, A.; Zingoni, A. Detuning CD8+ T lymphocytes by down-regulation of the activating receptor NKG2D: Role of NKG2D ligands released by activated T cells. Blood 2009, 113, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Stenqvist, A.C.; Nagaeva, O.; Kjellberg, L.; Wulff, M.; Baranov, V.; Mincheva-Nilsson, L. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: Evidence for immunosuppressive function. J. Immunol. 2009, 183, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Champsaur, M.; Beilke, J.N.; Ogasawara, K.; Koszinowski, U.H.; Jonjic, S.; Lanier, L.L. Intact NKG2D-independent function of NK cells chronically stimulated with the NKG2D ligand Rae-1. J. Immunol. 2010, 185, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Coudert, J.D.; Zimmer, J.; Tomasello, E.; Cebecauer, M.; Colonna, M.; Vivier, E.; Held, W. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood 2005, 106, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, N.; Jabri, B.; Dai, Z.; Ciszewski, C.; Stevens, A.M.; Yee, C.; Nakakuma, H.; Spies, T.; Groh, V. NKG2D initiates caspase-mediated CD3ζ degradation and lymphocyte receptor impairments associated with human cancer and autoimmune disease. J. Immunol. 2010, 185, 5732–5742. [Google Scholar] [CrossRef] [PubMed]
- Coudert, J.D.; Scarpellino, L.; Gros, F.; Vivier, E.; Held, W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood 2008, 111, 3571–3578. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, K.M.; Kim, D.W.; Heo, S.D. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.P.; Choi, S.C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the γc cytokines and TGF-β1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef] [PubMed]
- Roda-Navarro, P.; Reyburn, H.T. The traffic of the NKG2D/Dap10 receptor complex during natural killer (NK) cell activation. J. Biol. Chem. 2009, 284, 16463–16472. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, L.; Molfetta, R.; Zitti, B.; Peruzzi, G.; Fionda, C.; Capuano, C.; Galandrini, R.; Cippitelli, M.; Santoni, A.; Paolini, R. Ubiquitin-dependent endocytosis of NKG2D-DAP10 receptor complexes activates signaling and functions in human NK cells. Sci. Signal. 2015, 8, ra108. [Google Scholar] [CrossRef] [PubMed]
- Horng, T.; Bezbradica, J.S.; Medzhitov, R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat. Immunol. 2007, 8, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Galandrini, R.; Capuano, C.; Santoni, A. Activation of Lymphocyte Cytolytic Machinery: Where are we? Front. Immunol. 2013, 4, 390. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Tsvetanova, N.G.; Lobingier, B.T.; von Zastrow, M. Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol. 2015, 35, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, J.J.; di Guglielmo, G.M.; Dahan, S.; Dominguez, M.; Posner, B.I. Spatial and Temporal Regulation of Receptor Tyrosine Kinase Activation and Intracellular Signal Transduction. Annu. Rev. Biochem. 2016, 85, 573–597. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Martz, R.; Dorward, D.; Waisberg, M.; Pierce, S.K. Endocytosed BCRs sequentially regulate MAPK and Akt signaling pathways from intracellular compartments. Nat. Immunol. 2011, 12, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Staron, M.; Ferguson, S.M.; de Camilli, P.; Flavell, R.A. Dynamin 2-dependent endocytosis sustains T-cell receptor signaling and drives metabolic reprogramming in T lymphocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 4423–4428. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Bryceson, Y.T.; Kuppusamy, S.P.; Geraghty, D.E.; van der Meer, A.; Joosten, I.; Long, E.O. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol. 2006, 4, e9. [Google Scholar] [CrossRef] [PubMed]
- Miah, S.M.; Purdy, A.K.; Rodin, N.B.; MacFarlane IV, A.W.; Oshinsky, J.; Alvarez-Arias, D.A.; Campbell, K.S. Ubiquitylation of an internalized killer cell Ig-like receptor by Triad3A disrupts sustained NF-κB signaling. J. Immunol. 2011, 186, 2959–2969. [Google Scholar] [CrossRef] [PubMed]
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007, 109, 323–330. [Google Scholar]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.G.; Kiessling, R.; Malmberg, K.J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Gil-Krzewska, A.; Peruzzi, G.; Borrego, F. Matrix metalloproteinases inhibition promotes the polyfunctionality of human natural killer cells in therapeutic antibody-based anti-tumour immunotherapy. Clin. Exp. Immunol. 2013, 173, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology 2015, 4, e1001224. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, M.; Rusakiewicz, S.; Minard-Colin, V.; Delahaye, N.F.; Enot, D.; Vély, F.; Marabelle, A.; Papoular, B.; Piperoglou, C.; Ponzoni, M.; et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci. Transl. Med. 2015, 7, 283ra55. [Google Scholar] [CrossRef] [PubMed]
- Capuano, C.; Romanelli, M.; Pighi, C.; Cimino, G.; Rago, A.; Molfetta, R.; Paolini, R.; Santoni, A.; Galandrini, R. Anti-CD20 Therapy Acts via FcγRIIIA to Diminish Responsiveness of Human Natural Killer Cells. Cancer Res. 2015, 75, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Capuano, C.; Pighi, C.; Molfetta, R.; Paolini, R.; Battella, S.; Palmieri, G.; Giannini, G.; Belardinilli, F.; Santoni, A.; Galandrini, R. Obinutuzumab-mediated high-affinity ligation of FcγRIIIA/CD16 primes NK cells for IFNγ production. Oncoimmunology 2017, 6, e1290037. [Google Scholar] [CrossRef] [PubMed]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–44. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Weissman, A.M.; Kennedy, I.C.; Ortaldo, J.R. Engagement of the natural killer cell IgG Fc receptor results in tyrosine phosphorylation of the zeta chain. Proc. Natl. Acad. Sci. USA 1991, 88, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Galandrini, R.; Palmieri, G.; Paolini, R.; Piccoli, M.; Frati, L.; Santoni, A. Selective binding of shc-SH2 domain to tyrosine-phosphorylated ζ but not γ-chain upon CD16 ligation on human NK cells. J. Immunol. 1997, 159, 3767–3773. [Google Scholar] [PubMed]
- Natsume, A.; Niwa, R.; Satoh, M. Improving effector functions of antibodies for cancer treatment: Enhancing ADCC and CDC. Drug Des. Dev. Ther. 2009, 3, 7–16. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Díaz-Rubio, E. Progress in metastatic colorectal cancer: Growing role of cetuximab to optimize clinical outcome. Clin. Transl. Oncol. 2010, 12, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P.; Keating, G.M.L.; Scott, J. Trastuzumab: A review of its use as adjuvant treatment in human epidermal growth factor receptor 2 (HER2)-positive early breast cancer. Drugs 2010, 70, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Borrego, F.; Lopez-Beltran, A.; Pena, J.; Solana, R. Downregulation of Fcγ receptor IIIAα (CD16-II) on natural killer cells induced by anti-CD16 mAb is independent of protein tyrosine kinases and protein kinase C. Cell Immunol. 1994, 158, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Grzywacz, B.; Kataria, N.; Verneris, M.R. CD56dimCD16+ NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia 2007, 21, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, G.; Femnou, L.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Membrane-type 6 matrix metalloproteinase regulates the activation-induced downmodulation of CD16 in human primary NK cells. J. Immunol. 2013, 191, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Mota, G.; Moldovan, I.; Calugaru, A.; Hirt, M.; Kozma, E.; Galatiuc, C.; Brasoveanu, L.; Boltz-Nitulescu, G. Interaction of human immunoglobulin G with CD16 on natural killer cells: Ligand clearance, FcgammaRIIIA turnover and effects of metalloproteinases on FcγRIIIA-mediated binding, signal transduction and killing. Scand. J. Immunol. 2004, 59, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, S.; Spadaro, F.; Lugini, L.; Podo, F.; Ramoni, C. Functional role of phosphatidylcholine-specific phospholipase C in regulating CD16 membrane expression in natural killer cells. Eur. J. Immunol. 2007, 37, 2912–2922. [Google Scholar] [CrossRef] [PubMed]
- Paolini, R.; Serra, A.; Molfetta, R.; Piccoli, M.; Frati, L.; Santoni, A. Tyrosine kinase-dependent ubiquitination of CD16 zeta subunit in human NK cells following receptor engagement. Eur. J. Immunol. 1999, 29, 3179–3187. [Google Scholar] [CrossRef]
- Paolini, R.; Molfetta, R.; Piccoli, M.; Frati, L.; Santoni, A. Ubiquitination and degradation of Syk and ZAP-70 protein tyrosine kinases in human NK cells upon CD16 engagement. Proc. Natl. Acad. Sci. USA 2001, 98, 9611–9616. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Experimental Mouse Model | Findings | Reference |
|---|---|---|
| Nonobese diabetic (NOD) mice |
| [47] |
| FVB transgenic mice overexpressing Rae-1ε ligand ubiquitously or localized in normal epithelium |
| [48] |
| C57BL/6 transgenic mice constitutively and ubiquitously overexpressing MICA |
| [49] |
| C57BL/6 transgenic mice ubiquitously overexpressing Rae-1ε ligand |
| [60] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molfetta, R.; Quatrini, L.; Santoni, A.; Paolini, R. Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis. Int. J. Mol. Sci. 2017, 18, 1677. https://doi.org/10.3390/ijms18081677
Molfetta R, Quatrini L, Santoni A, Paolini R. Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis. International Journal of Molecular Sciences. 2017; 18(8):1677. https://doi.org/10.3390/ijms18081677
Chicago/Turabian StyleMolfetta, Rosa, Linda Quatrini, Angela Santoni, and Rossella Paolini. 2017. "Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis" International Journal of Molecular Sciences 18, no. 8: 1677. https://doi.org/10.3390/ijms18081677
APA StyleMolfetta, R., Quatrini, L., Santoni, A., & Paolini, R. (2017). Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis. International Journal of Molecular Sciences, 18(8), 1677. https://doi.org/10.3390/ijms18081677