Multi-Targeting Andrographolide, a Novel NF-κB Inhibitor, as a Potential Therapeutic Agent for Stroke


 ,
, 

Abstract
{kind=link}
{kind=link}
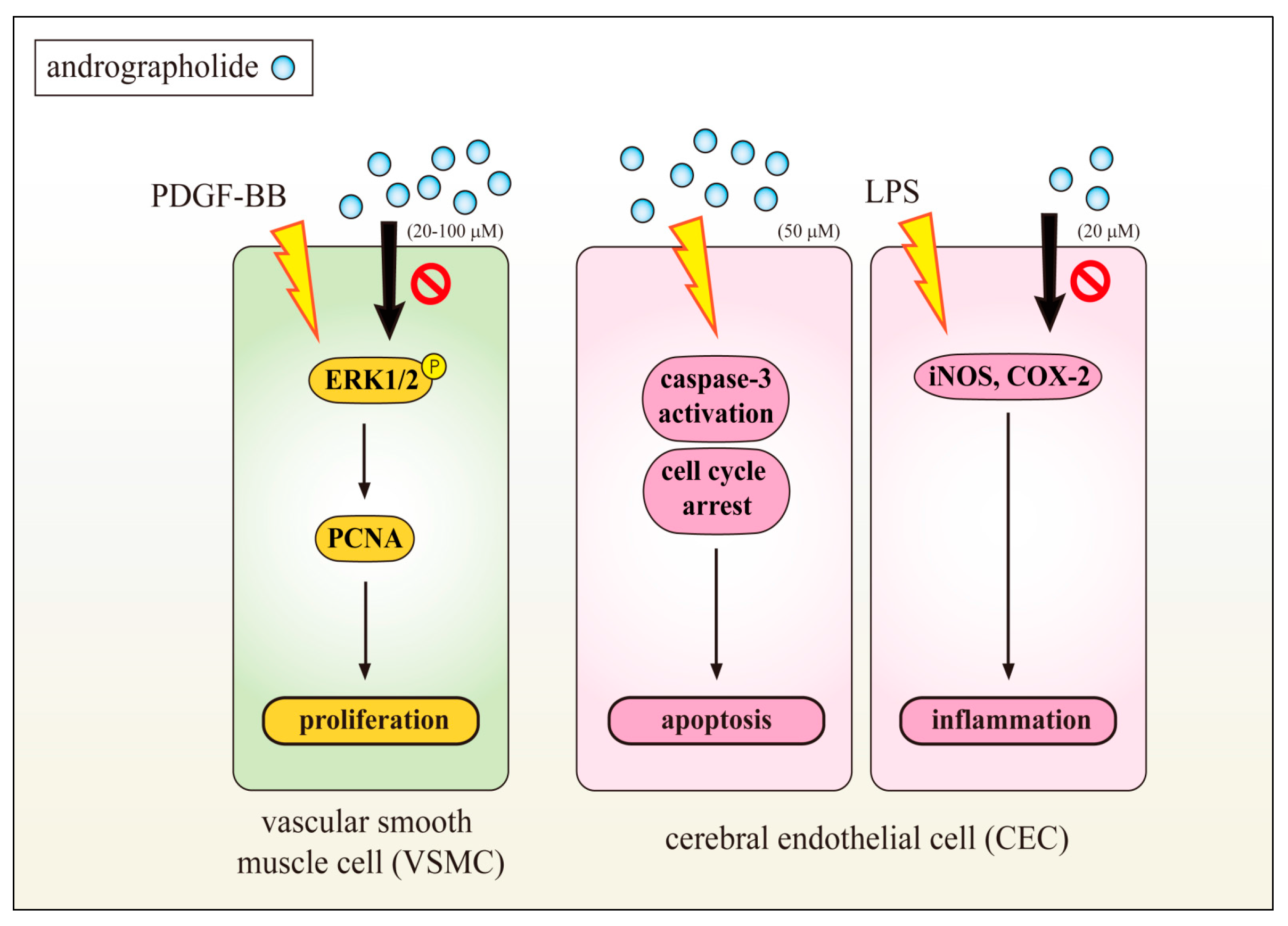{kind=link}
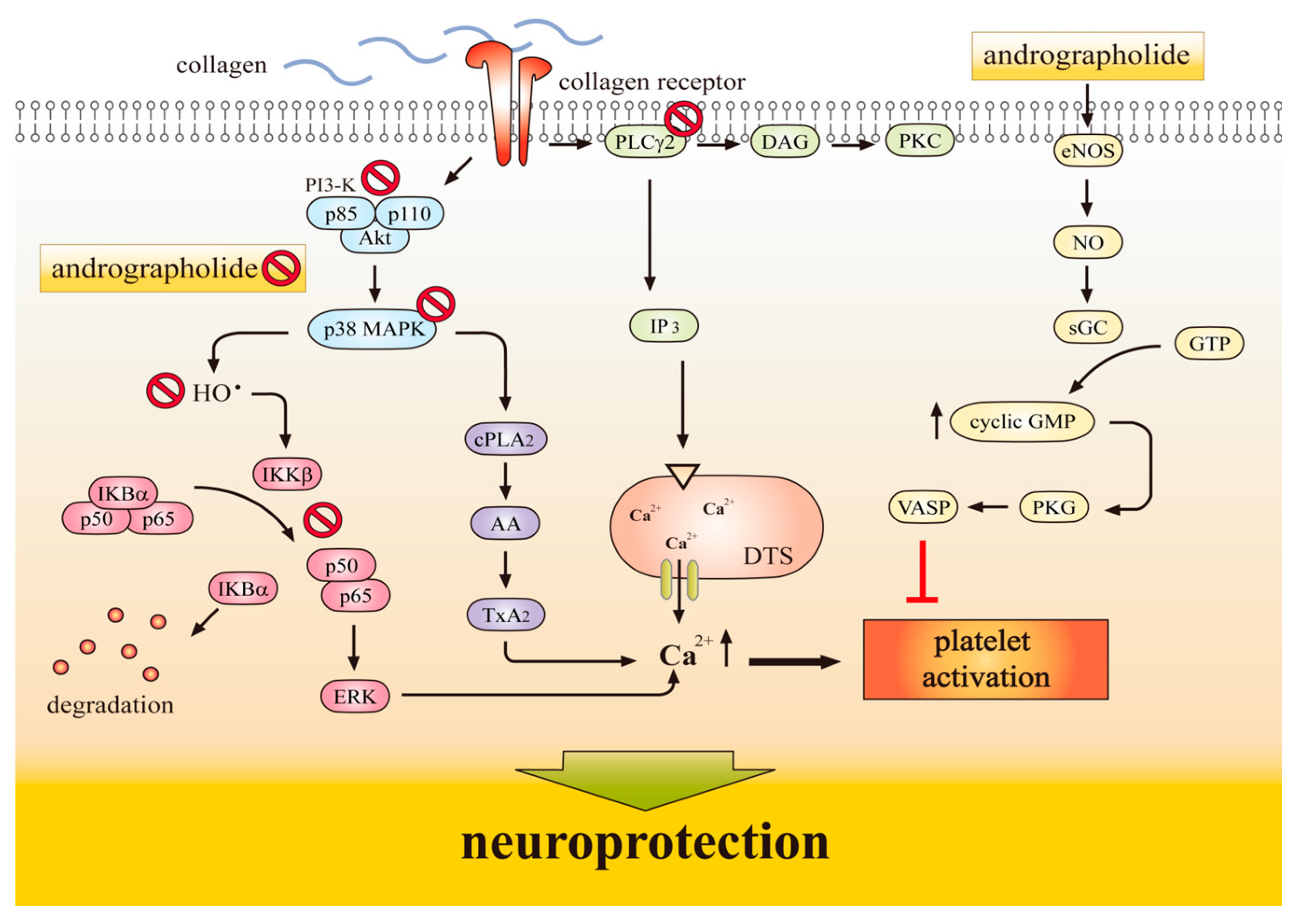{kind=link}
1. Introduction
1.1. Androrapholide in Neuroprotection
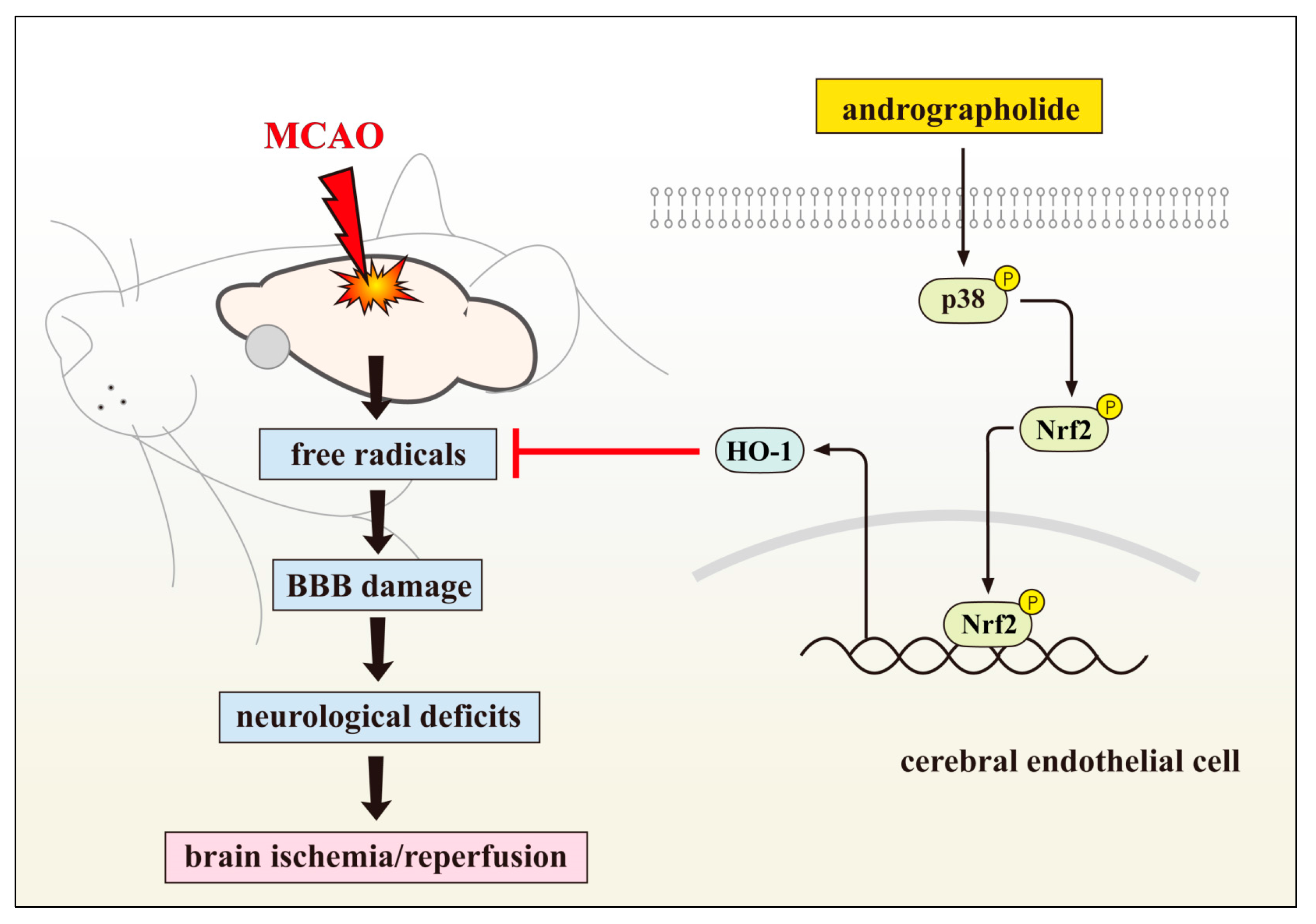
1.2. Inhibition of VSMC and CEC Dysfunction Represents Neuroprotection of Andrographolide
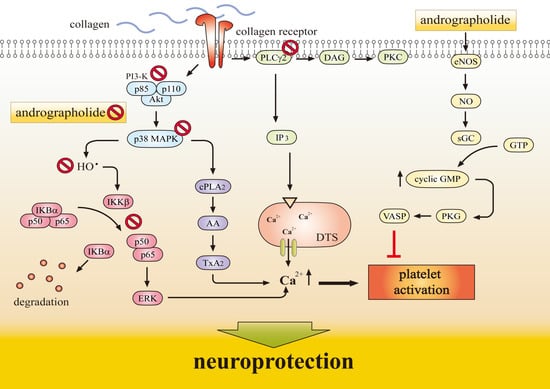
1.3. Antiplatelet Action of Andrographolide Conferring Neuroprotection
2. Molecular Mechanisms Underlying the Neuroprotective Effects of Andrographolide
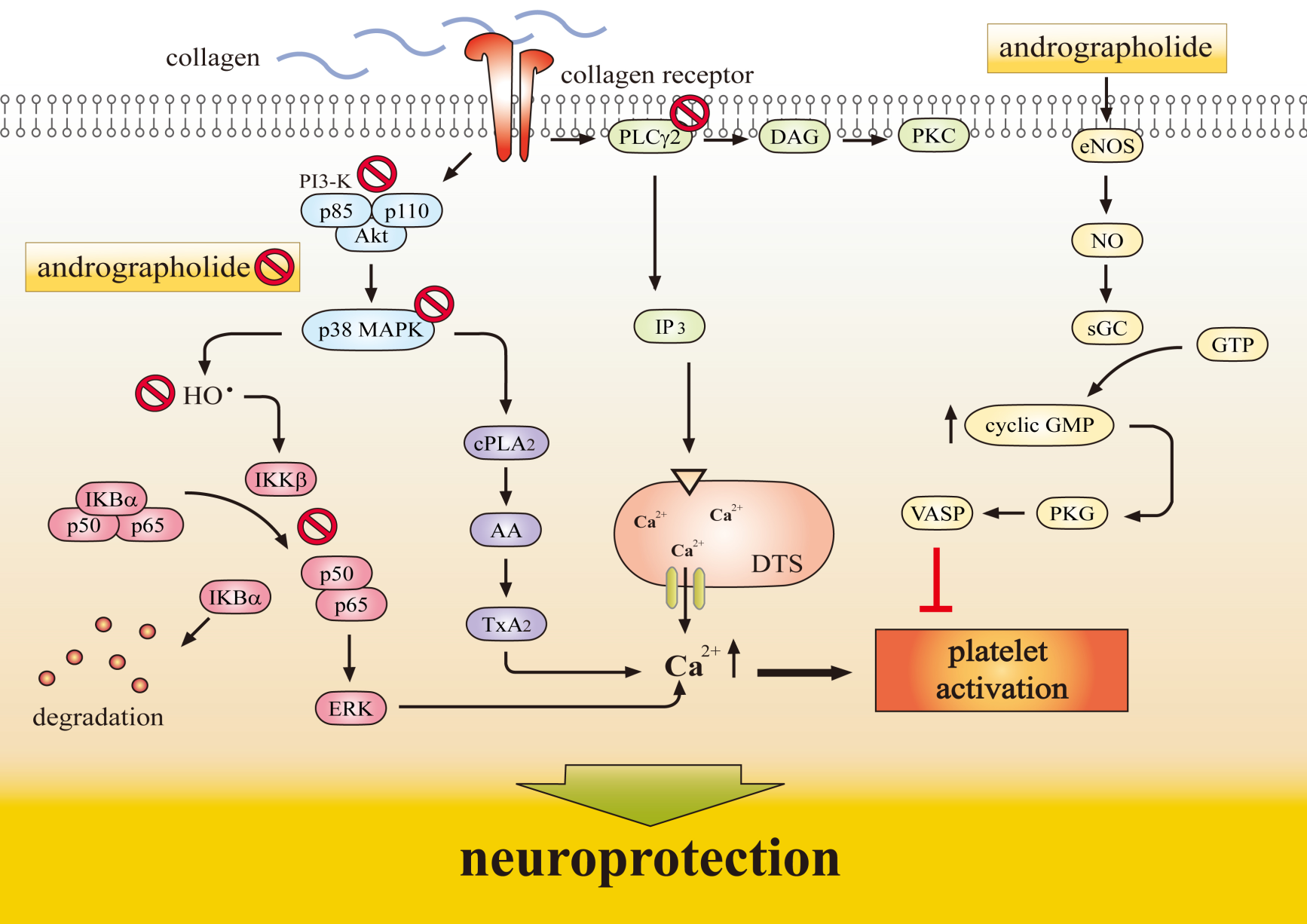
2.1. Neuroprotective Role of Andrographolide via HO-1 Induction
2.2. Andrographolide’s on Stroke Treatment via Inhibiting MAPK Signalling
2.3. Andrographolide’s Neuroprotection via Nrf2 Activation
2.4. NF-κB Inhibition Contributes Neuroprotection of Andrographolide
2.5. Andrographolide Improves Neuronal Functions via Induction of Apoptosis
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stoll, G.; Kleinschnitz, C.; Nieswandt, B. Molecular mechanisms of thrombus formation in ischemic stroke: Novel insights and targets for treatment. Blood 2008, 112, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R. Pharmacological approaches to acute ischaemic stroke: Reperfusion certainly, neuroprotection possibly. Br. J. Pharmacol. 2008, 153, S325–S338. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.V.; Zavala, J.A.; Donnan, G.A. Neuroprotection and thrombolysis: Combination therapy in acute ischaemic stroke. Exp. Opin. Pharmacother. 2006, 7, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, A.W. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Doré, S. Unique properties of polyphenol stilbenes in the brain: More than direct antioxidant actions; gene/protein regulatory activity. Neurosignals 2005, 14, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H. Exercise and the brain: Something to chew on. Trends Neurosci. 2009, 32, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C.; Chen, C.F.; Chiou, W.F. Andrographolide prevents oxygen radical production by human neutrophils: Possible mechanism(s) involved in its anti-inflammatory effect. Br. J. Pharmacol. 2002, 135, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wang, D.X.; Zhang, W.; Liao, X.Q.; Guan, X.; Bo, H.; Sun, J.Y.; Huang, N.W.; He, J.; Zhang, Y.K.; et al. Andrographolide protects against LPS-induced acute lung injury by inactivation of NF-κB. PLoS ONE 2013, 8, e56407. [Google Scholar] [CrossRef] [PubMed]
- Abu-Ghefreh, A.A.; Canatan, H.; Ezeamuzie, C.I. In vitro and in vivo anti-inflammatory effects of andrographolide. Int. Immunopharmacol. 2009, 9, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.J.; Wong, W.S.; Wong, P.T.; Bian, J.S. Neuroprotective effects of andrographolide in a rat model of permanent cerebral ischaemia. Br. J. Pharmacol. 2010, 161, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, B.; Zhang, W.; Wilson, B.; Hong, J.S. Andrographolide reduces inflammationmediated dopaminergic neurodegeneration in mesencephalic neuron-glia cultures by inhibiting microglial activation. J. Pharmacol. Exp. Ther. 2004, 308, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.L.; Chen, R.J.; Jayakumar, T.; Lu, W.J.; Hsieh, C.Y.; Hsu, M.J.; Yang, C.H.; Chang, C.C.; Lin, Y.K.; Lin, K.H.; et al. Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 2016, 170, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Hsieh, C.Y.; Jayakumar, T.; Lin, K.H.; Chou, D.S.; Lu, W.J.; Hsu, M.J.; Sheu, J.R. Andrographolide induces vascular smooth muscle cell apoptosis through a SHP-1-PP2A-p38MAPK-p53 cascade. Sci. Rep. 2014, 4, 5651. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Hsu, M.J.; Hsieh, C.Y.; Lee, L.W.; Chen, Z.C.; Sheu, J.R. Andrographolide inhibits nuclear factor-𝜅B activation through JNK-Akt-p65 signaling cascade in tumor necrosis factor-𝛼-stimulated vascular smooth muscle cells. Sci. World J. 2014, 2014, 1–10. [Google Scholar]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J. Structure and function of small arteries in hypertension. J. Hypertens. 1990, 8, 225–232. [Google Scholar]
- Ross, R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Duann, Y.F.; Yen, T.L.; Chen, Y.Y.; Jayakumar, T.; Ong, E.T.; Sheu, J.R. Andrographolide, a novel NF-κB inhibitor, inhibits vascular smooth muscle cell proliferation and cerebral endothelial cell inflammation. Acta Cardiol. Sin. 2014, 30, 308–315. [Google Scholar] [PubMed]
- Yen, T.L.; Hsu, W.H.; Huang, S.K.H.; Lu, W.J.; Chang, C.C.; Lien, L.M.; Hsiao, G.; Sheu, J.R.; Lin, K.H. A novel bioactivity of andrographolide from andrographis paniculata on cerebral ischemia/reperfusion-induced brain injury through induction of cerebral endothelial cell apoptosis. Pharm. Biol. 1150, 51, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Hsu, M.J.; Sheu, J.R.; Lee, L.W.; Hsie, C.Y. Andrographolide, a novel NF-κB inhibitor, induces vascular smooth muscle cell apoptosis via a ceramide-p47phox-ROS signaling cascade. Evid. Based Complement. Altern. Med. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, G.; Lin, K.H.; Chang, Y.; Chen, T.L.; Tzu, N.H.; Chou, D.S.; Sheu, J.R. Protective mechanisms of inosine in platelet activation and cerebral ischemic damage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Amroyan, E.; Gabrielian, E.; Panossian, A.; Wikman, G.; Wagner, H. Inhibitory effect of andrographolide from Andrographis paniculata on PAF-induced platelet aggregation. Phytomedicine 1999, 6, 27–31. [Google Scholar] [CrossRef]
- Thisoda, P.; Rangkadilok, N.; Pholphana, N.; Worasuttayangkurn, L.; Ruchirawat, S.; Satayavivad, J. Inhibitory effect of Andrographis paniculata extract and its active diterpenoids on platelet aggregation. Eur. J. Pharmacol. 2006, 553, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lin, K.H.; Hsu, M.J.; Chou, D.S.; Hsiao, G.; Sheu, J.R. Suppression of NF-κB signaling by andrographolide with a novel mechanism in human platelets: Regulatory roles of the p38MAPK- hydroxyl radical-ERK2 cascade. Biochem. Pharmacol. 2012, 84, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lee, J.J.; Chou, D.S.; Jayakumar, T.; Fong, T.H.; Hsiao, G.; Sheu, J.R. A novel role of andrographolide, an NF-κB inhibitor, on inhibition of platelet activation: The pivotal mechanisms of endothelial nitric oxide synthase/cyclic GMP. J. Mol. Med. 2011, 89, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Wang, J.T.; Fan, Q.X.; Geng, J.G. Andrographolide inhibits NF-κB activation and attenuates neointimal hyperplasia in arterial restenosis. Cell Res. 2007, 17, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.D.; Ye, B.Q.; Zheng, S.X.; Wang, J.T.; Wang, J.G.; Chen, M.; Liu, J.G.; Pei, X.H.; Wang, L.J.; Lin, Z.X.; et al. NF-κB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J. Biol. Chem. 2009, 284, 4473–4483. [Google Scholar] [CrossRef] [PubMed]
- Plesnila, N.; Zinkel, S.; Amin-Hanjani, S.; Qiu, J.; Korsmeyer, S.J.; Moskowitz, M.A. Function of BID—A molecule of the bcl-2 family in ischemic cell death in the brain. Eur. Surg. Res. 2002, 34, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Lien, L.M.; Su, C.C.; Hsu, W.H.; Lu, W.J.; Chung, C.L.; Yen, T.L.; Chiu, H.C.; Sheu, J.R.; Lin, K.H. Mechanisms of andrographolide-induced platelet apoptosis in human platelets: Regulatory roles of the extrinsic apoptotic pathway. Phytother. Res. 2013, 27, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 2010, 11, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Dore, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Pae, H.O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.H.; Choi, Y.K.; Lee, B.S.; Kim, S.R.; Chung, H.T. Heme oxygenase in the regulation of vascular biology: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 14, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, M.; Wang, M.; Li, Y.; Wen, A. Posttreatment with 11-keto-beta-boswellic acid ameliorates cerebral ischemia-reperfusion injury: Nrf2/HO-1 pathway as a potential mechanism. Mol. Neurobiol. 2015, 52, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, J.; Wan, F.; Zhang, W.; Yang, H.; Wang, L.; Qi, H.; Wu, C. Panaxatriol saponins attenuated oxygen-glucose deprivation injury in PC12 cells via activation of PI3K/Akt and Nrf2 signaling pathway. Oxid. Med. Cell Longev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Cheng, W.; Yu, P.; Wang, L.; Zhou, L.; Zeng, L.; Yang, Q. Resveratrol pretreatment attenuates injury and promotes proliferation of neural stem cells following oxygen-glucose deprivation/reoxygenation by upregulating the expression of Nrf2, HO-1 and NQO1 in vitro. Mol. Med. Rep. 2016, 14, 3646–3654. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; Kong, Y.Y.; Li, G.Q.; Guan, Y.F.; Wang, P.; Miao, C.Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef] [PubMed]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 2015, 1254, 197–210. [Google Scholar] [PubMed]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Müller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell Signal. 2012, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Nishimura, M.; Hashimoto, N. Mitogen-activated protein kinases and cerebral ischemia. Mol. Neurobiol. 2001, 23, 1–19. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.; Zhang, Y.Q.; Xu, Z.H. JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.Y.; Whitmarsh, A.J.; Yang, D.D.; Liao, G.; Schloemer, A.J.; Dong, C.; Bao, J.; Banasiak, K.J.; Haddad, G.G.; Flavell, R.A.; et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15184–15189. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.C.; Irving, E.A.; Ray, A.M.; Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.M.; Legos, J.J.; Erhardt, J.A.; Ohlstein, E.H.; et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 2001, 21, 129–145. [Google Scholar] [CrossRef]
- Gladbach, A.; van Eersel, J.; Bi, M.; Ke, Y.D.; Ittner, L.M. ERK inhibition with PD184161 mitigates brain damage in a mouse model of stroke. J. Neural Transm. 2014, 121, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Maddahi, A.; Kruse, L.S.; Chen, Q.W.; Edvinsson, L. The role of tumor necrosis factor-α and TNF-α receptors in cerebral arteries following cerebral ischemia in rat. J. Neuroinflamm. 2011, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.L.; Choi, Y.; Seow, W.L.; Manzanero, S.; Sobey, C.G.; Jo, D.G.; Arumugam, T.V. Evidence that neuronal notch-1 promotes JNK/c-Jun activation and cell death following ischemic stress. Brain Res. 2014, 1586, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Maddahi, A.; Edvinsson, L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J. Neuroinflamm. 2010, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef] [PubMed]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; McDonald, P.R.; Liu, J.; Klaassen, C.D. Screening of natural compounds as activators of the keap1-nrf2 pathway. Planta Med. 2014, 80, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Oz, H.S.; Barve, S.; De Villiers, W.J.; McClain, C.J.; Varilek, G.W. The green tea polyphenol (−)-epigallocatechin-3-gallate blocks nuclear factor-κB activation by inhibiting IκB kinase activity in the intestinal epithelial cell line IEC-6. Mol. Pharmacol. 2001, 528–533. [Google Scholar]
- Begum, R.; Sheliya, M.A.; Mir, S.R.; Singh, E.; Sharma, M. Inhibition of proinflammatory mediators by coumaroyl lupendioic acid, a new lupane-type triterpene from Careya arborea, on inflammation-induced animal model. J. Ethnopharmacol. 2017, 206, 376–392. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Guan, S.; Cheng, C.; Wu, S.; Wong, S.H.; Kemeny, D.M.; Leung, B.P.; Wong, W.S. A novel anti-inflammatory role for andrographolide in asthma via inhibition of the nuclear factor-κb pathway. Am. J. Respir. Crit. Care Med. 2009, 179, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.Y.; Hsu, M.J.; Hsiao, G.; Wang, Y.H.; Huang, C.W.; Chen, S.W.; Jayakumar, T.; Chiu, P.T.; Chiu, Y.H.; Sheu, J.R. Andrographolide enhances nuclear factor-𝜅B subunit p65 Ser 536 dephosphorylation through activation of protein phosphatase 2A in vascular smooth muscle cells. J. Biol. Chem. 2011, 286, 5942–5955. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-κB system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ku, K.T.; Huang, Y.L.; Huang, Y.J.; Chiou, W.F. Miyabenol A inhibits LPS-induced NO production via IKK/IκB inactivation in RAW 264.7 macrophages: Possible involvement of the p38 and PI3K pathways. J. Agric. Food Chem. 2008, 56, 8911–8918. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Chao, W.W.; Chen, M.L.; Lin, B.F. Ethyl acetate extracts of alfalfa (Medicago sativa L.) sprouts inhibit lipopolysaccharide-induced inflammation in vitro and in vivo. J. Biomed. Sci. 2009, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.J.; Koh, D.J.; Kim, S.H.; Park, S.J.; Ryu, J.H.; Kim, D.G.; Lee, J.Y.; Lee, K.T. Anti-inflammatory effects of sinapic acid through the suppression of inducible nitric oxide synthase, cyclooxygase-2, and proinflammatory cytokines expressions via nuclear factor-𝜅B inactivation. J. Agric. Food Chem. 2008, 56, 10265–10272. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.A. The role of free radical generation in increasing cerebrovascular permeability. Free Radic. Biol. Med. 2011, 51, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.T.; Leaver, H.A. Brain endothelial cell death: Modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol. Neurobiol. 2010, 42, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Liu, D.; Griffin, J.H.; Fernández, J.A.; Castellino, F.; Rosen, E.D.; Fukudome, K.; Zlokovic, B.V. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat. Med. 2003, 9, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.J.; Hsu, C.Y.; Chen, B.C.; Chen, M.C.; Ou, G.; Lin, C.H. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J. Neurosci. 2007, 27, 5719–5729. [Google Scholar] [CrossRef] [PubMed]
- Chern, C.M.; Liou, K.T.; Wang, Y.H.; Liao, J.F.; Yen, J.C.; Shen, Y.C. Andrographolide inhibits PI3K/AKT-dependent NOX2 and iNOS expression protecting mice against hypoxia/ischemia-induced oxidative brain injury. Planta. Med. 2011, 77, 1669–1679. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-H.; Yen, T.-L.; Hsu, C.-Y.; Thomas, P.-A.; Sheu, J.-R.; Jayakumar, T. Multi-Targeting Andrographolide, a Novel NF-κB Inhibitor, as a Potential Therapeutic Agent for Stroke. Int. J. Mol. Sci. 2017, 18, 1638. https://doi.org/10.3390/ijms18081638
Yang C-H, Yen T-L, Hsu C-Y, Thomas P-A, Sheu J-R, Jayakumar T. Multi-Targeting Andrographolide, a Novel NF-κB Inhibitor, as a Potential Therapeutic Agent for Stroke. International Journal of Molecular Sciences. 2017; 18(8):1638. https://doi.org/10.3390/ijms18081638
Chicago/Turabian StyleYang, Chih-Hao, Ting-Lin Yen, Chia-Yuan Hsu, Philip-Aloysius Thomas, Joen-Rong Sheu, and Thanasekaran Jayakumar. 2017. "Multi-Targeting Andrographolide, a Novel NF-κB Inhibitor, as a Potential Therapeutic Agent for Stroke" International Journal of Molecular Sciences 18, no. 8: 1638. https://doi.org/10.3390/ijms18081638
APA StyleYang, C.-H., Yen, T.-L., Hsu, C.-Y., Thomas, P.-A., Sheu, J.-R., & Jayakumar, T. (2017). Multi-Targeting Andrographolide, a Novel NF-κB Inhibitor, as a Potential Therapeutic Agent for Stroke. International Journal of Molecular Sciences, 18(8), 1638. https://doi.org/10.3390/ijms18081638