Rapid and Direct VHH and Target Identification by Staphylococcal Surface Display Libraries

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
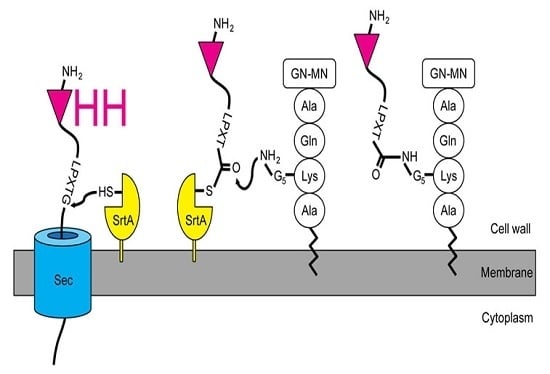
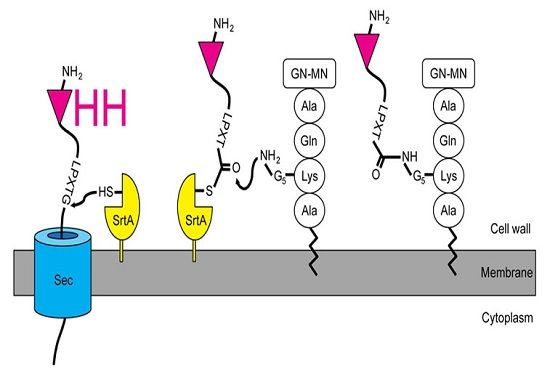
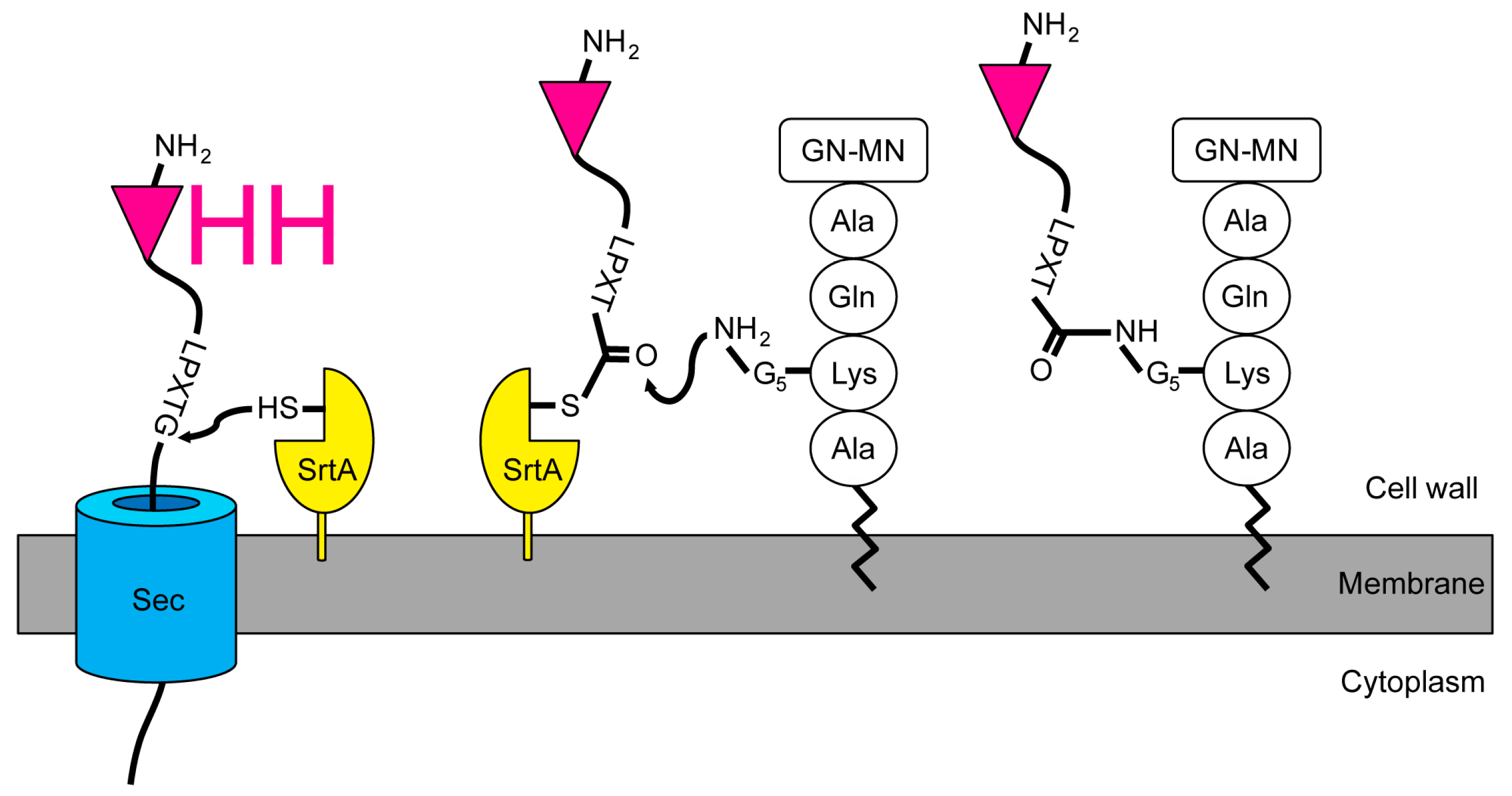
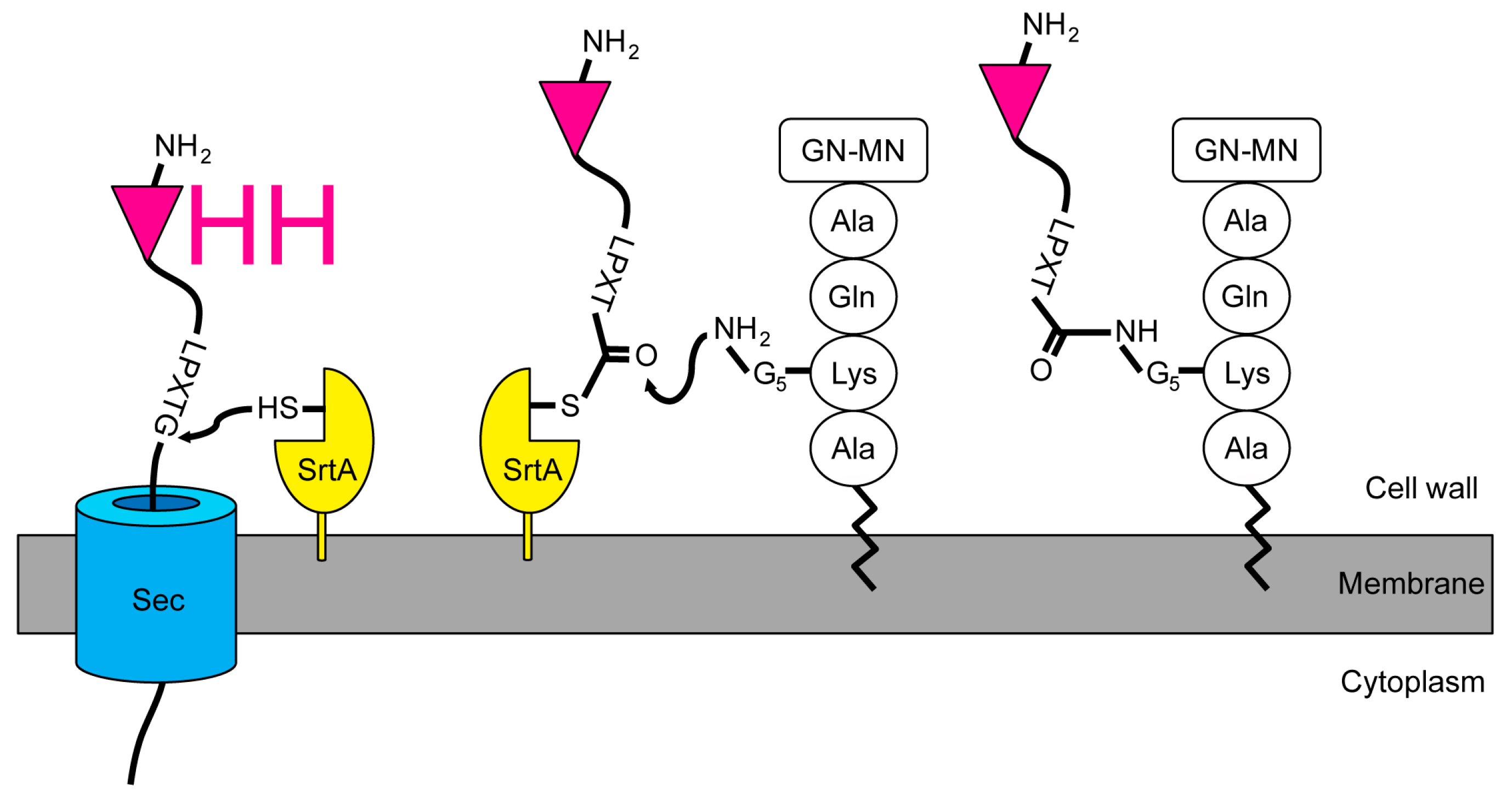
2.1. Expression and Attachment of Single-Domain Antigen-Binding Fragments from Camelid Heavy Chain-Only Antibodies (VHH) to the Peptidoglycan in S. aureus
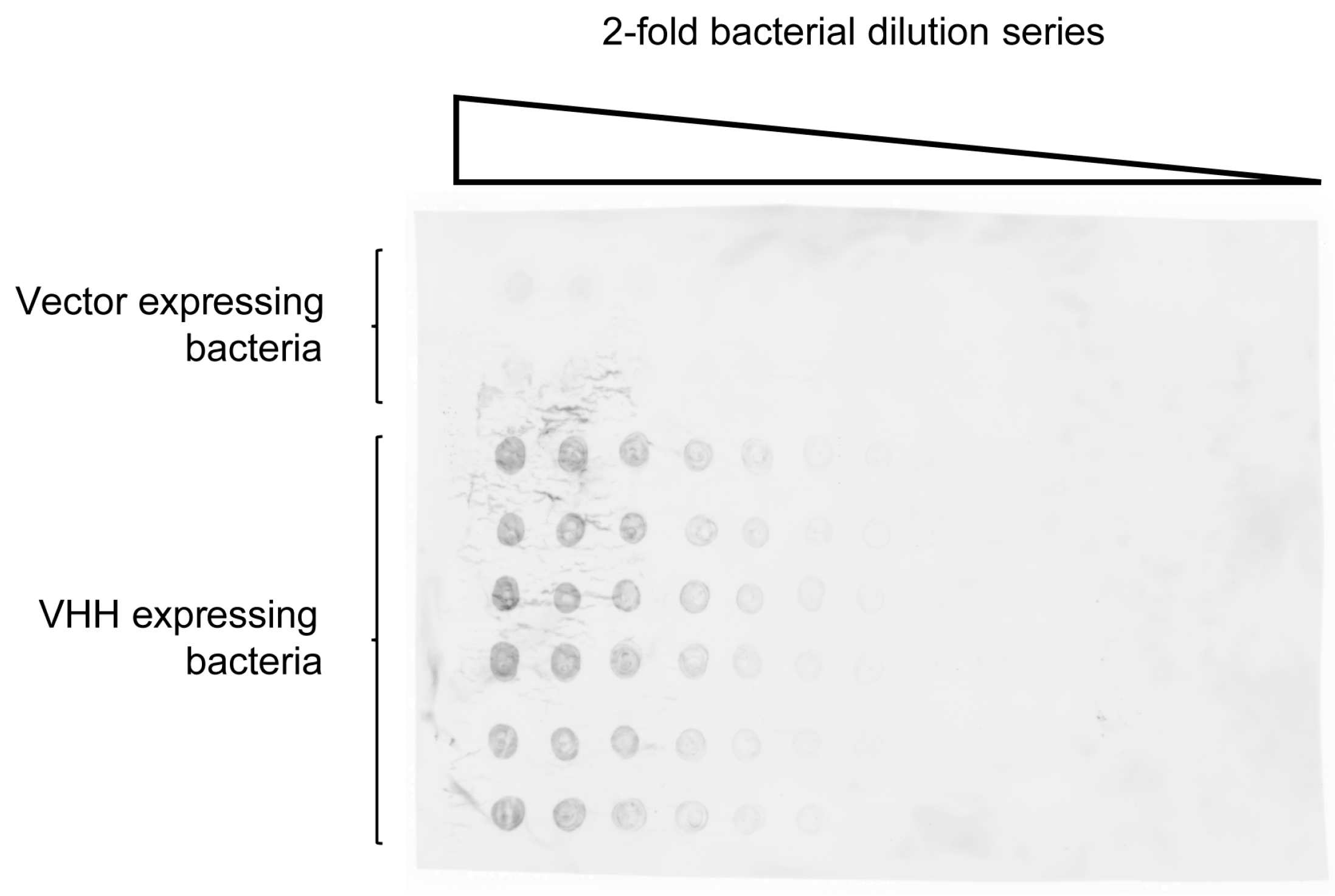
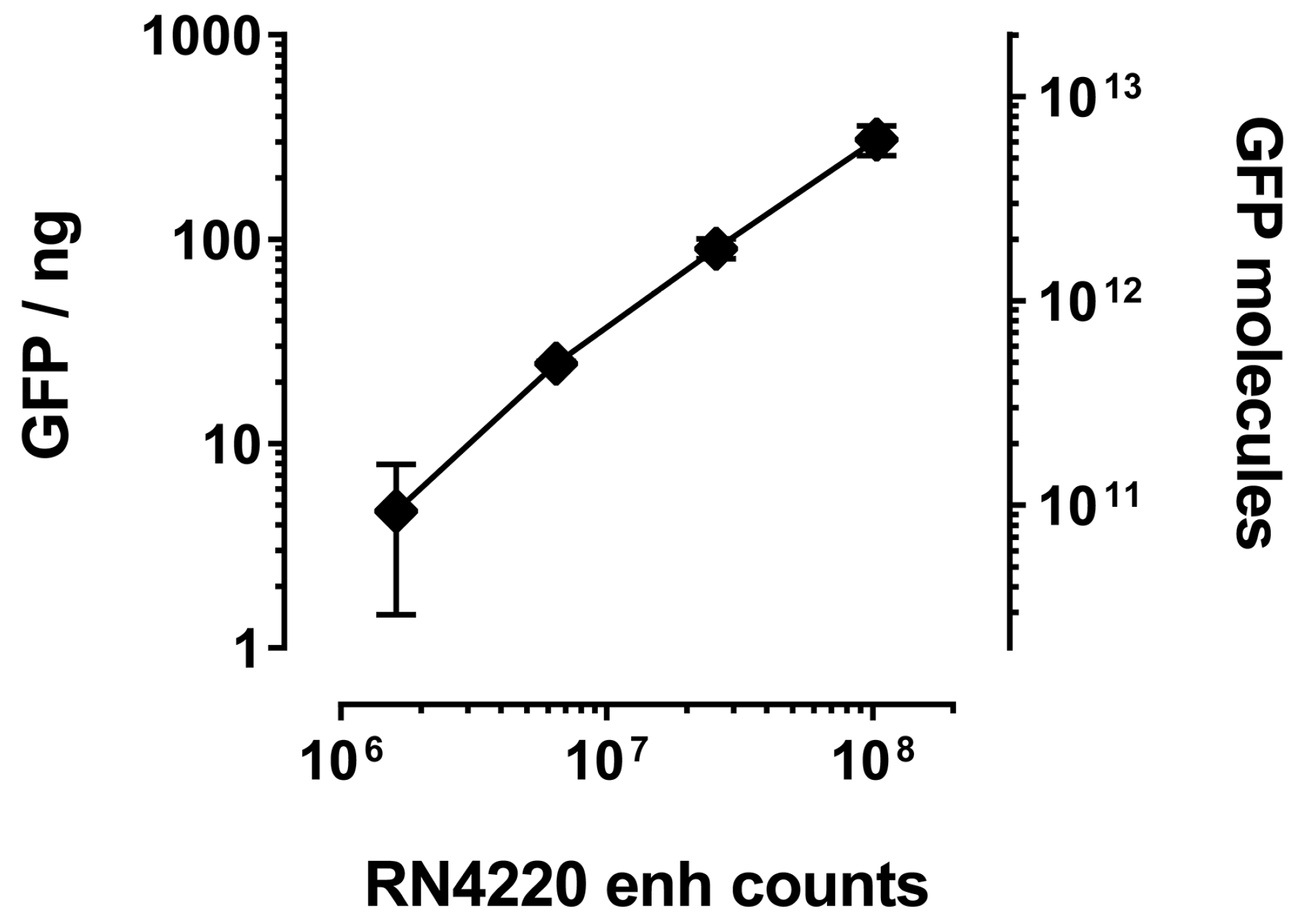
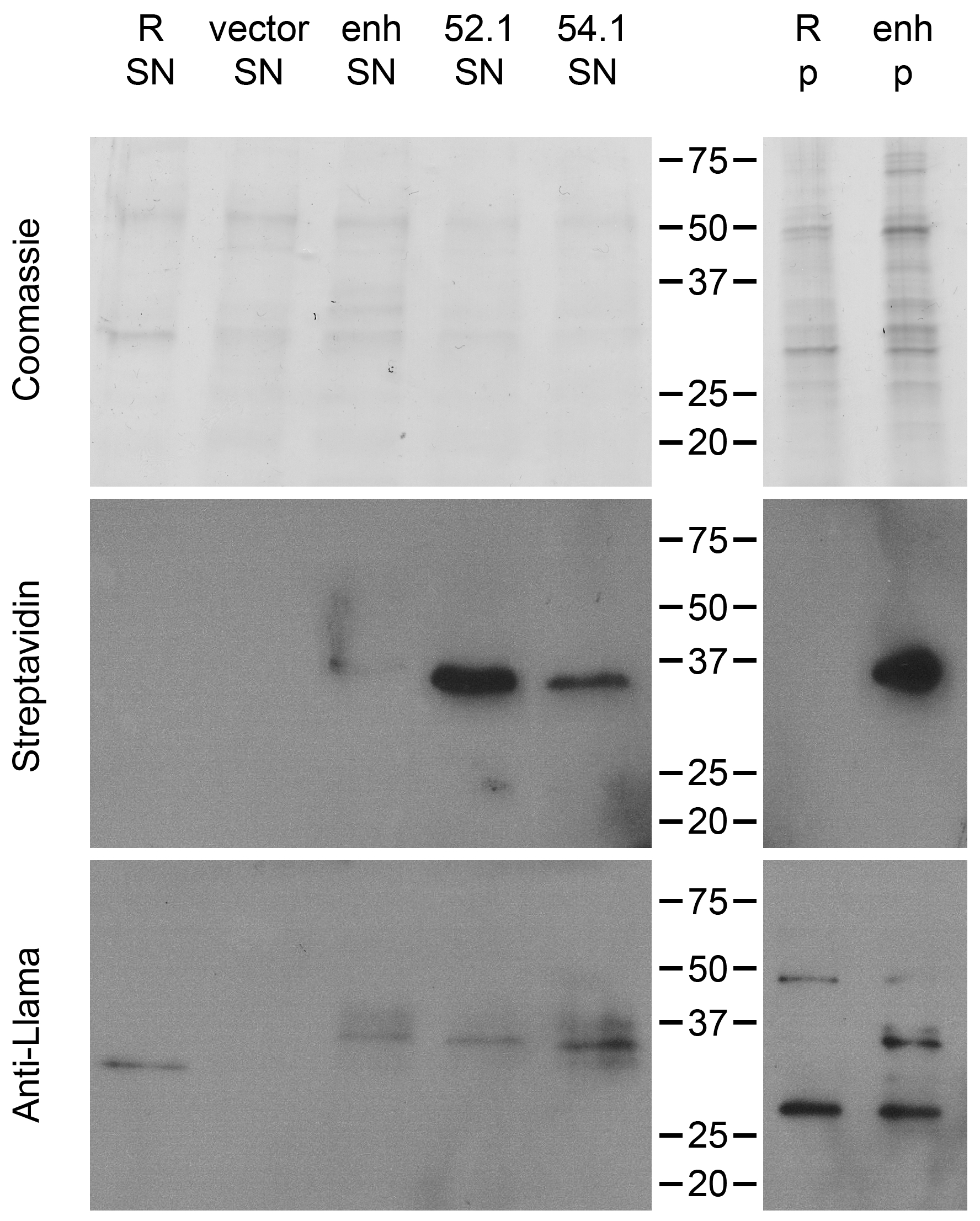
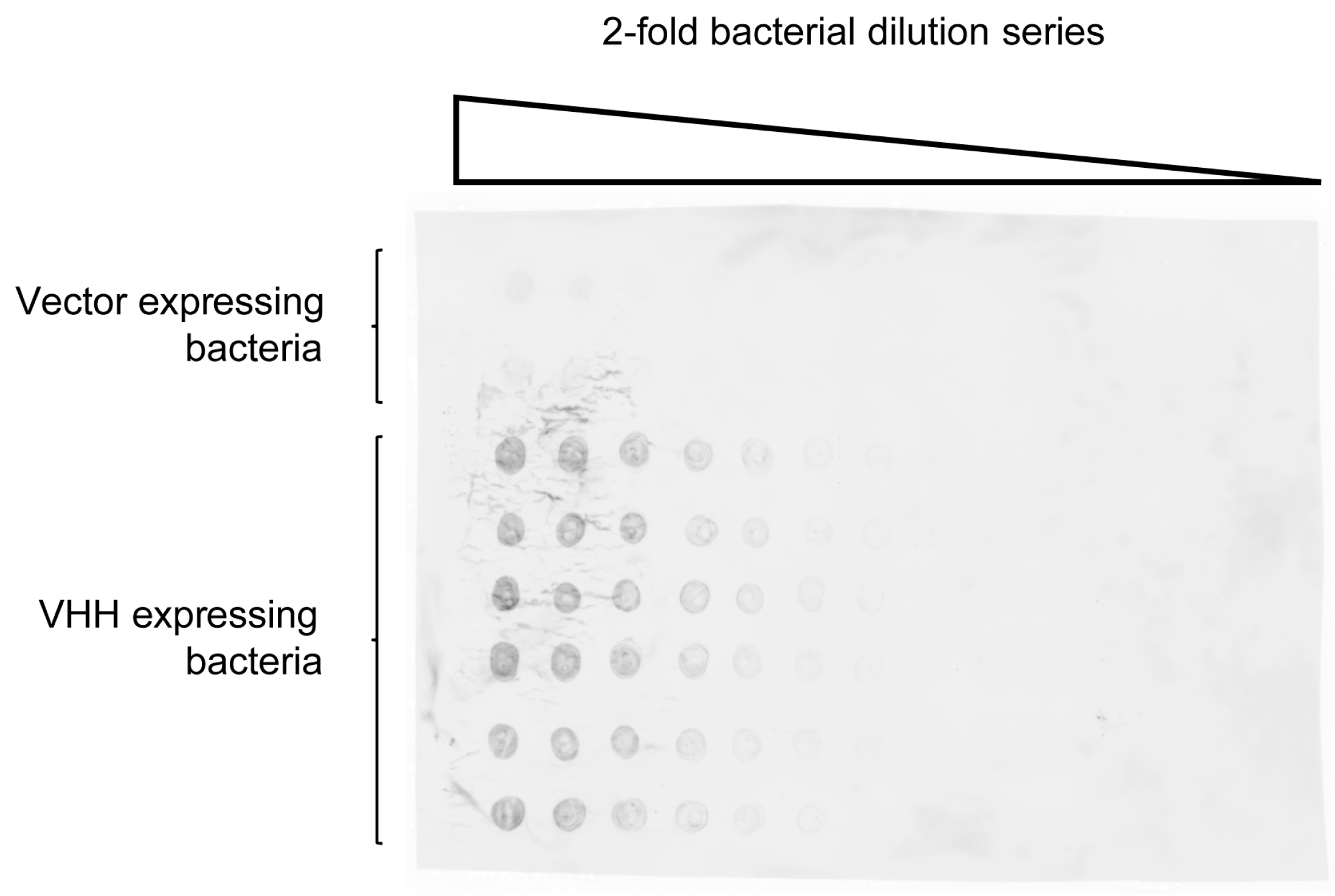
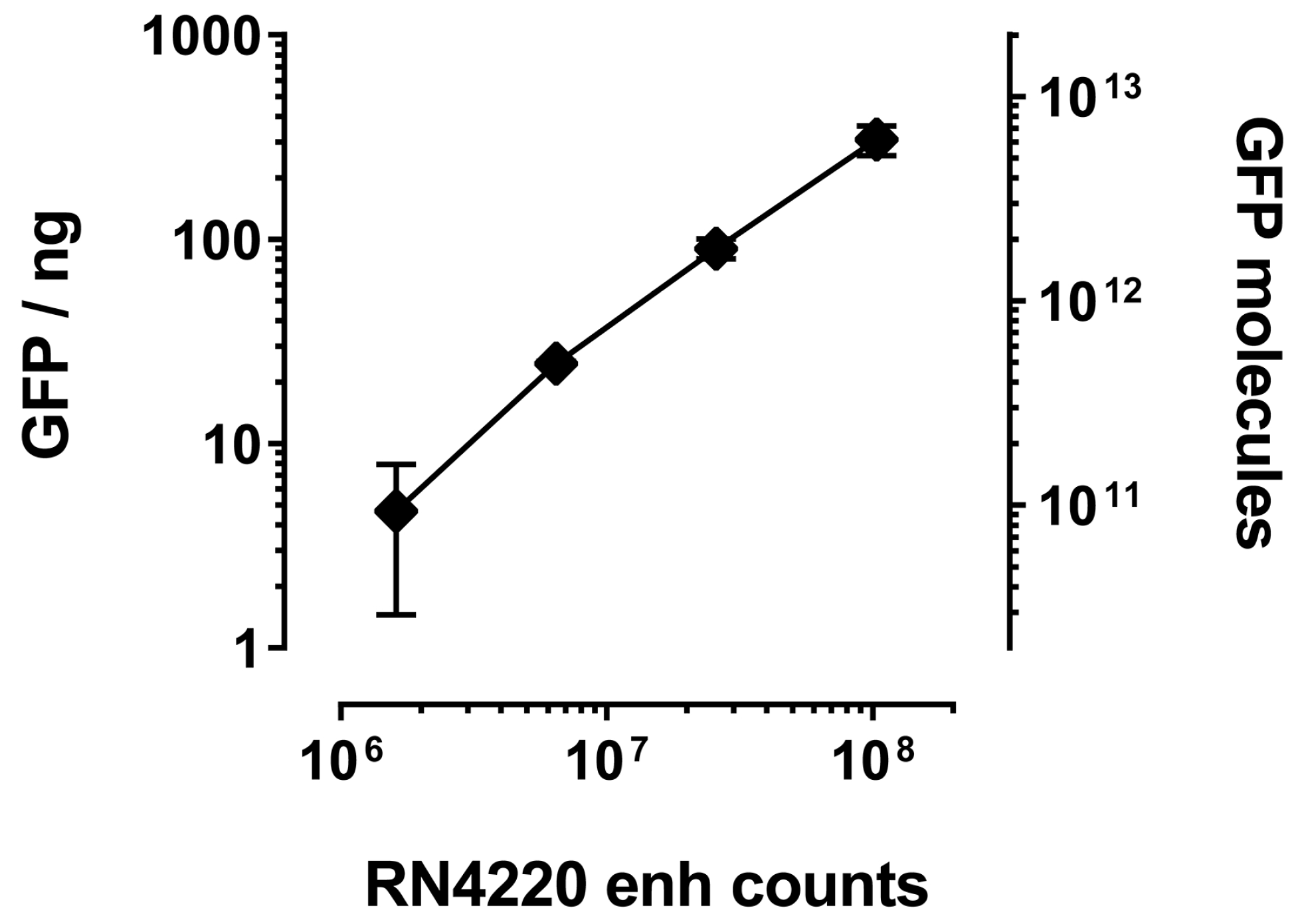
2.2. Quantification of VHH Surface Exposure and Immunoprecipitation (IP) Capacity
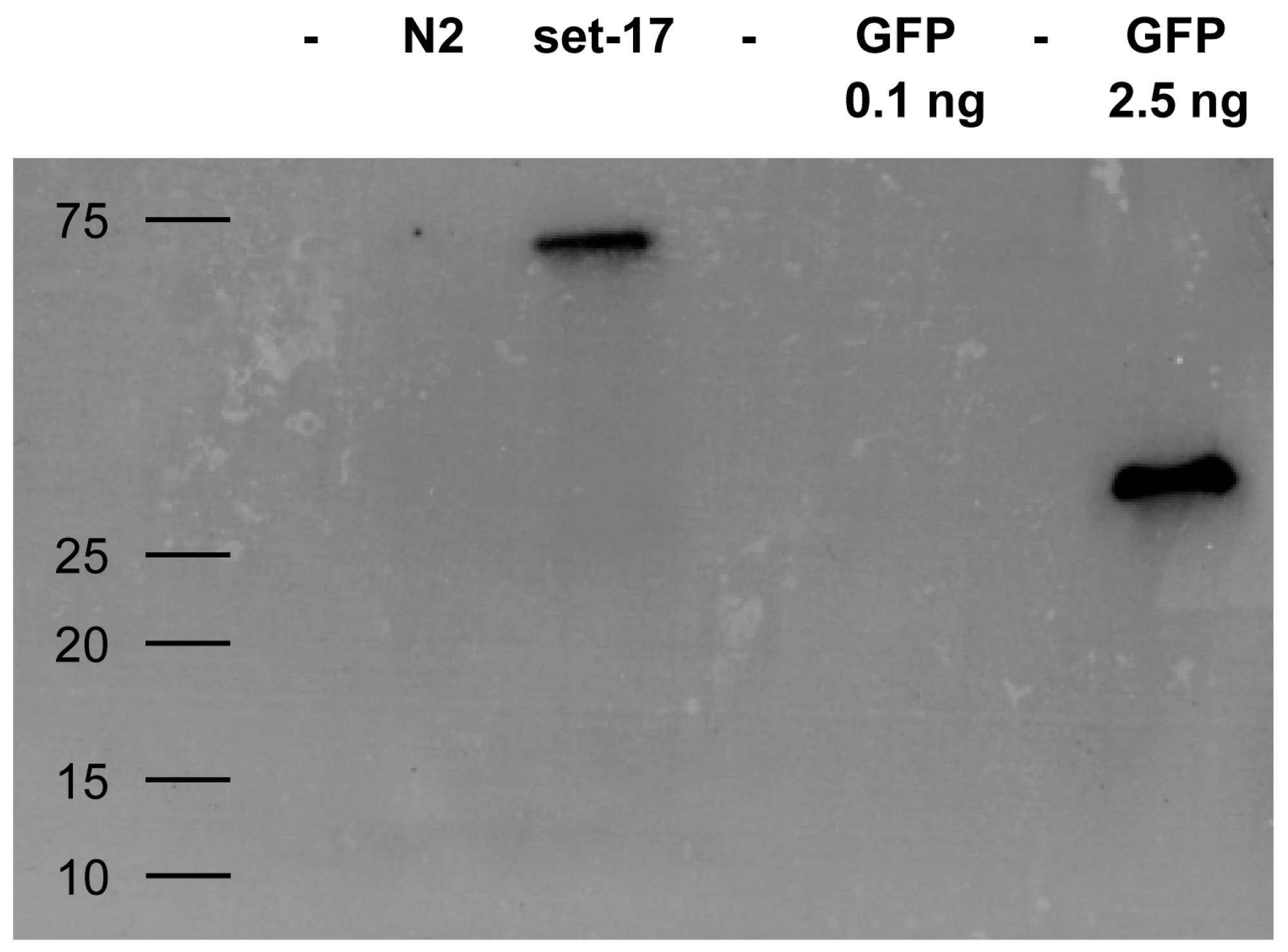
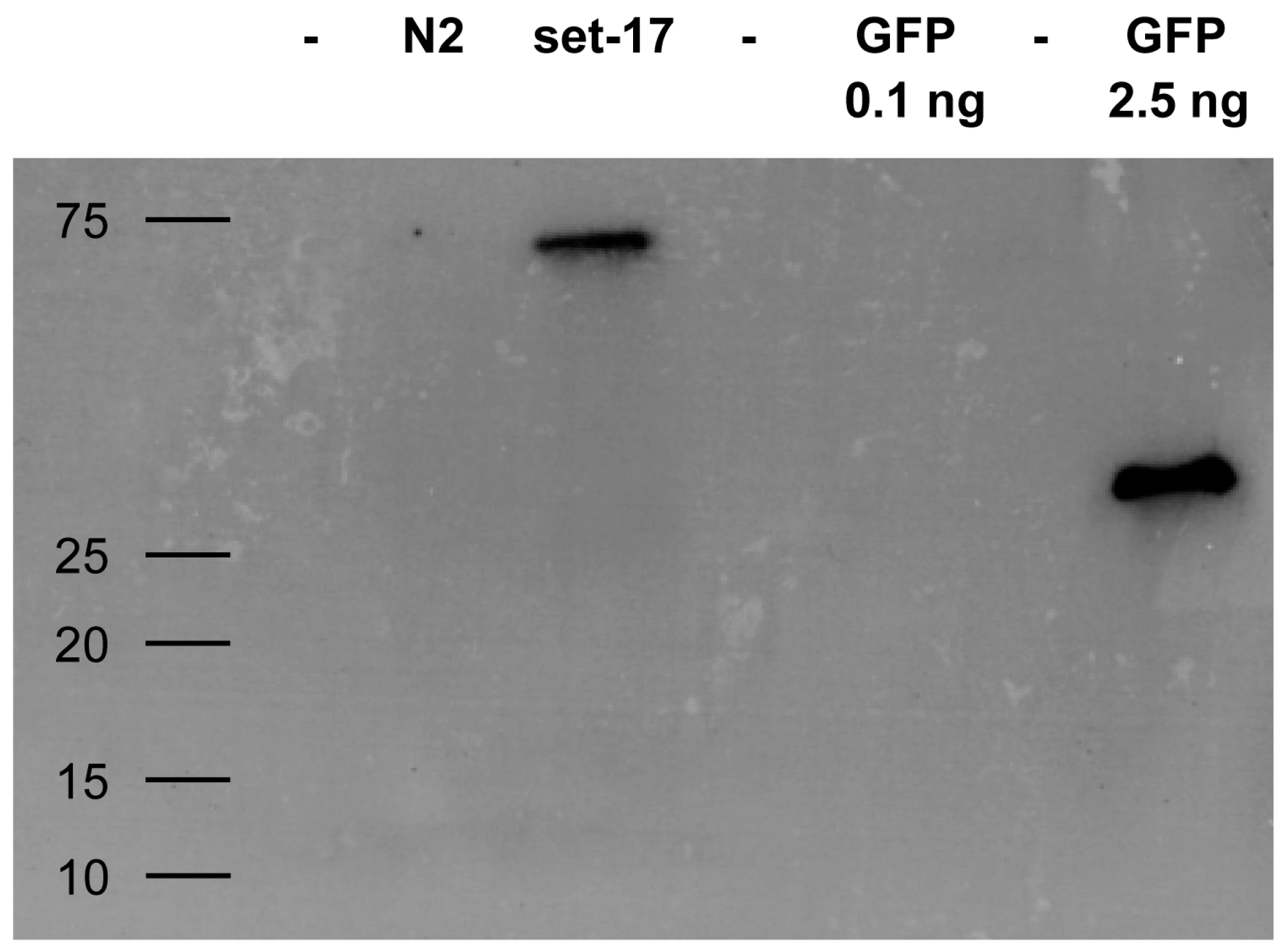
2.3. Detection of GFP-Tagged Proteins with Low Expression Levels in Cell Lysates
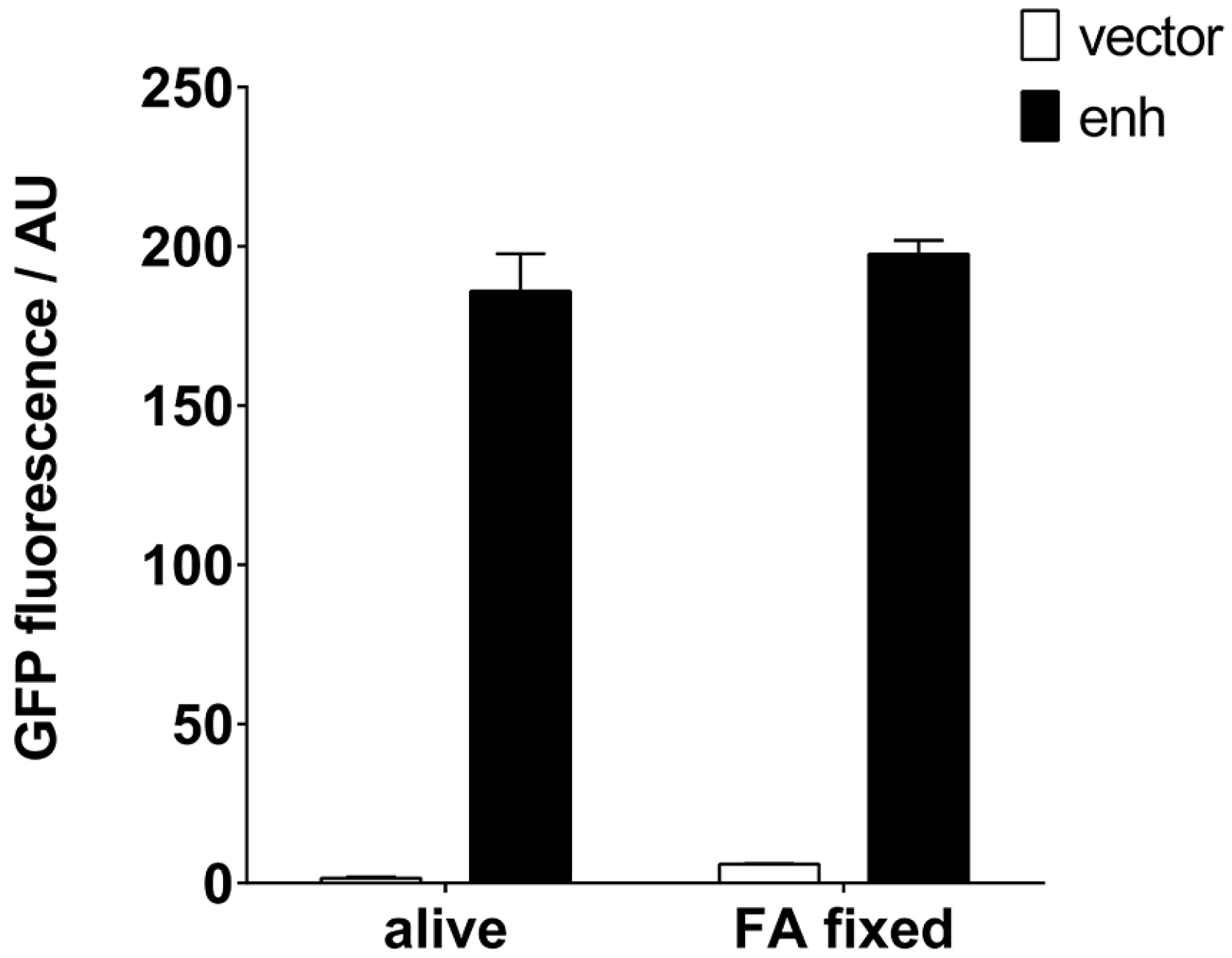
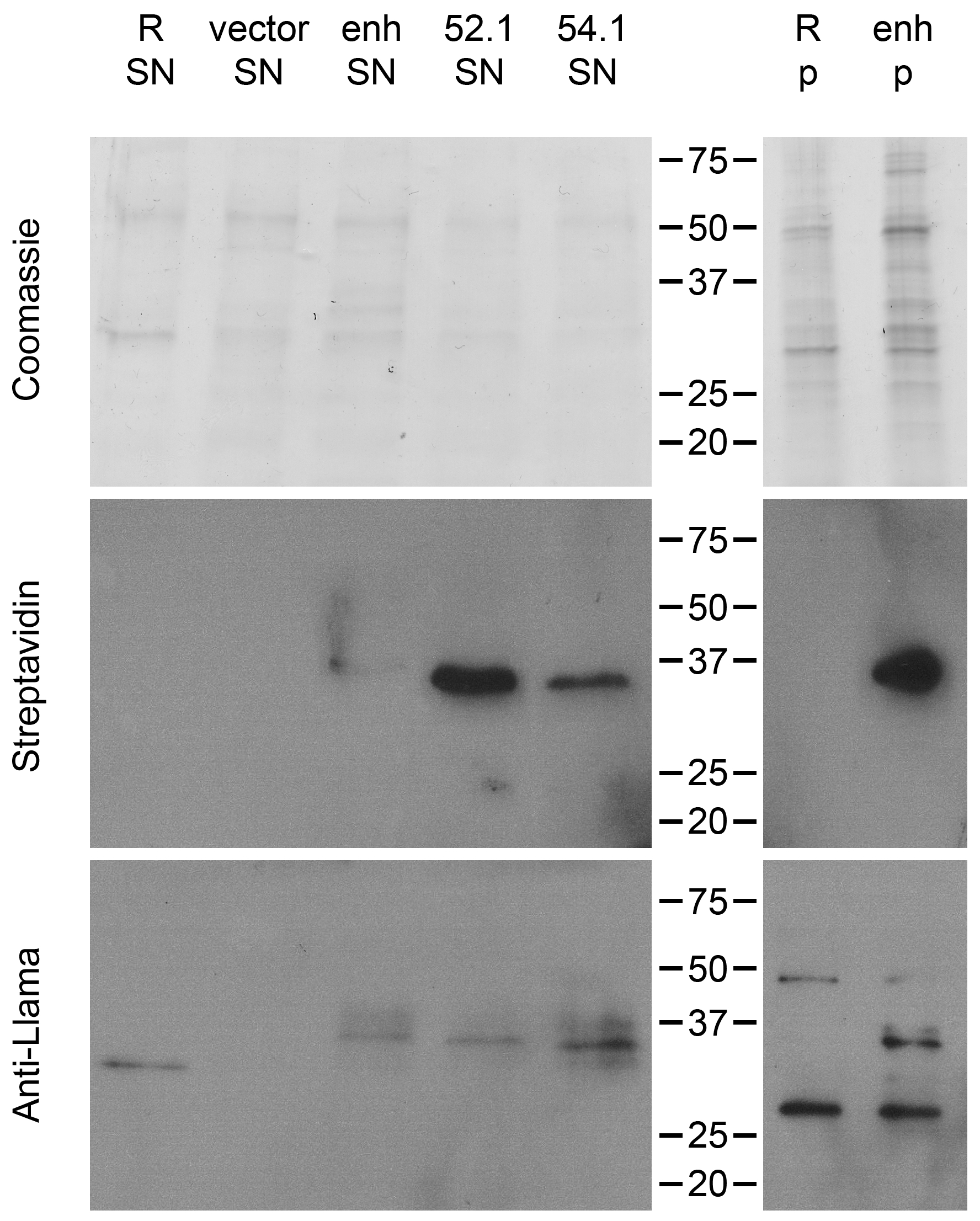
2.4. Release of the Displayed VHH by the Endogenous Staphylococcal SrtA Enzyme
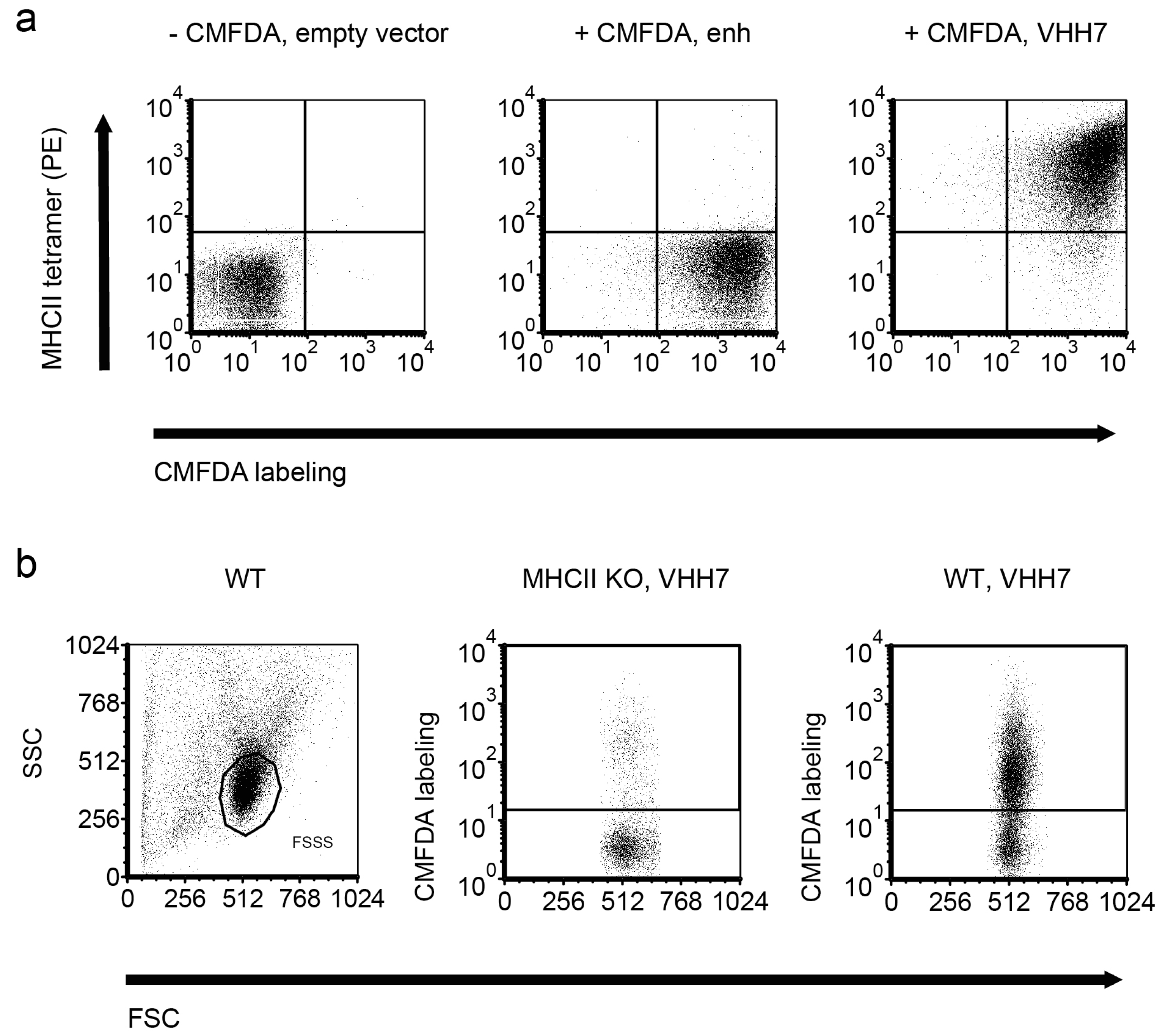
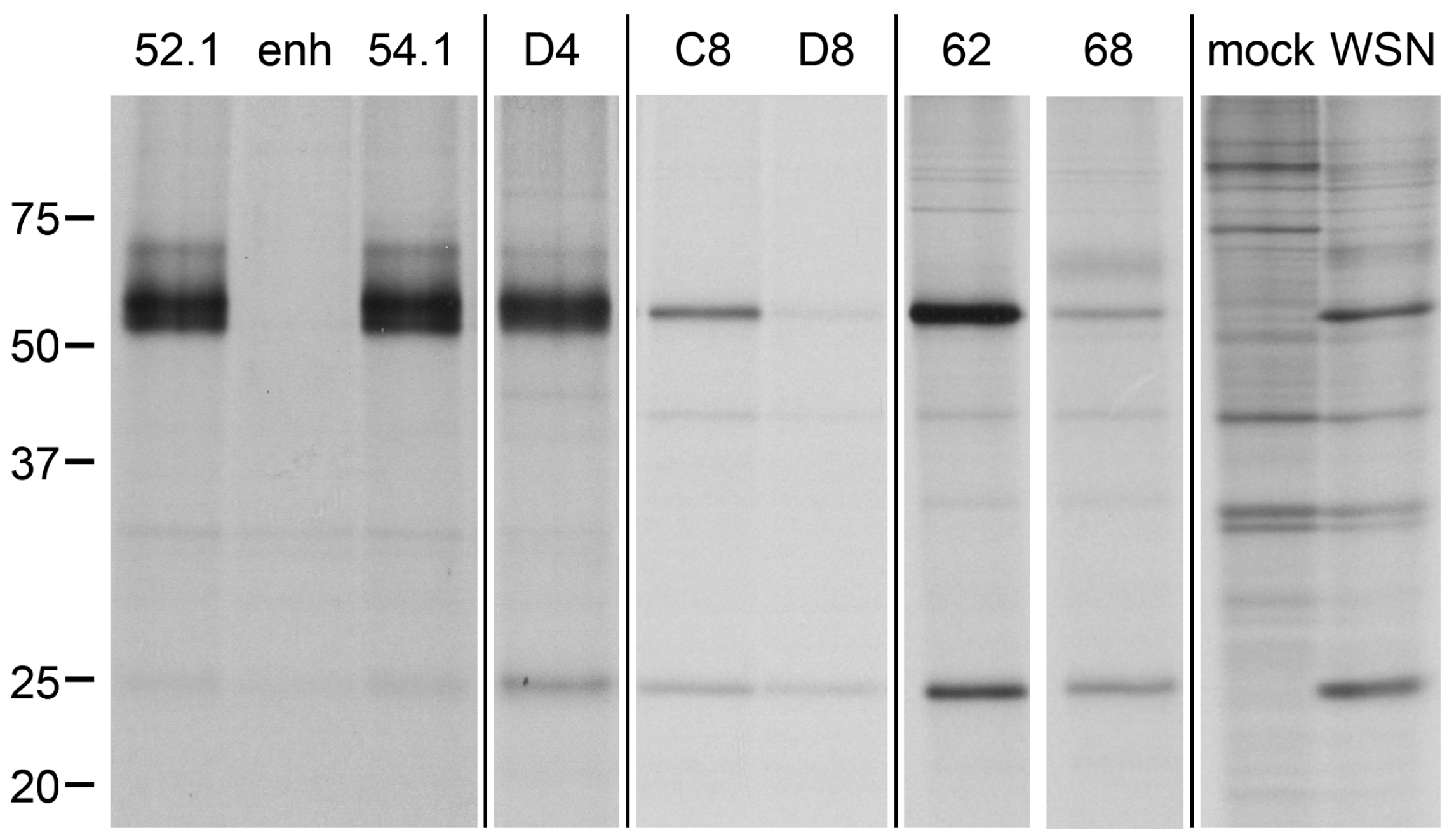
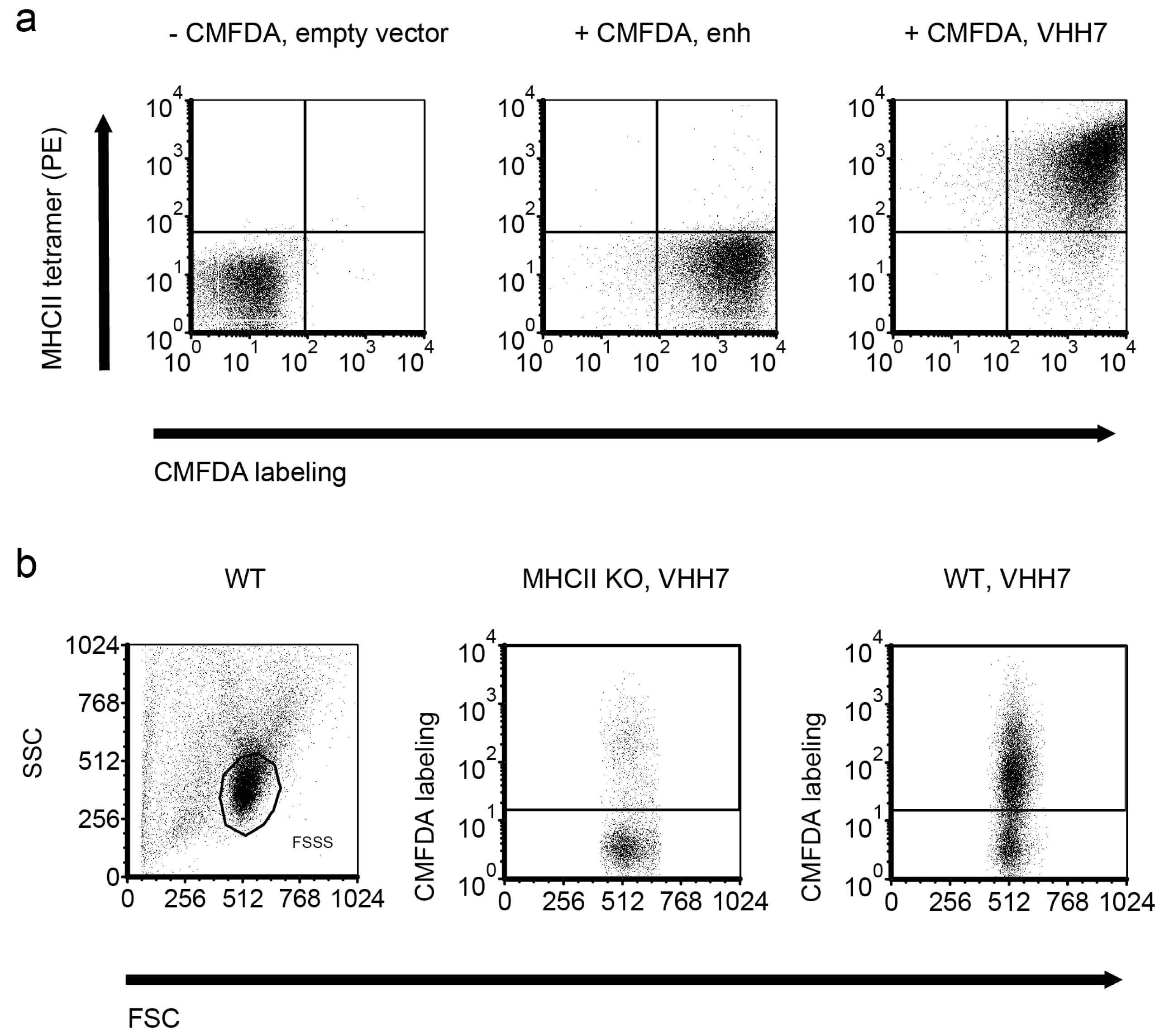
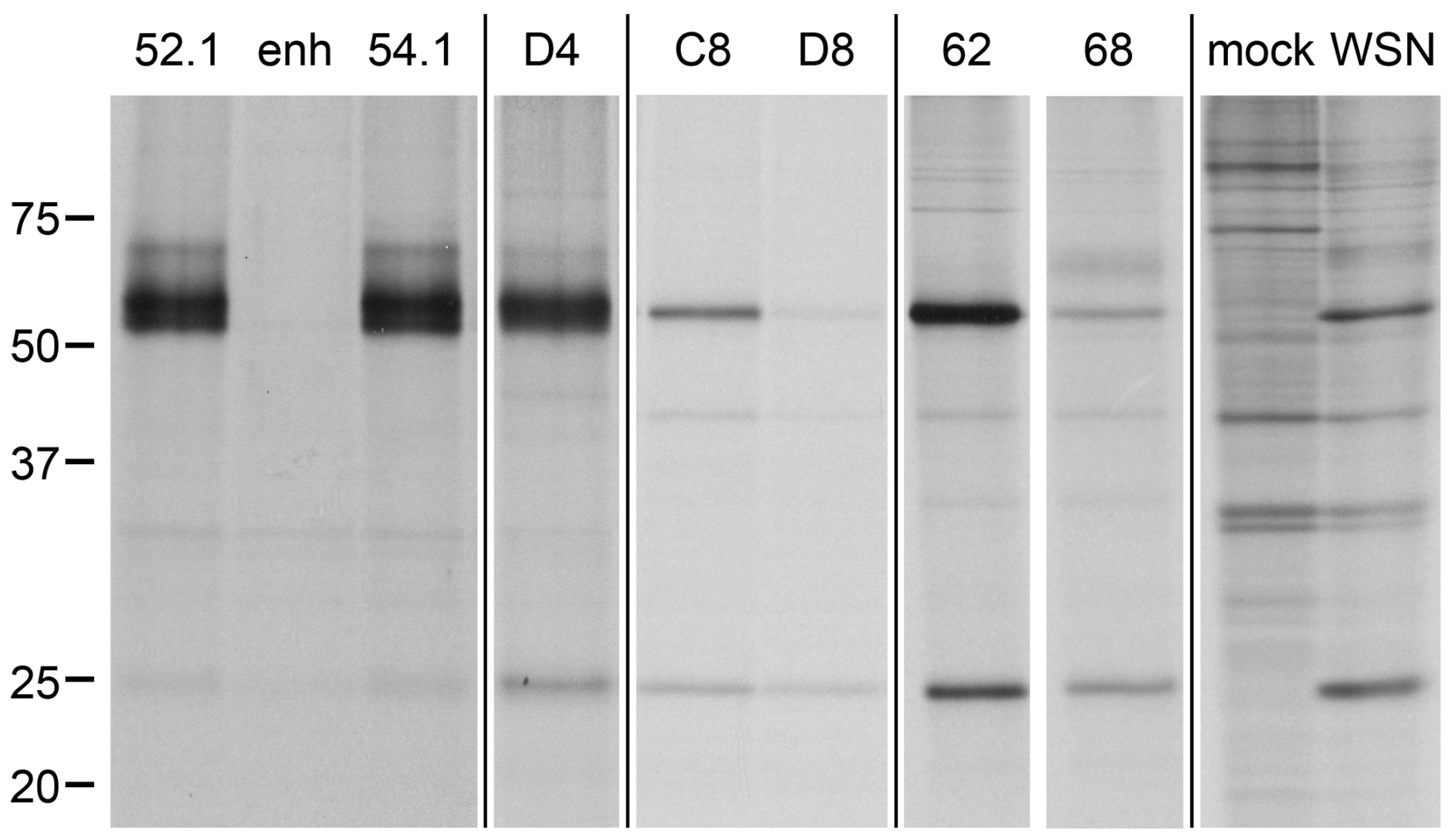
2.5. Direct Target Identification by Combined Staphylococcal Surface Display and IP
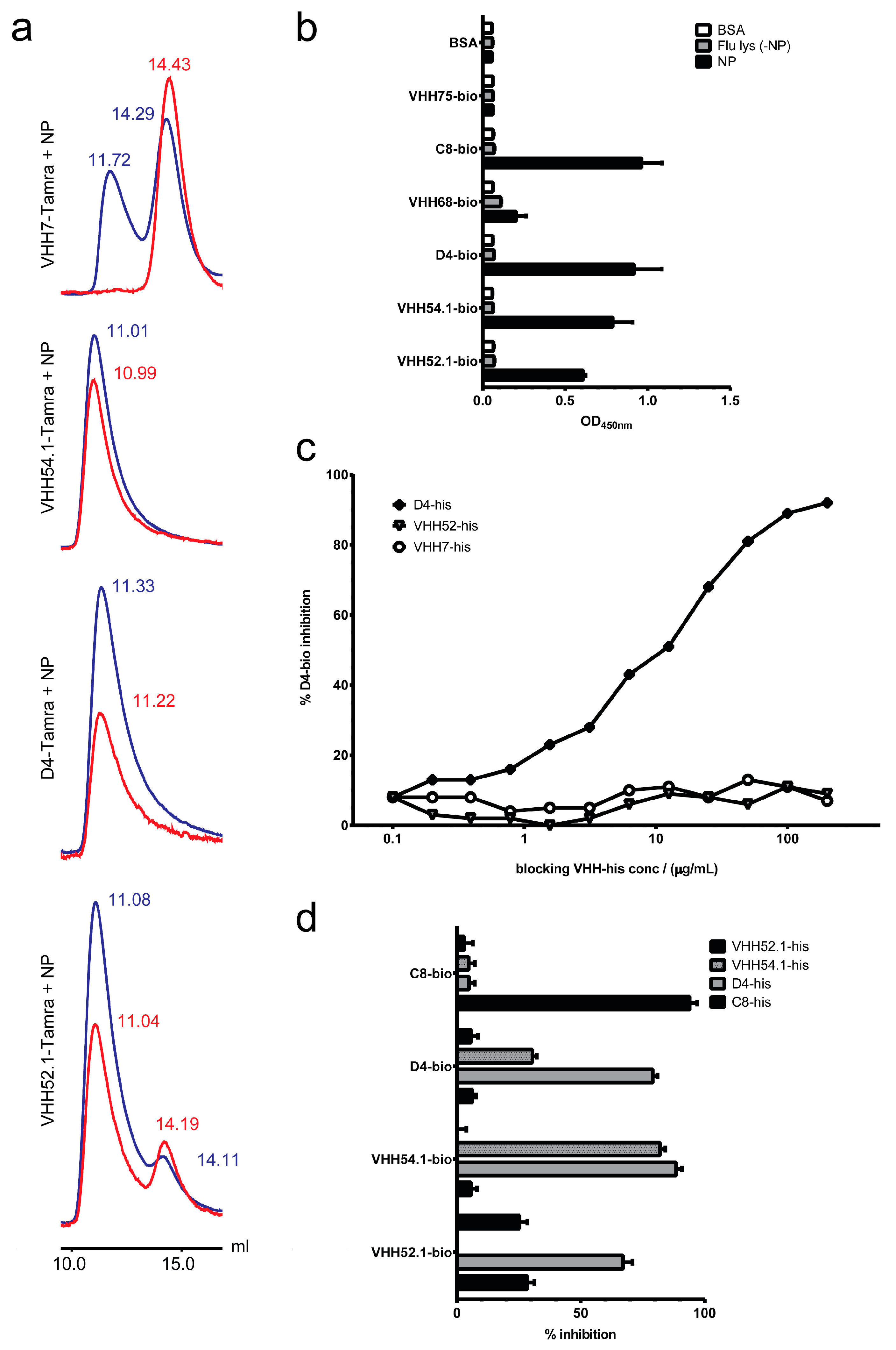
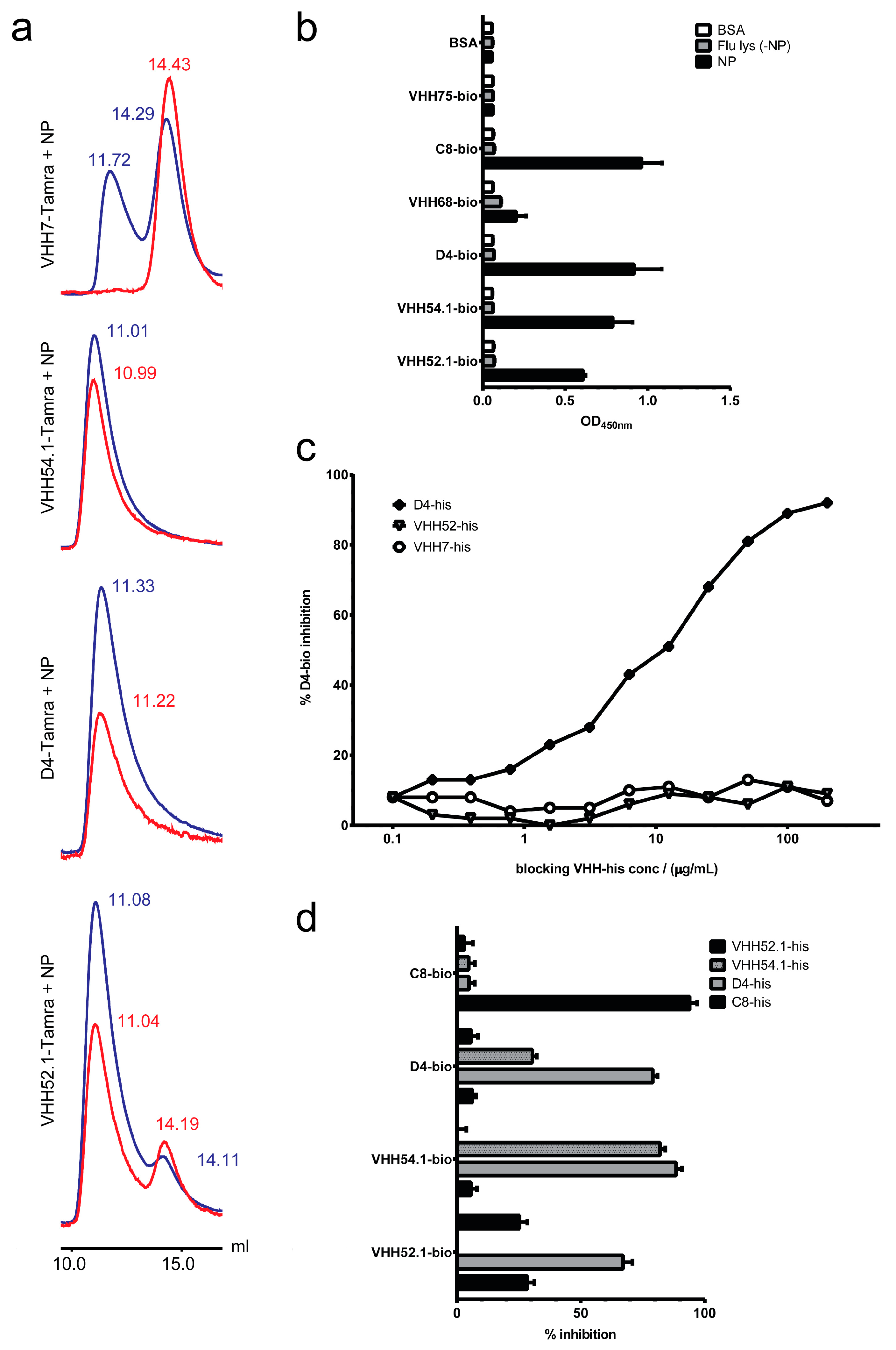
2.6. Target Verification and Characterization of VHH Identified by Staphylococcal Surface Display
3. Discussion
4. Materials and Methods
4.1. Generation and Mass Spectrometry of the pSA-VHH-SPAXrc Shuttle Vector Construct
4.2. Detection of Nitrocellulose Spotted S. aureus
4.3. Flow Cytometry and Fluorescence-Activated Cell Sorting (FACS) of S. aureus
4.4. IP with S. aureus
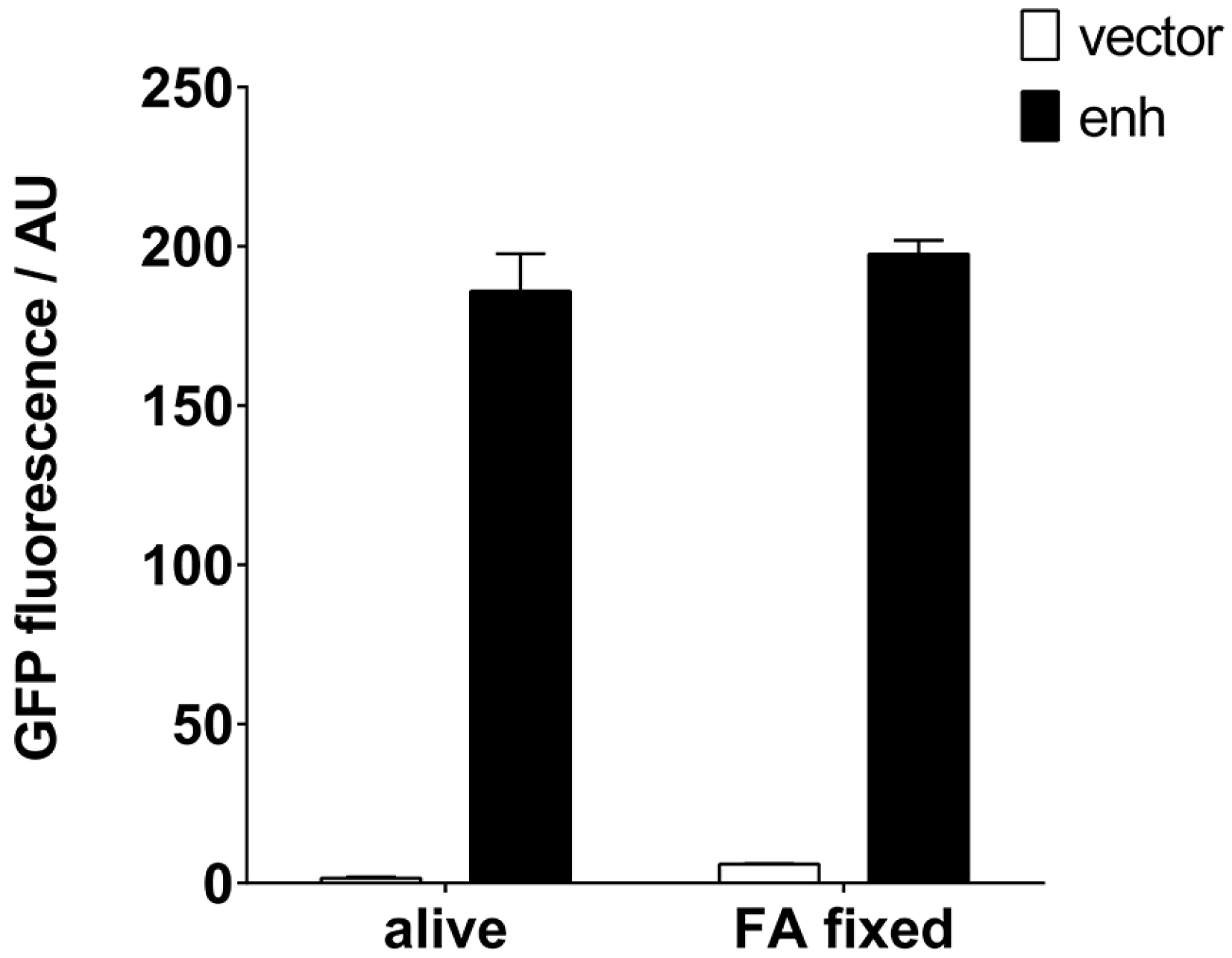
4.5. Fixation of S. aureus
4.6. Maintenance of C. elegans Strains and Lysate Preparation
4.7. GFP Western Blotting
4.8. VHH Release from S. aureus
4.9. Specificity and Competition Enzyme-Linked Immunosorbent Assay (ELISA) and Fast Protein Liquid Chromatography (FPLC)
Supplementary Materials:
Acknowledgments
Conflicts of Interest
Abbreviations
| CDR | complementarity-determining region(s) |
| CMFDA | 5-chloromethylfluorescein diacetate (CellTracker Green) |
| enh | VHH specific for GFP (enhancer) |
| FA | formaldehyde |
| FACS | fluorescence-activated cell sorting |
| FR | framework region(s) |
| GFP | green fluorescent protein |
| HA | hemagglutinin |
| HRP | horseradish peroxidase |
| IP | immunoprecipitation |
| KO | knockout |
| M1 | matrix protein 1 |
| MHCII | major histocompatibility complex class II |
| NP | nucleoprotein |
| PBS | phosphate buffered saline |
| PBSB | PBS, 2% BSA |
| PBSI | PBS, 2% ICS |
| PBSMT | PBS, 4% milk, 0.1% Tween-20 |
| PE | phycoerythrin |
| scFv | single-chain variable fragment(s) |
| SMM | 0.5 M sucrose, 20 mM maleate, 20 mM MgCl2 (pH 6.5) |
| SrtA | staphylococcal housekeeping sortase A |
| TAMRA | 5-carboxytetramethylrhodamine |
| TBSMT | Tris buffered saline, 5% milk, 0.1% Tween-20 |
| VHH | single-domain antigen recognition fragment(s) from camelid heavy chain-only antibodies |
| WT | wild type |
| Xc | staphylococcal protein A constant region (amino acids 421–516) |
| Xr | staphylococcal protein A repetitive region (amino acids 324–420) |
References
- Chan, C.E.Z.; Lim, A.P.C.; MacAry, P.A.; Hanson, B.J. The role of phage display in therapeutic antibody discovery. Int. Immunol. 2014, 26, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.T.; Raeeszadeh-Sarmazdeh, M.; Price, J.V. Engineering antibodies by yeast display. Arch. Biochem. Biophys. 2012, 526, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Scholler, N. Selection of antibody fragments by yeast display. Methods Mol. Biol. 2012, 907, 259–280. [Google Scholar] [PubMed]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [PubMed]
- Feldhaus, M.J.; Siegel, R.W. Yeast display of antibody fragments: A discovery and characterization platform. J. Immunol. Methods 2004, 290, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Doerner, A.; Rhiel, L.; Zielonka, S.; Kolmar, H. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett. 2014, 588, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Zahnd, C.; Amstutz, P.; Plückthun, A. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 2007, 4, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Dufner, P.; Jermutus, L.; Minter, R.R. Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 2006, 24, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, T.; Fujino, Y.; Ueda, T. PURE ribosome display and its application in antibody technology. Biochim. Biophys. Acta 2014, 1844, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Van Bloois, E.; Winter, R.T.; Kolmar, H.; Fraaije, M.W. Decorating microbes: Surface display of proteins on Escherichia coli. Trends Biotechnol. 2011, 29, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, T.; Vanderleyden, J.; Spaepen, S. Autotransporter-based cell surface display in Gram-negative bacteria. Crit. Rev. Microbiol. 2015, 41, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Jahns, A.C.; Rehm, B.H.A. Relevant uses of surface proteins-display on self-organized biological structures. Microb. Biotechnol. 2012, 5, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Galán, A.; Comor, L.; Horvatić, A.; Kuleš, J.; Guillemin, N.; Mrljaka, V.; Bhideabc, M. Library-based display technologies: Where do we stand? Mol. Biosyst. 2016, 12, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Schüürmann, J.; Quehl, P.; Festel, G.; Jose, J. Bacterial whole-cell biocatalysts by surface display of enzymes: Toward industrial application. Appl. Microbiol. Biotechnol. 2014, 98, 8031–8046. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S. Protein engineering with bacterial display. Curr. Opin. Struct. Biol. 2007, 17, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.D.; Du, J.; Reyes, P.R.; Rehm, B.H.A. In vivo polyester immobilized sortase for tagless protein purification. Microb. Cell Fact. 2015, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Löfblom, J. Bacterial display in combinatorial protein engineering. Biotechnol. J. 2011, 6, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Caldwell, C.W.; Sprouse, R.F.; Durham, J.B. Extraction and recovery of IgG antibodies using Staphylococcus aureus. Med. Lab. Sci. 1982, 39, 145–149. [Google Scholar] [PubMed]
- Jonsson, S.; Kronvall, G. The use of protein A-containing Staphylococcus aureus as a solid phase anti-IgG reagent in radioimmunoassays as exemplified in the quantitation of α-fetoprotein in normal human adult serum. Eur. J. Immunol. 1974, 4, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH2-Gly3 substrates. J. Biol. Chem. 2000, 275, 9876–9881. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Dedent, A.C.; Schneewind, O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 192–221. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Schneewind, O. The YSIRK-G/S motif of staphylococcal protein A and its role in efficiency of signal peptide processing. J. Bacteriol. 2003, 185, 2910–2919. [Google Scholar] [CrossRef] [PubMed]
- DeDent, A.; Bae, T.; Missiakas, D.M.; Schneewind, O. Signal peptides direct surface proteins to two distinct envelope locations of Staphylococcus aureus. EMBO J. 2008, 27, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Missiakas, D.M. Protein secretion and surface display in Gram-positive bacteria. Phil. Trans. R. Soc. B 2012, 367, 1123–1139. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Götz, F. Cell wall antibiotics provoke accumulation of anchored mCherry in the cross wall of Staphylococcus aureus. PLoS ONE 2012, 7, e30076. [Google Scholar] [CrossRef] [PubMed]
- Kronqvist, N.; Löfblom, J.; Jonsson, A.; Wernérus, H.; Ståhl, S. A novel affinity protein selection system based on staphylococcal cell surface display and flow cytometry. Protein Eng. Des. Sel. 2008, 21, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Vu, K.B.; Ghahroudi, M.A.; Wyns, L.; Muyldermans, S. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 1997, 34, 1121–1131. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Ruuls, R.C.; Nijman, I.J.; Niewold, T.H.; Frenken, L.G.J.; de Geus, B. Llama heavy-chain V regions consist of at least four distinct subfamilies revealing novel sequence features. Mol. Immunol. 2000, 37, 579–590. [Google Scholar] [CrossRef]
- Saerens, D.; Pellis, M.; Loris, R.; Pardon, E.; Dumoulin, M.; Matagne, A.; Wyns, L.; Muyldermans, S.; Conrath, K. Identification of a universal VHH framework to graft non-canonical antigen-binding loops of camel single-domain antibodies. J. Mol. Biol. 2005, 352, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juarez, K.; Urrutia, M.; Cauerhff, A.; Danquah, W.; Rissiek, B.; Scheuplein, F.; et al. Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; van Solt, C.B.; Fijten, H.P.D. Enhancement of toxin- and virus-neutralizing capacity of single-domain antibody fragments by N-glycosylation. Appl. Microbiol. Biotechnol. 2009, 84, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Govaert, J.; Pellis, M.; Deschacht, N.; Vincke, C.; Conrath, K.; Muyldermans, S.; Saerens, D. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J. Biol. Chem. 2012, 287, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Hussack, G.; Mackenzie, C.R.; Tanha, J. Characterization of single-domain antibodies with an engineered disulfide bond. Methods Mol. Biol. 2012, 911, 417–429. [Google Scholar] [PubMed]
- Desmyter, A.; Decanniere, K.; Muyldermans, S.; Wyns, L. Antigen specificity and high affinity binding provided by one single loop of a camel single-domain antibody. J. Biol. Chem. 2001, 276, 26285–26290. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.P.; Witte, M.D.; Theile, C.S.; Bozkurt, G.; Kundrat, L.; Blom, A.E.M.; Ploegh, H.L. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.D.; Wu, T.; Guimaraes, C.P.; Theile, C.S.; Blom, A.E.M.; Ingram, J.R.; Li, Z.; Kundrat, L.; Goldberg, S.D.; Ploegh, H.L. Site-specific protein modification using immobilized sortase in batch and continuous-flow systems. Nat. Protoc. 2015, 10, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2013, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Guss, B.; Uhlén, M.; Nilsson, B.; Lindberg, M.; Sjöquist, J.; Sjödahl, J. Region X, the cell-wall-attachment part of staphylococcal protein A. Eur. J. Biochem. 1984, 138, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-Delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2010, 17, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.; Memmi, G.; Hernandez, D.; Bard, J.; Beaume, M.; Gill, S.; Francois, P.; Cheung, A.L. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 2011, 193, 2332–2335. [Google Scholar] [CrossRef] [PubMed]
- Monk, I.R.; Shah, I.M.; Xu, M.; Tan, M.-W.; Foster, T.J. Transforming the untransformable: Application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio 2012. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Model, P.; Fischetti, V.A. Sorting of protein a to the staphylococcal cell wall. Cell 1992, 70, 267–281. [Google Scholar] [CrossRef]
- Schneewind, O.; Mihaylova-Petkov, D.; Model, P. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J. 1993, 12, 4803–4811. [Google Scholar] [PubMed]
- Witte, M.D.; Cragnolini, J.J.; Dougan, S.K.; Yoder, N.C.; Popp, M.W.; Ploegh, H.L. Preparation of unnatural N-to-N and C-to-C protein fusions. Proc. Natl. Acad. Sci. USA 2012, 109, 11993–11998. [Google Scholar] [CrossRef] [PubMed]
- Madsen, L.; Labrecque, N.; Engberg, J.; Dierich, A.; Svejgaard, A.; Benoist, C.; Mathis, D.; Fugger, L. Mice lacking all conventional MHC class II genes. Proc. Natl. Acad. Sci. USA 1999, 96, 338–343. [Google Scholar] [CrossRef]
- Avalos, A.M.; Bilate, A.M.; Witte, M.D.; Tai, A.K.; He, J.; Frushicheva, M.P.; Thill, P.D.; Meyer-Wentrup, F.; Theile, C.S.; Chakraborty, A.K.; et al. Monovalent engagement of the BCR activates ovalbumin-specific transnuclear B cells. J. Exp. Med. 2014, 211, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Kline, K.A.; Fälker, S.; Dahlberg, S.; Normark, S.; Henriques-Normark, B. Bacterial adhesins in host-microbe interactions. Cell Host Microbe 2009, 5, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Chamessian, A.G.; McEnaney, P.J.; Murelli, R.P.; Kazmiercak, B.I.; Spiegel, D.A. A biosynthetic strategy for re-engineering the Staphylococcus aureus cell wall with non-native small molecules. ACS Chem. Biol. 2010, 5, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, A.P.A.; Budzik, J.M.; Oh, S.-Y.; Schneewind, O. Architects at the bacterial surface-sortases and the assembly of pili with isopeptide bonds. Nat. Rev. Microbiol. 2011, 9, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Schmidt, F.I.; Hanke, L.; Cragnolini, J.; Cavallari, M.; Altenburg, A.; Brewer, R.; Ingram, J.; Shoemaker, C.; Pleogh, H.L. Intracellular expression of camelid single-domain antibodies specific for influenza virus nucleoprotein uncovers distinct features of its nuclear localization. J. Virol. 2015, 89, 2792–2800. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Chenavas, S.; Estrozi, L.F.; Slama-Schwok, A.; Delmas, B.; di Primo, C.; Baudin, F.; Li, X.; Crépin, T.; Ruigrok, R.W.H. Monomeric nucleoprotein of influenza A virus. PLoS Pathog. 2013, 9, e1003275. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Keliher, E.J.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Edens, J.G.; Tas, J.M.J.; Victora, G.; et al. Use of 18F-2-fluorodeoxyglucose to label antibody fragments for immuno-positron emission tomography of pancreatic cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Wang, L.; Eden, J.G.; Jacobsen, J.T.; Hossain, I.; Wang, Q.; Victora, G.D.; Vasdev, N.; Ploegh, H.; Liang, S.H. Enzyme-mediated modification of single-domain antibodies for imaging modalities with different characteristics. Angew. Chem. Int. Ed. Engl. 2016, 128, 538–543. [Google Scholar] [CrossRef]
- Gautam, S.; Gniadek, T.J.; Kim, T.; Spiegel, D.A. Exterior design: Strategies for redecorating the bacterial surface with small molecules. Trends Biotechnol. 2013, 31, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Löfblom, J.; Kronqvist, N.; Uhlén, M.; Stahl, S.; Wernerus, H. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J. Appl. Microbiol. 2007, 102, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Laddaga, R.A. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett. 1992, 73, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Saerens, D.; Muyldermans, S. Single Domain Antibodies. Methods Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Thys, B.; Saerens, D.; Schotte, L.; de Bleeser, G.; Muyldermans, S.; Hassanzadeh-Ghassabeh, G.; Rombaut, B. A simple quantitative affinity capturing assay of poliovirus antigens and subviral particles by single-domain antibodies using magnetic beads. J. Virol. Methods 2011, 173, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Hanke, L.; Morin, B.; Brewer, R.; Brusic, V.; Whelan, S.P.J.; Ploegh, H.L. Phenotypic lentivirus screens to identify functional single domain antibodies. Nat. Microbiol. 2016, 1, 16080. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Guss, B.; Nilsson, B.; Gatenbeck, S.; Philipsin, L.; Lindberg, M. Complete sequence of the staphylococcal gene encoding protein A. A gene evolved through multiple duplications. J. Biol. Chem. 1984, 259, 1695–1702. [Google Scholar] [PubMed]
- Nilsson, B.; Abrahmsén, L.; Uhlén, M. Immobilization and purification of enzymes with staphylococcal protein A gene fusion vectors. EMBO J. 1985, 4, 1075–1080. [Google Scholar] [PubMed]
- Patel, A.H.; Kornblum, J.; Kreiswirth, B.; Novick, R.; Foster, T.J. Regulation of the protein A-encoding gene in Staphylococcus aureus. Gene 1992, 114, 25–34. [Google Scholar] [CrossRef]
- Löfdahl, S.; Guss, B.; Uhlén, M.; Philipson, L.; Lindberg, M. Gene for staphylococcal protein A. Proc. Natl. Acad. Sci. USA 1983, 80, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R.; Griffiths, A.D.; Johnson, K.S.; Chiswell, D.J.; Hudson, P.; Winter, G. Multi-subunit proteins on the surface of filamentous phage: Methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res. 1991, 19, 4133–4137. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Popp, M.W.-L.; Ernst, R.; Chew, G.-L.; Spooner, E.; Ploegh, H.L. A straight path to circular proteins. J. Biol. Chem. 2009, 284, 16028–16036. [Google Scholar] [CrossRef] [PubMed]
- Wuethrich, I.; Peeters, J.G.C.; Blom, A.E.M.; Theile, C.S.; Li, Z.; Spooner, E.; Pleogh, H.L.; Guimaraes, C.P. Site-specific chemoenzymatic labeling of aerolysin enables the identification of new aerolysin receptors. PLoS ONE 2014, 9, e109883. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.K.; Ashour, J.; Karssemeijer, R.A.; Popp, M.W.; Avalos, A.M.; Barisa, M.; Altehburg, A.F.; Ingam, J.R.; Cragnolini, J.J.; et al. Antigen-specific B-cell receptor sensitizes B cells to infection by influenza virus. Nature 2013, 503, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Antos, J.M.; Chew, G.-L.; Guimaraes, C.P.; Yoder, N.C.; Grotenberg, G.M.; Popp, W.-L.M.; Ploegh, H. Site-specific N- and C-terminal labeling of a single polypeptide using sortases of different specificity. J. Am. Chem. Soc. 2009, 131, 10800–10801. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Lu, A.; Chen, J.W.; Ruan, J.; Tang, C.; Wu, H.; Pleogh, H.L. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 2016, 213, 771–790. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.R.; Knockenhauer, K.E.; Markus, B.M.; Mandelbaum, J.; Ramek, A.; Shan, Y.; Shaw, D.E.; Schwartz, T.U.; Pleogh, H.L.; Lourido, S. Allosteric activation of apicomplexan calcium-dependent protein kinases. Proc. Natl. Acad. Sci. USA 2015, 112, E4975–E4984. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallari, M. Rapid and Direct VHH and Target Identification by Staphylococcal Surface Display Libraries. Int. J. Mol. Sci. 2017, 18, 1507. https://doi.org/10.3390/ijms18071507
Cavallari M. Rapid and Direct VHH and Target Identification by Staphylococcal Surface Display Libraries. International Journal of Molecular Sciences. 2017; 18(7):1507. https://doi.org/10.3390/ijms18071507
Chicago/Turabian StyleCavallari, Marco. 2017. "Rapid and Direct VHH and Target Identification by Staphylococcal Surface Display Libraries" International Journal of Molecular Sciences 18, no. 7: 1507. https://doi.org/10.3390/ijms18071507
APA StyleCavallari, M. (2017). Rapid and Direct VHH and Target Identification by Staphylococcal Surface Display Libraries. International Journal of Molecular Sciences, 18(7), 1507. https://doi.org/10.3390/ijms18071507