Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension

 ,
,
Abstract
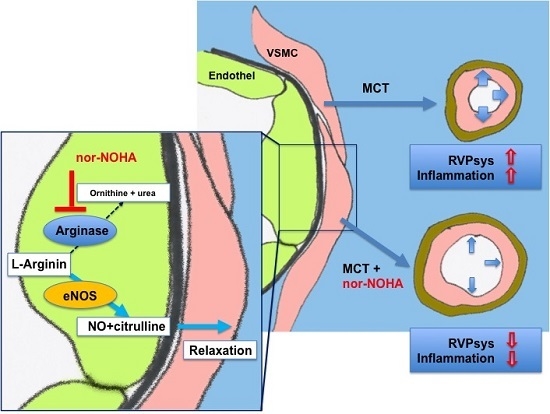
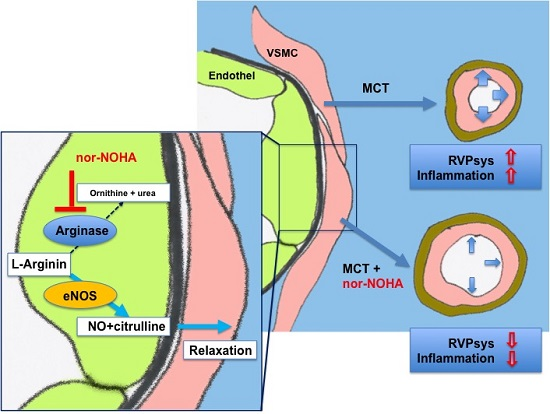
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
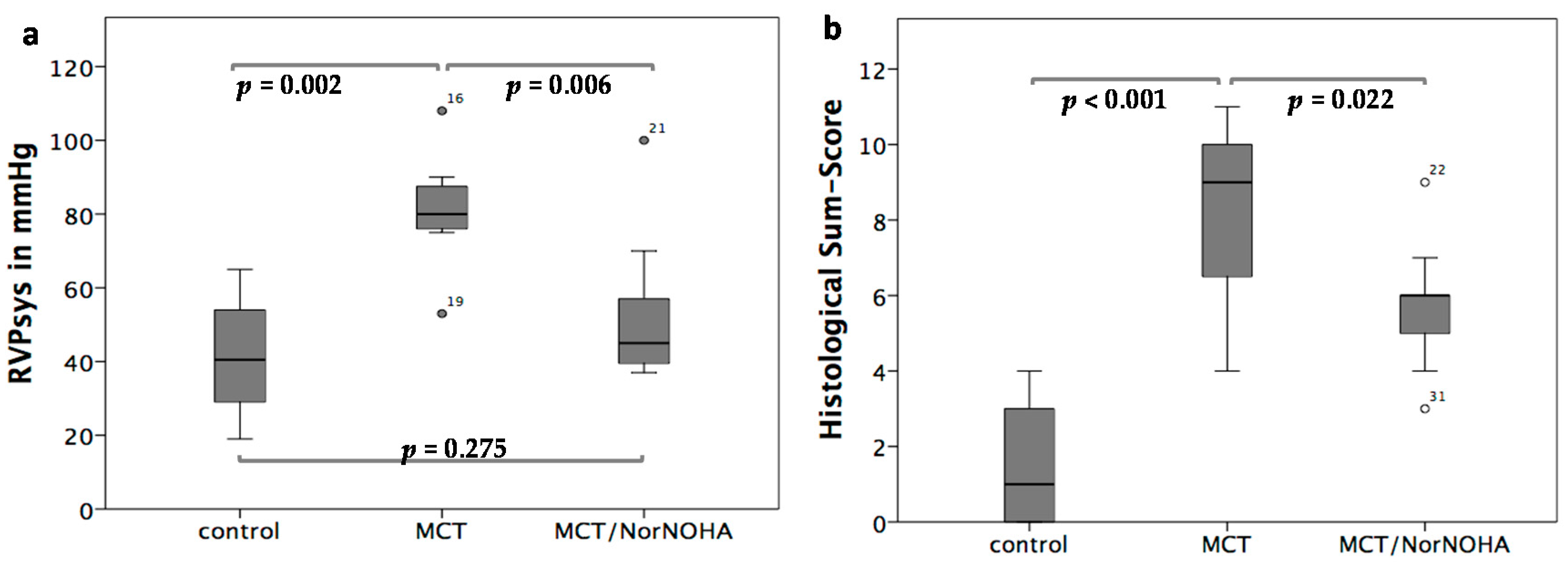
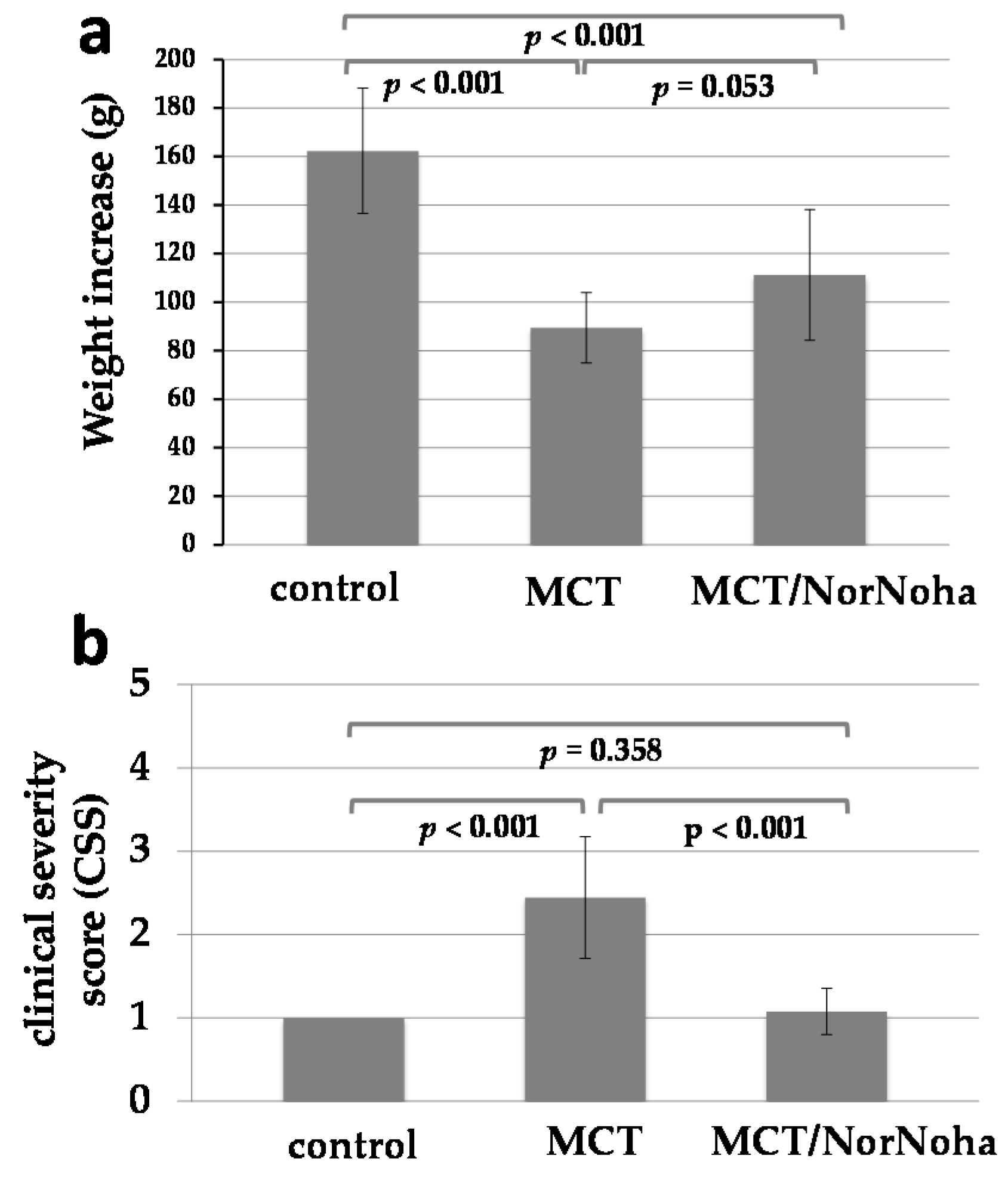
2.1. Clinical and Haemodynamic Characterization of the Different Experimental Groups
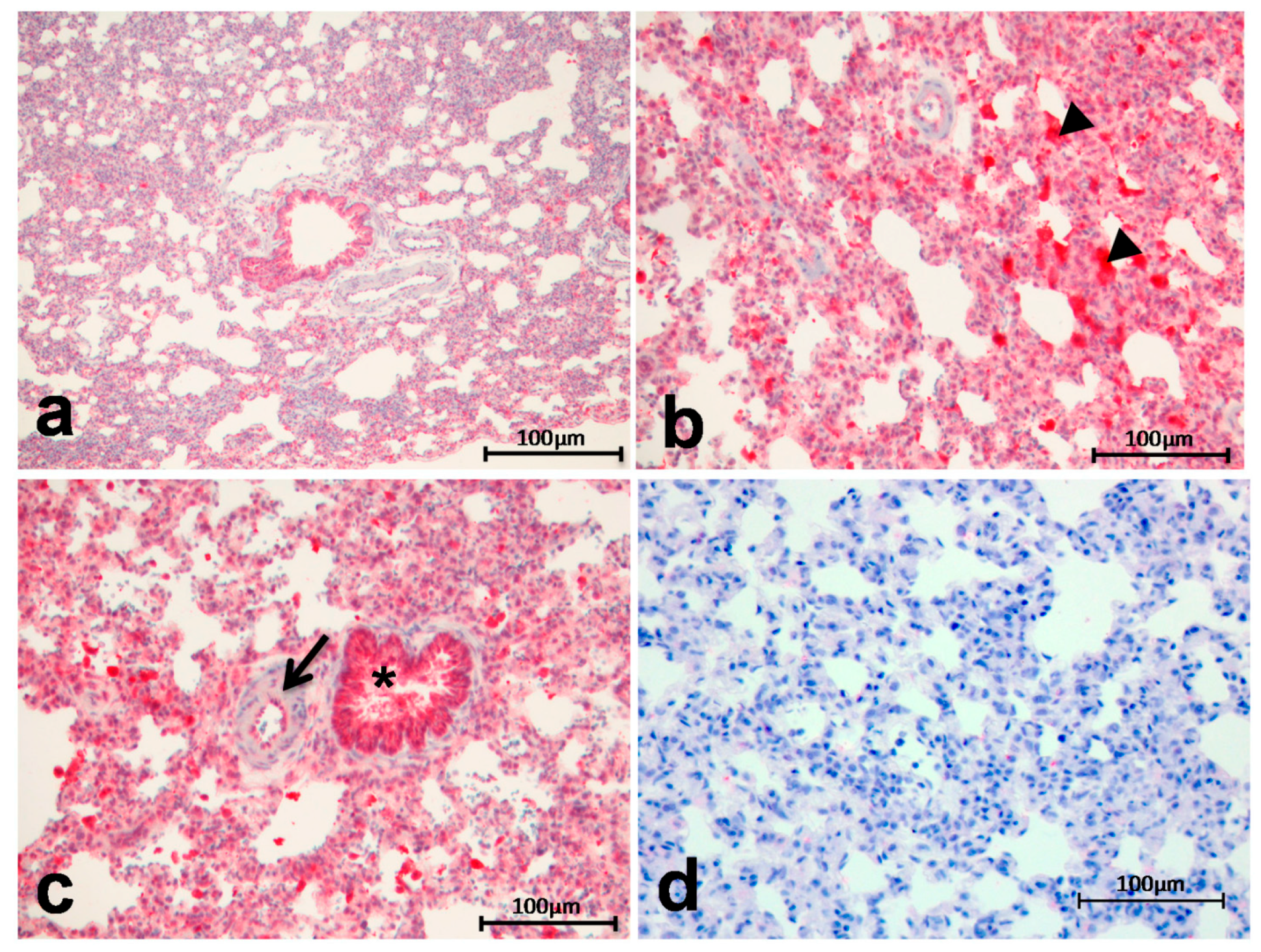
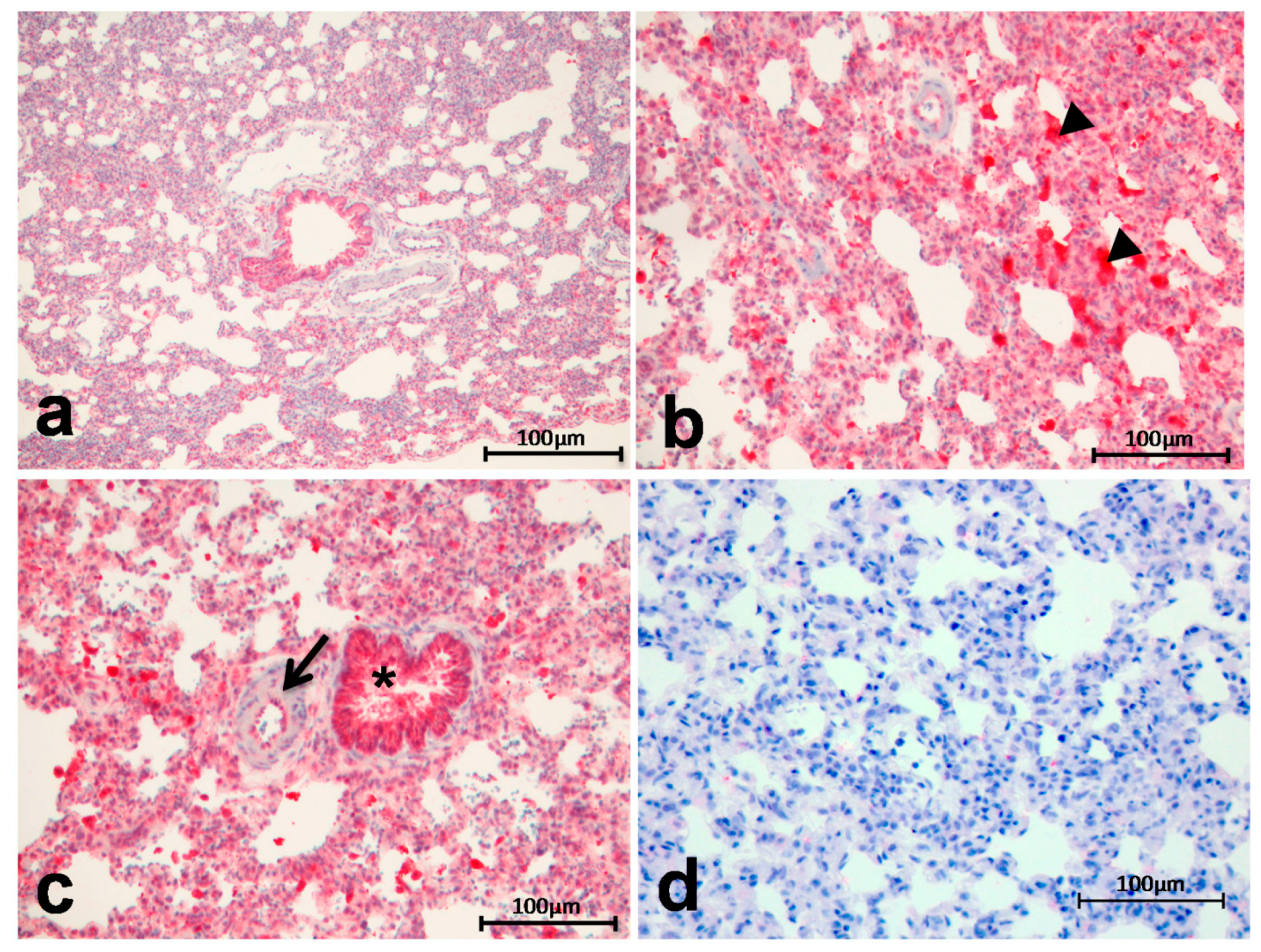
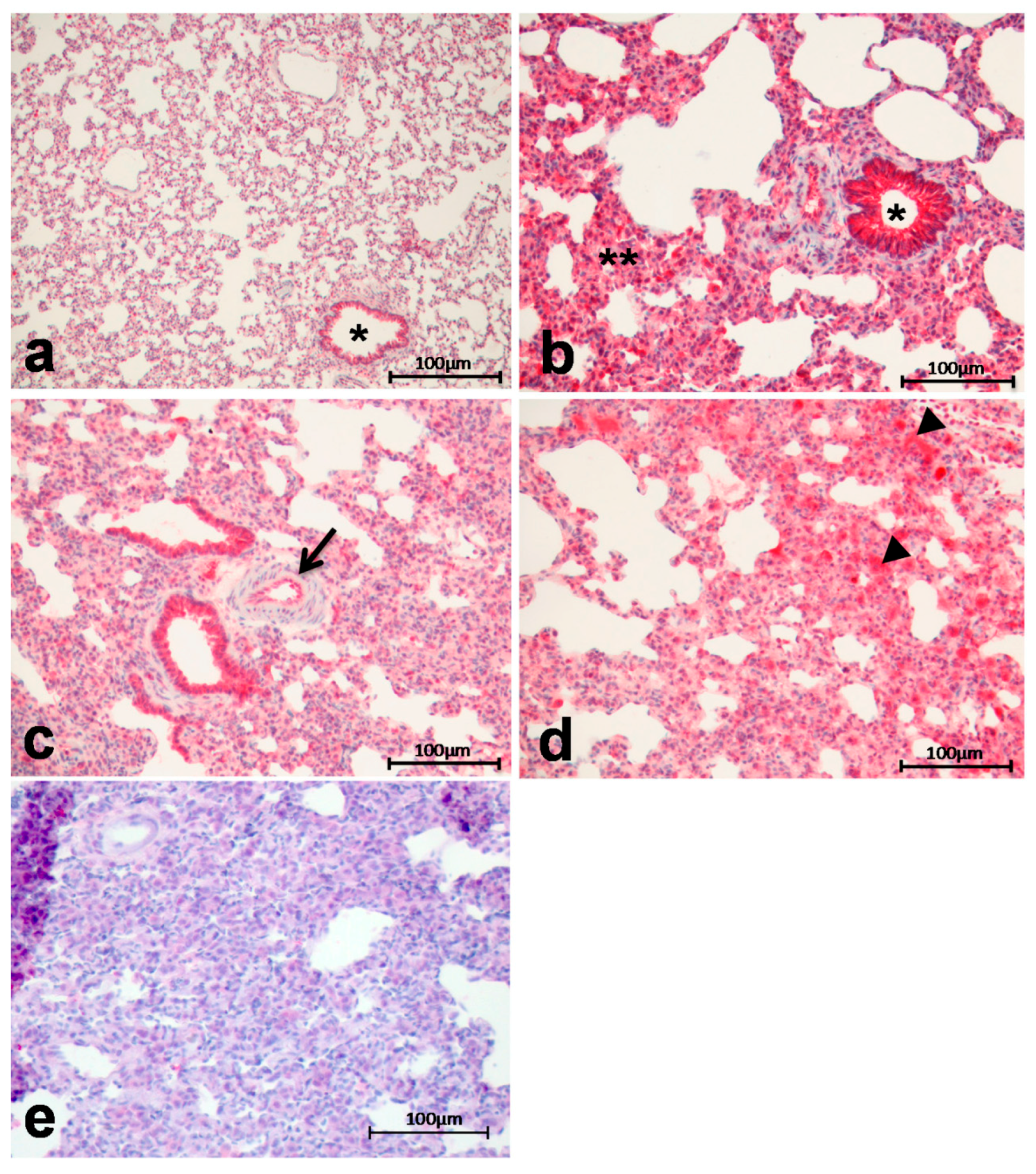
2.2. Immunohistochemistry
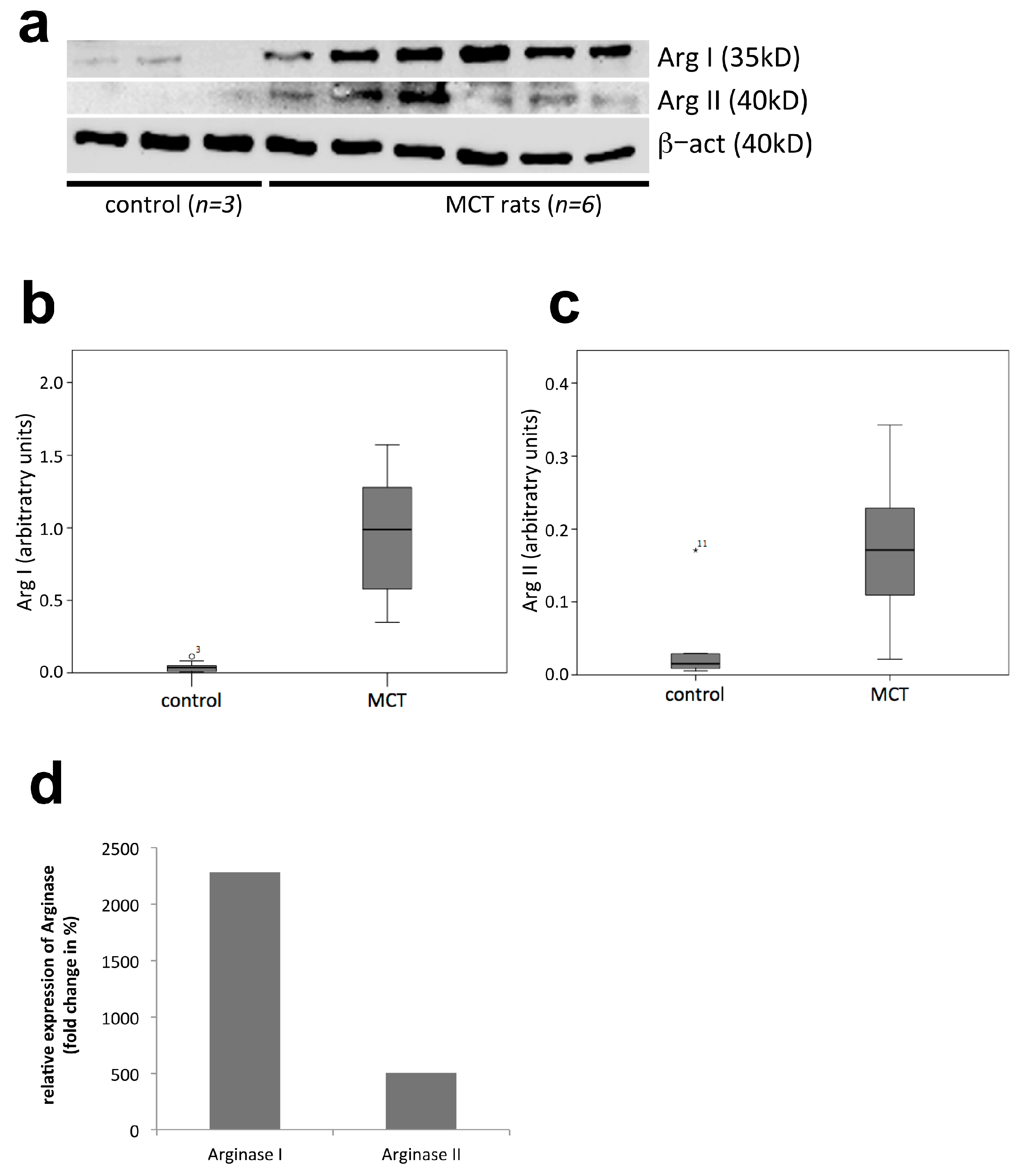
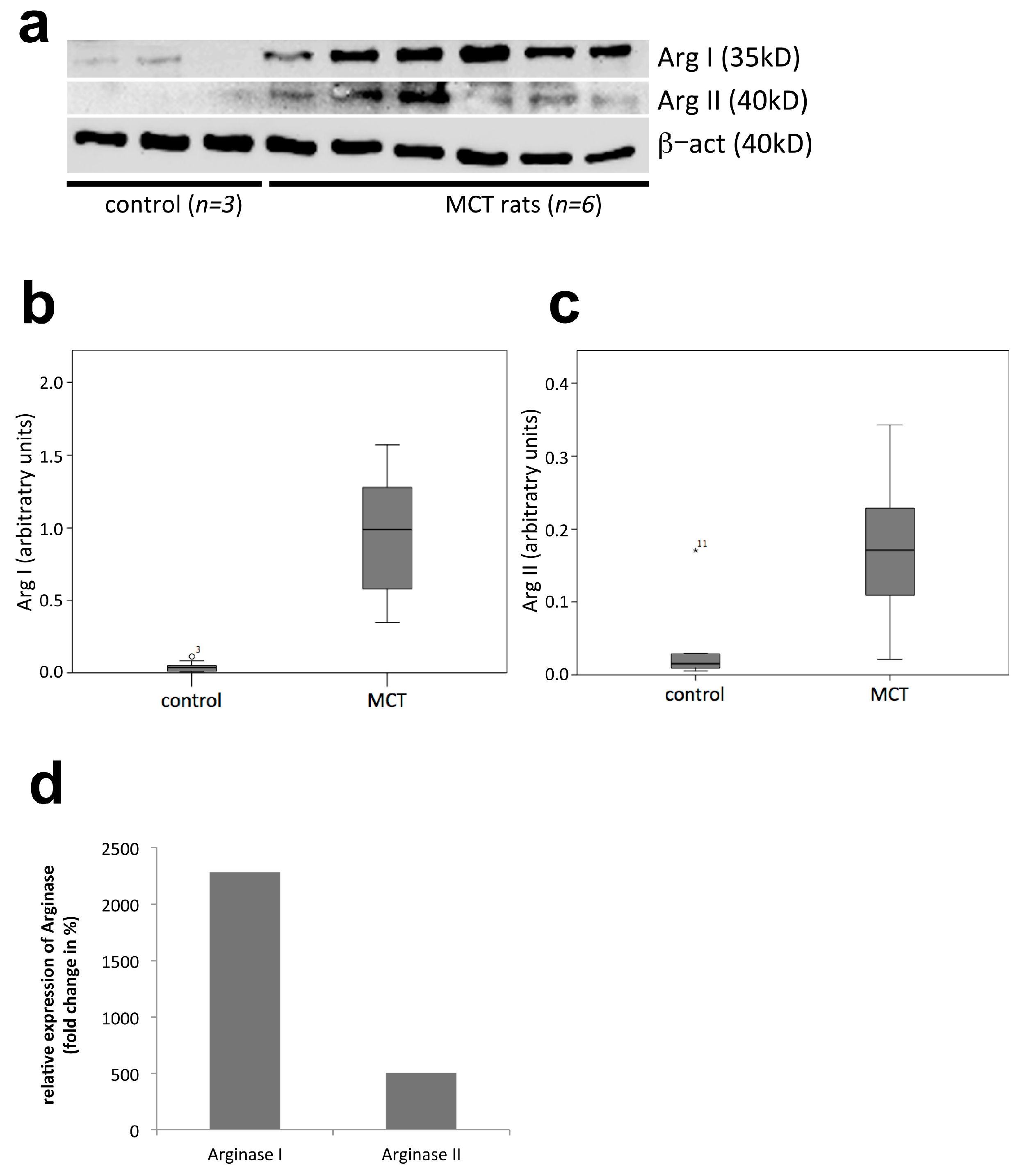
2.3. Western Blot Analysis
2.4. Haemodynamics
2.5. Histological Evaluation of Lung Tissue Remodeling Using a Sum-Score System
3. Discussion
4. Material and Methods
4.1. Animal Model of Induced Pulmonary Hypertension and Treatment Protocol
4.2. Haemodynamic Measurements
4.3. Histological Evaluation
4.4. Immunohistochemistry
4.5. Protein Isolation and Western Blotting
4.6. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Arg | Arginase |
| CTEPH | Chronic Thromboembolic Pulmonary Hypertension |
| MACI | Macitentan |
| MCT | Monocrotaline |
| Nor-NOHA | Nω-hydroxy-nor-l-arginine |
| p.i. | post injectionem |
| PAH | Pulmonary Arterial Hypertension |
| PAP | Pulmonary Artery Pressure |
| PH | Pulmonary Hypertension |
| RVPsys | Systolic Right Ventricular Pressure |
References
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.C.; Bye, H.; Brock, M.; Swiss Society of Pulmonary Hypertension. The pathogenesis of pulmonary hypertension—An update. Swiss Med. Wkly. 2015, 145, w14202. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. The Emerging Role of Arginase in Endothelial Dysfunction in Diabetes. Curr. Vasc. Pharmacol. 2016, 14, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Role of arginase in vessel wall remodeling. Front. Immunol. 2013, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Reyna, S.V.; Peyton, K.J.; Schafer, A.I. Transforming growth factor-beta(1) stimulates L-arginine transport and metabolism in vascular smooth muscle cells: Role in polyamine and collagen synthesis. Circulation 2001, 103, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.H.; Jacobs, A.T.; Morris, S.M., Jr.; Ignarro, L.J. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2000, 279, C248–C256. [Google Scholar] [PubMed]
- Chen, B.; Calvert, A.E.; Cui, H.; Nelin, L.D. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1151–L1159. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Lee, C.M.; Kang, M.J.; Huang, Y.; Giordano, F.J.; Lee, P.J.; Trow, T.K.; Homer, R.J.; Sessa, W.C.; Elias, J.A.; et al. IL-13 receptor alpha2-arginase 2 pathway mediates IL-13-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L112–L124. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meininger, C.J.; Kelly, K.A.; Hawker, J.R., Jr.; Morris, S.M., Jr.; Wu, G. Activities of arginase I and II are limiting for endothelial cell proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R64–R69. [Google Scholar] [PubMed]
- Toby, I.T.; Chicoine, L.G.; Cui, H.; Chen, B.; Nelin, L.D. Hypoxia-induced proliferation of human pulmonary microvascular endothelial cells depends on epidermal growth factor receptor tyrosine kinase activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L600–L606. [Google Scholar] [CrossRef] [PubMed]
- Kobs, R.W.; Chesler, N.C. The mechanobiology of pulmonary vascular remodeling in the congenital absence of eNOS. Biomech. Model. Mechanobiol. 2006, 5, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Dzik, J.M. Evolutionary roots of arginase expression and regulation. Front. Immunol. 2014, 5, 544. [Google Scholar] [CrossRef] [PubMed]
- Waisbren, S.E.; Gropman, A.L.; Batshaw, M.L. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J. Inherit. Metab. Dis. 2016, 39, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Pera, T.; Zuidhof, A.B.; Smit, M.; Menzen, M.H.; Klein, T.; Flik, G.; Zaagsma, J.; Meurs, H.; Maarsingh, H. Arginase inhibition prevents inflammation and remodeling in a guinea pig model of chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 2014, 349, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Nara, A.; Nagai, H.; Shintani-Ishida, K.; Ogura, S.; Shimosawa, T.; Kuwahira, I.; Shirai, M.; Yoshida, K. Pulmonary arterial hypertension in rats due to age-related arginase activation in intermittent hypoxia. Am. J. Respir. Cell Mol. Biol. 2015, 53, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Li, X.Y.; Niu, H.; Wang, H.; Jia, P.; Gong, W.; Wu, D.; Qin, W.; Xing, C. Arginase inhibitor attenuates pulmonary artery hypertension induced by hypoxia. Mol. Cell. Biochem. 2016, 412, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef] [PubMed]
- Na, S.; Kim, O.S.; Ryoo, S.; Kweon, T.D.; Choi, Y.S.; Shim, H.S.; Oh, Y.J. Cervical ganglion block attenuates the progression of pulmonary hypertension via nitric oxide and arginase pathways. Hypertension 2014, 63, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.C.; Wedes, S.H.; Hsu, J.W.; Bohren, K.M.; Comhair, S.A.; Jahoor, F.; Erzurum, S.C. Arginine metabolic endotypes in pulmonary arterial hypertension. Pulm. Circ. 2015, 5, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Kim, H.Y.; Klings, E.S.; Wood, J.; Porter, J.B.; Trachtenberg, F.; Sweeters, N.; Olivieri, N.F.; Kwiatkowski, J.L.; Virzi, L.; et al. Dysregulated arginine metabolism and cardiopulmonary dysfunction in patients with thalassaemia. Br. J. Haematol. 2015, 169, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Calvert, T.J.; Chen, B.; Chicoine, L.G.; Joshi, M.; Bauer, J.A.; Liu, Y.; Nelin, L.D. Mice deficient in Mkp-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1518–H1528. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Jin, Y.; Chen, B.; Tipple, T.E.; Nelin, L.D. Arginase II is a target of miR-17-5p and regulates miR-17-5p expression in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L197–L204. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Strauch, K.; Jin, Y.; Cui, H.; Nelin, L.D.; Chicoine, L.G. Asymmetric dimethylarginine does not inhibit arginase activity and is pro-proliferative in pulmonary endothelial cells. Clin. Exp. Pharmacol. Physiol. 2014, 41, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Arginase: Structure, mechanism, and physiological role in male and female sexual arousal. Acc. Chem. Res. 2005, 38, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Park, C.; Ahn, M.; Lee, J.H.; Shin, T. Immunohistochemical study of arginase 1 and 2 in various tissues of rats. Acta Histochem. 2012, 114, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Sun, B.; Song, X.; Zheng, Y.; Wang, L.; Wang, T.; Liu, S. Arginase inhibition protects against hypoxiainduced pulmonary arterial hypertension. Mol. Med. Rep. 2015, 12, 4743–4749. [Google Scholar] [PubMed]
- Chen, B.; Calvert, A.E.; Meng, X.; Nelin, L.D. Pharmacologic agents elevating cAMP prevent arginase II expression and proliferation of pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2012, 47, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Reyna, S.V.; Peyton, K.J.; Schafer, A.I. Physiological cyclic stretch directs L-arginine transport and metabolism to collagen synthesis in vascular smooth muscle. FASEB J. 2000, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chandrasekharan, U.M.; Bandyopadhyay, S.; Morris, S.M., Jr.; DiCorleto, P.E.; Kashyap, V.S. Thrombin induces endothelial arginase through AP-1 activation. Am. J. Physiol. Cell Physiol. 2010, 298, C952–C960. [Google Scholar] [CrossRef] [PubMed]
- Krotova, K.; Patel, J.M.; Block, E.R.; Zharikov, S. Hypoxic upregulation of arginase II in human lung endothelial cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1541–C1548. [Google Scholar] [CrossRef] [PubMed]
- Krotova, K.; Patel, J.M.; Block, E.R.; Zharikov, S. Endothelial arginase II responds to pharmacological inhibition by elevation in protein level. Mol. Cell. Biochem. 2010, 343, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.F.; Barandier, C.; Viswambharan, H.; Kwak, B.R.; Mach, F.; Mazzolai, L.; Hayoz, D.; Ruffieux, J.; Rusconi, S.; Montani, J.P.; et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: Implications for atherosclerotic endothelial dysfunction. Circulation 2004, 110, 3708–3714. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ming, X.F. Endothelial arginase: A new target in atherosclerosis. Curr. Hypertens. Rep. 2006, 8, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Steppan, J.; Tran, H.T.; Bead, V.R.; Oh, Y.J.; Sikka, G.; Bivalacqua, T.J.; Burnett, A.L.; Berkowitz, D.E.; Santhanam, L. Arginase Inhibition Reverses Endothelial Dysfunction, Pulmonary Hypertension, and Vascular Stiffness in Transgenic Sickle Cell Mice. Anesth. Analg. 2016, 123, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Grün, K.; Betge, S.; Rohm, I.; Ndongson-Dongmo, B.; Bauer, R.; Schulze, P.C.; Lichtenauer, M.; Petersen, I.; Neri, D.; et al. Lung tissue remodelling in MCT-induced pulmonary hypertension: A proposal for a novel scoring system and changes in extracellular matrix and fibrosis associated gene expression. Oncotarget 2016, 7, 81241. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, C.; Grün, K.; Betge, S.; Pernow, J.; Kelm, M.; Muessig, J.; Masyuk, M.; Kuethe, F.; Ndongson-Dongmo, B.; Bauer, R.; et al. Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension. Int. J. Mol. Sci. 2017, 18, 1609. https://doi.org/10.3390/ijms18081609
Jung C, Grün K, Betge S, Pernow J, Kelm M, Muessig J, Masyuk M, Kuethe F, Ndongson-Dongmo B, Bauer R, et al. Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension. International Journal of Molecular Sciences. 2017; 18(8):1609. https://doi.org/10.3390/ijms18081609
Chicago/Turabian StyleJung, Christian, Katja Grün, Stefan Betge, John Pernow, Malte Kelm, Johanna Muessig, Maryna Masyuk, Friedhelm Kuethe, Bernadin Ndongson-Dongmo, Reinhard Bauer, and et al. 2017. "Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension" International Journal of Molecular Sciences 18, no. 8: 1609. https://doi.org/10.3390/ijms18081609
APA StyleJung, C., Grün, K., Betge, S., Pernow, J., Kelm, M., Muessig, J., Masyuk, M., Kuethe, F., Ndongson-Dongmo, B., Bauer, R., Lauten, A., Schulze, P. C., Berndt, A., & Franz, M. (2017). Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension. International Journal of Molecular Sciences, 18(8), 1609. https://doi.org/10.3390/ijms18081609