Overcoming Oncogenic Mediated Tumor Immunity in Prostate Cancer


{kind=link}
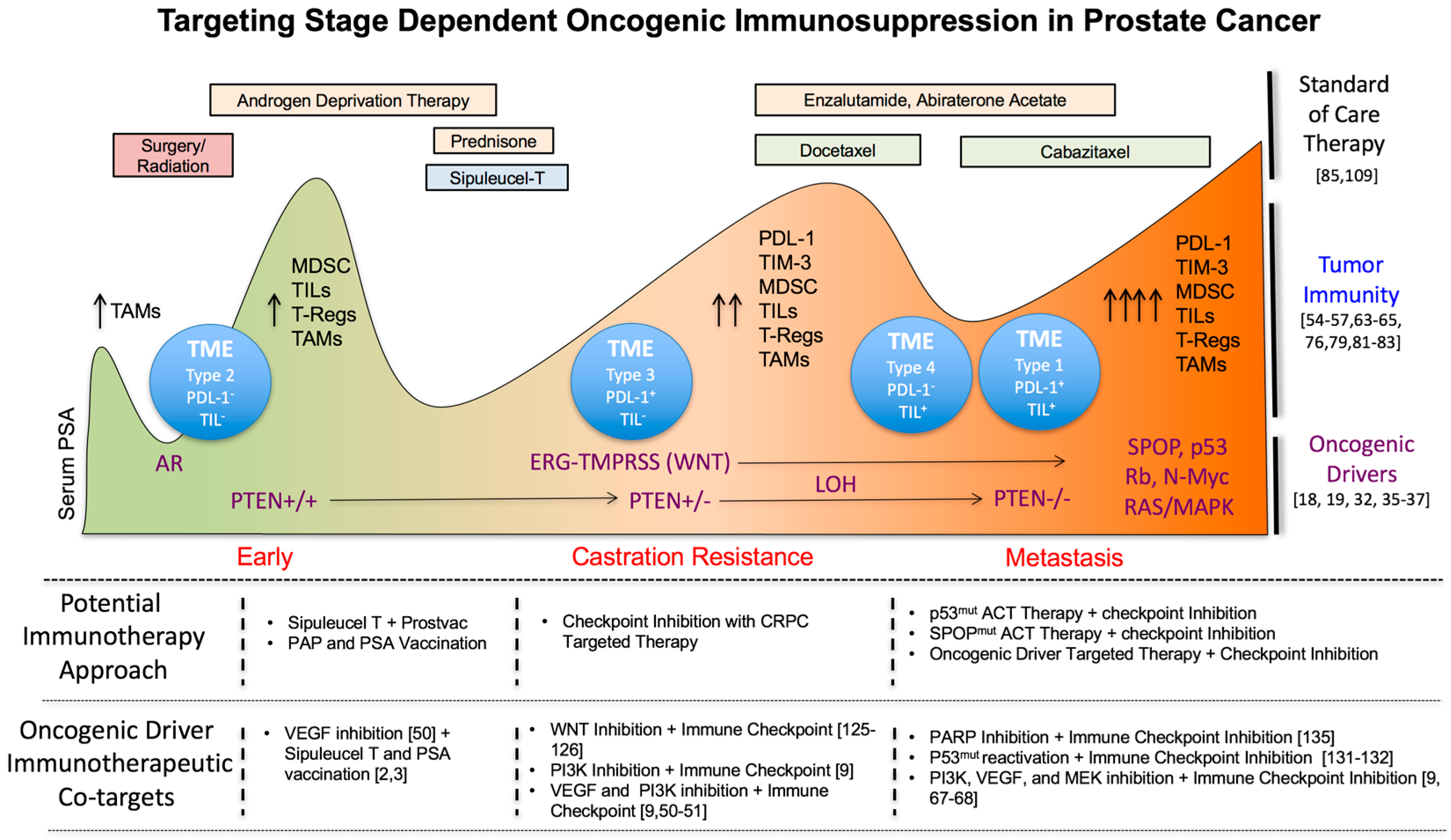
Abstract
1. Introduction
2. Prostate Cancer Heterogeneity: A Challenge for Targeted Therapies
Progression-Dependent Changes in Oncogenic Driver Pathways
3. Changes in Tumor Immunity during Prostate Cancer Progression
3.1. Effects of Inflammation on Disease Initiation and Tumor Immunity
3.2. Correlation of an Immunosuppressive Tumor Microenvironment with Prostate Cancer Progression
3.3. Targeting Immunosuppressive TME Subtypes in PC
3.4. The Impact of Standard of Care Treatments on Prostate Tumor Immunity
3.4.1. Effects on Nuclear Receptor Signaling
3.4.2. The Impact of Chemotherapy and Radiotherapy on Immunotherapy
4. Tumor Autonomous Oncogenic Drivers in Prostate Cancer Progression as Novel Immunotherapeutic Co-Targets during Early, Mid and Late Stage Disease
4.1. Early Stage Disease
4.2. Castration Resistant Prostate Cancer (CRPC)
4.3. Metastasis
5. Discussion
Abbreviations
| ACTs | adoptive cell transfer therapies |
| APCs | antigen presenting cells |
| AR | androgen receptor |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CRPC | castration resistant prostate cancer |
| EBRT | external bean radiation therapy |
| EMT | epithelial-mesenchymal transition |
| ERG | ETS-related gene; GR glucocorticoid receptor |
| mCRPC | metastatic castration resistant prostate cancer |
| MDSC | myeloid derived suppressor cell |
| NE-CRPC | neuroendocrine castration resistant prostate cancer |
| NK | natural killer cell |
| NSAIDs | non-steroidal anti- inflammatory drugs; |
| OS | overall survival |
| PAP | prostatic acid phosphatase |
| PCPT | prostate cancer prevention trial |
| PILS | prostate infiltrating lymphocytes |
| PSA | prostate specific antigen |
| RT | radiotherapy |
| SPOP | speckle type poz protein |
| TAM | tumor-associated macrophage |
| TILs | tumor infiltrating lymphocytes |
| TME | tumor microenvironment |
| TNBC | triple negative breast cancer |
Acknowledgments
Conflicts of Interest
References and Notes
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based psa-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Higano, C.S. Provenge (sipuleucel-t) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Barbieri, C.E. Clinical variability and molecular heterogeneity in prostate cancer. Asian J. Androl. 2016, 18, 543–548. [Google Scholar] [PubMed]
- Dong, Y.; Richards, J.A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2014, 33, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. PTEN null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Marrero, L.; Rodriguez, P.; Del Valle, L.; Ochoa, A.; Cui, Y. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res. 2013, 73, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic braf(v600e) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, M.; Wang, S.; Hong, B.; Wang, Z.; Li, H.; Zheng, Y.; Yang, J.; Davis, R.E.; Qian, J.; et al. P38 mapk-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat. Commun. 2014, 5, 4229. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of pten promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to braf inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in braf-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.K.; Macintyre, G.; Wedge, D.C.; van Loo, P.; Patel, K.; Lunke, S.; Alexandrov, L.B.; Sloggett, C.; Cmero, M.; Marass, F.; et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 2015, 6, 6605. [Google Scholar] [CrossRef] [PubMed]
- Danila, D.C.; Heller, G.; Gignac, G.A.; Gonzalez-Espinoza, R.; Anand, A.; Tanaka, E.; Lilja, H.; Schwartz, L.; Larson, S.; Fleisher, M.; et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin. Cancer Res. 2007, 13, 7053–7058. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Shin, H.W.; Jung, A.R.; Kwon, O.S.; Choi, Y.J.; Park, J.; Lee, J.Y. Prostate-specific extracellular vesicles as a novel biomarker in human prostate cancer. Sci. Rep. 2016, 6, 30386. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Hackenberg, M.; de Menezes, R.; Misovic, B.; Wachalska, M.; Geldof, A.; Zini, N.; de Reijke, T.; Wurdinger, T.; Vis, A.; et al. Noninvasive prostate cancer detection by measuring miRNA variants (isomiRs) in urine extracellular vesicles. Oncotarget 2016, 7, 22566–22578. [Google Scholar] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Zijlstra, A.; Rubin, M.A.; Di Vizio, D. Extracellular vesicles for liquid biopsy in prostate cancer: Where are we and where are we headed? Prostate Cancer Prostatic Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Punnoose, E.A.; Ferraldeschi, R.; Szafer-Glusman, E.; Tucker, E.K.; Mohan, S.; Flohr, P.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rodrigues, D.N.; et al. Pten loss in circulating tumour cells correlates with pten loss in fresh tumour tissue from castration-resistant prostate cancer patients. Br. J. Cancer 2015, 113, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.V.; Bingham, C.; Fittipaldi, P.; Austin, L.; Palazzo, J.; Palmer, G.; Alpaugh, K.; Cristofanilli, M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res. 2014, 16, 445. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Kantoff, P.W.; Armstrong, A.J.; Bahnson, R.R.; Cohen, M.; D’Amico, A.V.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate cancer, version 2.2014. J. Natl. Compr. Cancer Netw. 2014, 12, 686–718. [Google Scholar] [CrossRef]
- Chen, Y.; Sawyers, C.L.; Scher, H.I. Targeting the androgen receptor pathway in prostate cancer. Curr. Opin. Pharmacol. 2008, 8, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.; Collazo, J.; Kyprianou, N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int. J. Biol. Sci. 2014, 10, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. PTEN dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Helsen, C.; Prekovic, S.; van den Broeck, T.; Spans, L.; Van Poppel, H.; Joniau, S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Madan, R.A.; Tsang, K.Y.; Pazdur, M.P.; Skarupa, L.; Jones, J.L.; Poole, D.J.; Higgins, J.P.; Hodge, J.W.; et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based psa vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol. Immunother. 2010, 59, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, P.F.; Chodak, G.; Whitmore, J.B.; Sims, R.; Frohlich, M.W.; Kantoff, P.W. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-t in the immunotherapy for prostate adenocarcinoma treatment (impact) trial. Urology 2013, 81, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Gurel, B.; Lucia, M.S.; Thompson, I.M., Jr.; Goodman, P.J.; Tangen, C.M.; Kristal, A.R.; Parnes, H.L.; Hoque, A.; Lippman, S.M.; Sutcliffe, S.; et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2014, 23, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Lolli, C.; Caffo, O.; Scarpi, E.; Aieta, M.; Conteduca, V.; Maines, F.; Bianchi, E.; Massari, F.; Veccia, A.; Chiuri, V.E.; et al. Systemic immune-inflammation index predicts the clinical outcome in patients with mcrpc treated with abiraterone. Front. Pharmacol. 2016, 7, 376. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Howard, L.E.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freedland, S.J. Aspirin, nsaids, and risk of prostate cancer: Results from the reduce study. Clin. Cancer Res. 2015, 21, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Gollapudi, K.; Galet, C.; Grogan, T.; Zhang, H.; Said, J.W.; Huang, J.; Elashoff, D.; Freedland, S.J.; Rettig, M.; Aronson, W.J. Association between tumor-associated macrophage infiltration, high grade prostate cancer, and biochemical recurrence after radical prostatectomy. Am. J. Cancer Res. 2013, 3, 523–529. [Google Scholar] [PubMed]
- Lundholm, M.; Hagglof, C.; Wikberg, M.L.; Stattin, P.; Egevad, L.; Bergh, A.; Wikstrom, P.; Palmqvist, R.; Edin, S. Secreted factors from colorectal and prostate cancer cells skew the immune response in opposite directions. Sci. Rep. 2015, 5, 15651. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Brakenhielm, E.; Burton, J.B.; Johnson, M.; Chavarria, N.; Morizono, K.; Chen, I.; Alitalo, K.; Wu, L. Modulating metastasis by a lymphangiogenic switch in prostate cancer. Int. J. Cancer 2007, 121, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lalani, A.S.; Harding, T.C.; Luan, B.; Koprivnikar, K.; Huan Tu, G.; Prell, R.; VanRoey, M.J.; Simmons, A.D.; Jooss, K. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin. Cancer Res. 2006, 12, 6808–6816. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: An overview of rationale, preclinical evidence, and initial clinical data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Zurita, A.J.; Werner, L.; Bruce, J.Y.; Carducci, M.A.; Stein, M.N.; Heath, E.I.; Hussain, A.; Tran, H.T.; Sweeney, C.J.; et al. A randomized phase ii trial of short-course androgen deprivation therapy with or without bevacizumab for patients with recurrent prostate cancer after definitive local therapy. J. Clin. Oncol. 2016, 34, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Docetaxel with or without low-dose, short course sunitinib in refractory solid tumors, NCT01803503.
- Strasner, A.; Karin, M. Immune infiltration and prostate cancer. Front. Oncol. 2015, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Kiniwa, Y.; Miyahara, Y.; Wang, H.Y.; Peng, W.; Peng, G.; Wheeler, T.M.; Thompson, T.C.; Old, L.J.; Wang, R.F. CD8+ FOXP3+ regulatory t cells mediate immunosuppression in prostate cancer. Clin. Cancer Res. 2007, 13, 6947–6958. [Google Scholar] [CrossRef] [PubMed]
- Yuri, P.; Hendri, A.Z.; Danarto, R. Association between tumor-associated macrophages and microvessel density on prostate cancer progression. Prostate Int. 2015, 3, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Qian, Y.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Hao, W. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol. Lett. 2015, 10, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Izumi, K.; Lee, S.O.; Lin, W.J.; Yeh, S.; Chang, C. Anti-androgen receptor asc-j9 versus anti-androgens MDV3100 (enzalutamide) or casodex (bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell Death Dis. 2013, 4, e764. [Google Scholar] [CrossRef] [PubMed]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.; Tan, Z.; Ma, K.; Bao, L.; Yun, Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int. J. Clin. Exp. Med. 2014, 7, 3181–3192. [Google Scholar] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-dependent mdsc infiltration impairs tumor progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Komarova, E.A.; Krivokrysenko, V.; Wang, K.; Neznanov, N.; Chernov, M.V.; Komarov, P.G.; Brennan, M.L.; Golovkina, T.V.; Rokhlin, O.W.; Kuprash, D.V.; et al. p53 Is a suppressor of inflammatory response in mice. FASEB J. 2005, 19, 1030–1032. [Google Scholar] [CrossRef] [PubMed]
- Gevensleben, H.; Dietrich, D.; Golletz, C.; Steiner, S.; Jung, M.; Thiesler, T.; Majores, M.; Stein, J.; Uhl, B.; Muller, S.; et al. The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin. Cancer Res. 2016, 22, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Sio, A.; Angeles, A.; Roberts, M.E.; Azad, A.A.; Chi, K.N.; Zoubeidi, A. PD-L1 is highly expressed in enzalutamide resistant prostate cancer. Oncotarget 2015, 6, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.R.; Jin, Z.H.; Yuan, K.C.; Jin, X.S. Analysis of TIM-3 as a therapeutic target in prostate cancer. Tumour Biol. 2014, 35, 11409–11414. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. Pdl1 regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. Ras/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between mek and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The activation of mapk in melanoma cells resistant to braf inhibition promotes PD-L1 expression that is reversible by mek and PI3K inhibition. Clin. Cancer Res. 2013, 19, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Atefi, M.; Avramis, E.; Lassen, A.; Wong, D.J.; Robert, L.; Foulad, D.; Cerniglia, M.; Titz, B.; Chodon, T.; Graeber, T.G.; et al. Effects of mapk and PI3K pathways on PD-L1 expression in melanoma. Clin. Cancer Res. 2014, 20, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Lee, M.J.; Shin, D.C.; Kim, J.S.; Chwae, Y.J.; Kwon, M.H.; Kim, K.; Park, S. Activation of mitogen activated protein kinase-erk kinase (MEK) increases t cell immunoglobulin mucin domain-3 (TIM-3) transcription in human t lymphocytes and a human mast cell line. Mol. Immunol. 2011, 48, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.M.; Nirschl, T.R.; Nirschl, C.J.; Francica, B.J.; Kochel, C.M.; van Bokhoven, A.; Meeker, A.K.; Lucia, M.S.; Anders, R.A.; DeMarzo, A.M.; et al. Paucity of pd-l1 expression in prostate cancer: Innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying cancers based on t-cell infiltration and pd-l1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.; Carroll, P.; Weinberg, V.; Chan, S.; Lewis, J.; Corman, J.; Amling, C.L.; Stephenson, R.A.; Simko, J.; Sheikh, N.A.; et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Madan, R.A.; Tsang, K.Y.; Jochems, C.; Marte, J.L.; Farsaci, B.; Tucker, J.A.; Hodge, J.W.; Liewehr, D.J.; Steinberg, S.M.; et al. Immune impact induced by prostvac (PSA-tricom), a therapeutic vaccine for prostate cancer. Cancer Immunol. Res. 2014, 2, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Valdman, A.; Jaraj, S.J.; Comperat, E.; Charlotte, F.; Roupret, M.; Pisa, P.; Egevad, L. Distribution of foxp3-, CD4- and CD8-positive lymphocytic cells in benign and malignant prostate tissue. APMIS 2010, 118, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Flammiger, A.; Weisbach, L.; Huland, H.; Tennstedt, P.; Simon, R.; Minner, S.; Bokemeyer, C.; Sauter, G.; Schlomm, T.; Trepel, M. High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur. J. Cancer 2013, 49, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Wang, L.; Dai, J.; Kryczek, I.; Wei, S.; Vatan, L.; Altuwaijri, S.; Sparwasser, T.; Wang, G.; Keller, E.T.; et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology 2012, 1, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, N.; Takayama, H.; Nakayama, M.; Nakai, Y.; Kawashima, A.; Mukai, M.; Nagahara, A.; Aozasa, K.; Tsujimura, A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011, 107, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Moore, M.L.; Grayson, J.M.; Dubey, P. Increased CD8+ T-cell function following castration and immunization is countered by parallel expansion of regulatory t cells. Cancer Res. 2012, 72, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; Bruno, T.C.; Maris, C.H.; Xu, L.; Thoburn, C.J.; DeMarzo, A.M.; Meeker, A.K.; Isaacs, W.B.; Drake, C.G. Phenotypic analysis of prostate-infiltrating lymphocytes reveals Th17 and treg skewing. Clin. Cancer Res. 2008, 14, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Kibel, A.S.; Yu, E.Y.; Karsh, L.I.; Elfiky, A.; Shore, N.D.; Vogelzang, N.J.; Corman, J.M.; Millard, F.E.; Maher, J.C.; et al. Sequencing of sipuleucel-t and androgen deprivation therapy in men with hormone-sensitive biochemically recurrent prostate cancer: A phase ii randomized trial. Clin. Cancer Res. 2017, 23, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.J.; Lai, K.P.; Zeng, W.; Chuang, K.H.; Altuwaijri, S.; Chang, C. Androgen receptor influences on body defense system via modulation of innate and adaptive immune systems: Lessons from conditional ar knockout mice. Am. J. Pathol. 2012, 181, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Xu, M.; Liang, Y.; Yang, K.; Guo, Y.; Yang, X.; Fu, Y.X. Androgen receptor antagonists compromise t cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med. 2016, 8, 333ra347. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397.e9–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Girndt, M.; Sester, U.; Kaul, H.; Hunger, F.; Kohler, H. Glucocorticoids inhibit activation-dependent expression of costimulatory molecule B7–1 in human monocytes. Transplantation 1998, 66, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, M.; Verhaar, A.P.; van den Brink, G.R.; Hommes, D.W. Glucocorticoid signaling: A nongenomic mechanism for T-cell immunosuppression. Trends Mol. Med. 2007, 13, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Gasser, J.; Feige, U. Dexamethasone enhances CTLA-4 expression during t cell activation. Cell Mol. Life Sci. 1999, 55, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Gu, B.; Zhang, P.; Wu, X. Dexamethasone enhances programmed cell death 1 (pd-1) expression during t cell activation: An insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 2015, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.E.; et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Diederich, S.; Eigendorff, E.; Burkhardt, P.; Quinkler, M.; Bumke-Vogt, C.; Rochel, M.; Seidelmann, D.; Esperling, P.; Oelkers, W.; Bahr, V. 11β-hydroxysteroid dehydrogenase types 1 and 2: An important pharmacokinetic determinant for the activity of synthetic mineralo- and glucocorticoids. J. Clin. Endocrinol. Metab. 2002, 87, 5695–5701. [Google Scholar] [CrossRef] [PubMed]
- Kotsakis, A.; Sarra, E.; Peraki, M.; Koukourakis, M.; Apostolaki, S.; Souglakos, J.; Mavromanomakis, E.; Vlachonikolis, J.; Georgoulias, V. Docetaxel-induced lymphopenia in patients with solid tumors: A prospective phenotypic analysis. Cancer 2000, 89, 1380–1386. [Google Scholar] [CrossRef]
- Kodumudi, K.N.; Woan, K.; Gilvary, D.L.; Sahakian, E.; Wei, S.; Djeu, J.Y. A novel chemoimmunomodulating property of docetaxel: Suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010, 16, 4583–4594. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Duan, X.F.; Wang, L.P.; Xu, Y.J.; Huang, L.; Zhang, T.F.; Liu, J.Y.; Li, F.; Zhang, Z.; Yue, D.L.; et al. Selective depletion of regulatory t cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J. Immunol. Res. 2014, 2014, 286170. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.T.; Schlom, J.; Hodge, J.W. Combination of docetaxel and recombinant vaccine enhances t-cell responses and antitumor activity: Effects of docetaxel on immune enhancement. Clin. Cancer Res. 2008, 14, 3536–3544. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Carson, W.E., 3rd; Shapiro, C.L.; Crespin, T.R.; Thornton, L.M.; Andersen, B.L. Cellular immunity in breast cancer patients completing taxane treatment. Clin. Cancer Res. 2004, 10, 3401–3409. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.J. Immune modulation by ionizing radiation and its implications for cancer immunotherapy. Curr. Pharm. Des. 2002, 8, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Chang, J.S.; Rha, K.H.; Hong, S.J.; Choi, Y.D.; Ham, W.S.; Kim, J.W.; Cho, J. Does radiotherapy for the primary tumor benefit prostate cancer patients with distant metastasis at initial diagnosis? PLoS ONE 2016, 11, e0147191. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Hipkiss, E.L.; Borzillary, S.; Wada, S.; Grosso, J.F.; Yen, H.R.; Getnet, D.; Bruno, T.C.; Goldberg, M.V.; Pardoll, D.M.; et al. Radiotherapy augments the immune response to prostate cancer in a time-dependent manner. Prostate 2008, 68, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase i/ii study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Thompson, I.; Thrasher, J.B.; Aus, G.; Burnett, A.L.; Canby-Hagino, E.D.; Cookson, M.S.; D’Amico, A.V.; Dmochowski, R.R.; Eton, D.T.; Forman, J.D.; et al. Guideline for the management of clinically localized prostate cancer: 2007 update. J. Urol. 2007, 177, 2106–2131. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Arlen, P.M.; Mohebtash, M.; Hodge, J.W.; Gulley, J.L. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin. Investig. Drugs 2009, 18, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Genentech, Inc. Study of ipatasertib or GDC-0980 with abiraterone acetate versus coralie in participants with castration-resistant prostate cancer previously treated with docetaxel chemotherapy, NCT02215096.
- GlaxoSmithKline. Dose-finding study of GSK2636771 when administered in combination with enzalutamide in male subjects with metastatic castration-resistant prostate cancer, NCT02215096, 2018.
- AstraZeneca. AZD8186 first time in patient ascending dose study, NCT01884285, 2017.
- Institute of Cancer Research, UK; Royal Marsden, NHS Foundation Trust. A study of enzalutamide in combination with AZD5363 in patients with MCRPC, NCT02525068, 2018.
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Nam, R.K.; Sugar, L.; Yang, W.; Srivastava, S.; Klotz, L.H.; Yang, L.Y.; Stanimirovic, A.; Encioiu, E.; Neill, M.; Loblaw, D.A.; et al. Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br. J. Cancer 2007, 97, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Iljin, K.; Wolf, M.; Edgren, H.; Gupta, S.; Kilpinen, S.; Skotheim, R.I.; Peltola, M.; Smit, F.; Verhaegh, G.; Schalken, J.; et al. TMPRSS2 fusions with oncogenic ets factors in prostate cancer involve unbalanced genomic rearrangements and are associated with hdac1 and epigenetic reprogramming. Cancer Res. 2006, 66, 10242–10246. [Google Scholar] [CrossRef] [PubMed]
- Adamo, P.; Ladomery, M.R. The oncogene erg: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yi, Y.; Yull, F.E.; Blackwell, T.S.; Clark, P.E.; Koyama, T.; Smith, J.A., Jr.; Matusik, R.J. NF-κB gene signature predicts prostate cancer progression. Cancer Res. 2014, 74, 2763–2772. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. NF-κB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhao, J.C.; Kim, J.; Jin, H.J.; Wang, C.Y.; Yu, J. ERG is a critical regulator of WNT/LEF1 signaling in prostate cancer. Cancer Res. 2013, 73, 6068–6079. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, A.; Tadagavadi, R.K.; Swafford, D.; Manicassamy, S. Modulation of inflammatory responses by WNT/β-catenin signaling in dendritic cells: A novel immunotherapy target for autoimmunity and cancer. Front. Immunol. 2016, 7, 460. [Google Scholar] [CrossRef] [PubMed]
- Novartis, Pharmaceuticals. A study of LGK974 in patients with malignancies dependent on wnt ligands, NCT01351103, 2018.
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Demichelis, F. Prostate cancer: Intrapatient heterogeneity in prostate cancer. Nat. Rev. Urol. 2015, 12, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Cui, Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J. Immunother. Cancer 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Bjorklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 AND MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 blockade synergizes therapeutically with parp inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. Study of olaparib (lynparza™) versus enzalutamide or abiraterone acetate in men with metastatic castration-resistant prostate cancer (profound study), NCT02987543, 2020.
- Clovis Oncology, Inc.; Foundation, Medicine. A study of rucaparib in patients with metastatic castration-resistant prostate cancer and homologous recombination gene deficiency, NCT02952534, 2019.
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. Parp inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-cell transfer therapy targeting mutant kras in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; McGuinness, D.H.; McCall, P.; Underwood, M.A.; Seywright, M.; Orange, C.; Edwards, J. Upregulation of MAPK pathway is associated with survival in castrate-resistant prostate cancer. Br. J. Cancer 2011, 104, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote emt and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, J.; Qian, J.; Wezeman, M.; Kwak, L.W.; Yi, Q. Tumor evasion of the immune system: Inhibiting p38 MAPK signaling restores the function of dendritic cells in multiple myeloma. Blood 2006, 107, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.J.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes t cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Effector, Therapeutics. A dose escalation and cohort-expansion study of oral EFT508 in subjects with advanced solid tumors, NCT02605083, 2017.
- Jonsson Comprehensive Cancer Center; National Cancer, Institute. Trametinib in treating patients with progressive metastatic hormone-resistant prostate cancer, NCT02881242, 2018.
- Van Rhee, F.; Szymonifka, J.; Anaissie, E.; Nair, B.; Waheed, S.; Alsayed, Y.; Petty, N.; Shaughnessy, J.D., Jr.; Hoering, A.; Crowley, J.; et al. Total therapy 3 for multiple myeloma: Prognostic implications of cumulative dosing and premature discontinuation of vtd maintenance components, bortezomib, thalidomide, and dexamethasone, relevant to all phases of therapy. Blood 2010, 116, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-CAS9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryant, G.; Wang, L.; Mulholland, D.J. Overcoming Oncogenic Mediated Tumor Immunity in Prostate Cancer. Int. J. Mol. Sci. 2017, 18, 1542. https://doi.org/10.3390/ijms18071542
Bryant G, Wang L, Mulholland DJ. Overcoming Oncogenic Mediated Tumor Immunity in Prostate Cancer. International Journal of Molecular Sciences. 2017; 18(7):1542. https://doi.org/10.3390/ijms18071542
Chicago/Turabian StyleBryant, Geoffrey, Lin Wang, and David J. Mulholland. 2017. "Overcoming Oncogenic Mediated Tumor Immunity in Prostate Cancer" International Journal of Molecular Sciences 18, no. 7: 1542. https://doi.org/10.3390/ijms18071542
APA StyleBryant, G., Wang, L., & Mulholland, D. J. (2017). Overcoming Oncogenic Mediated Tumor Immunity in Prostate Cancer. International Journal of Molecular Sciences, 18(7), 1542. https://doi.org/10.3390/ijms18071542