Licochalcone A Prevents Platelet Activation and Thrombus Formation through the Inhibition of PLCγ2-PKC, Akt, and MAPK Pathways

Abstract
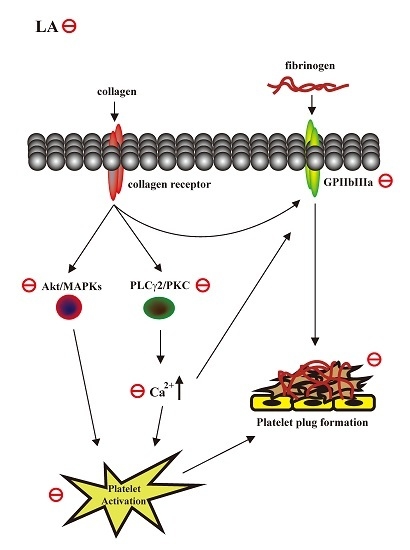
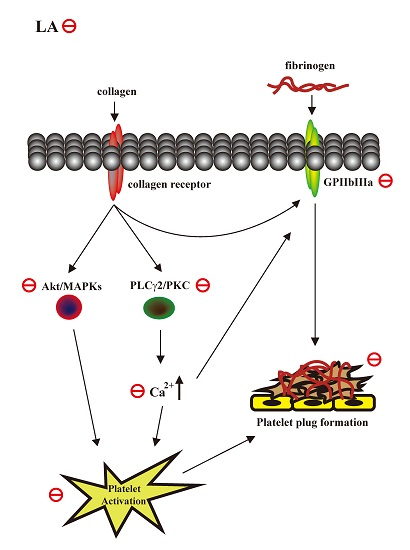
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
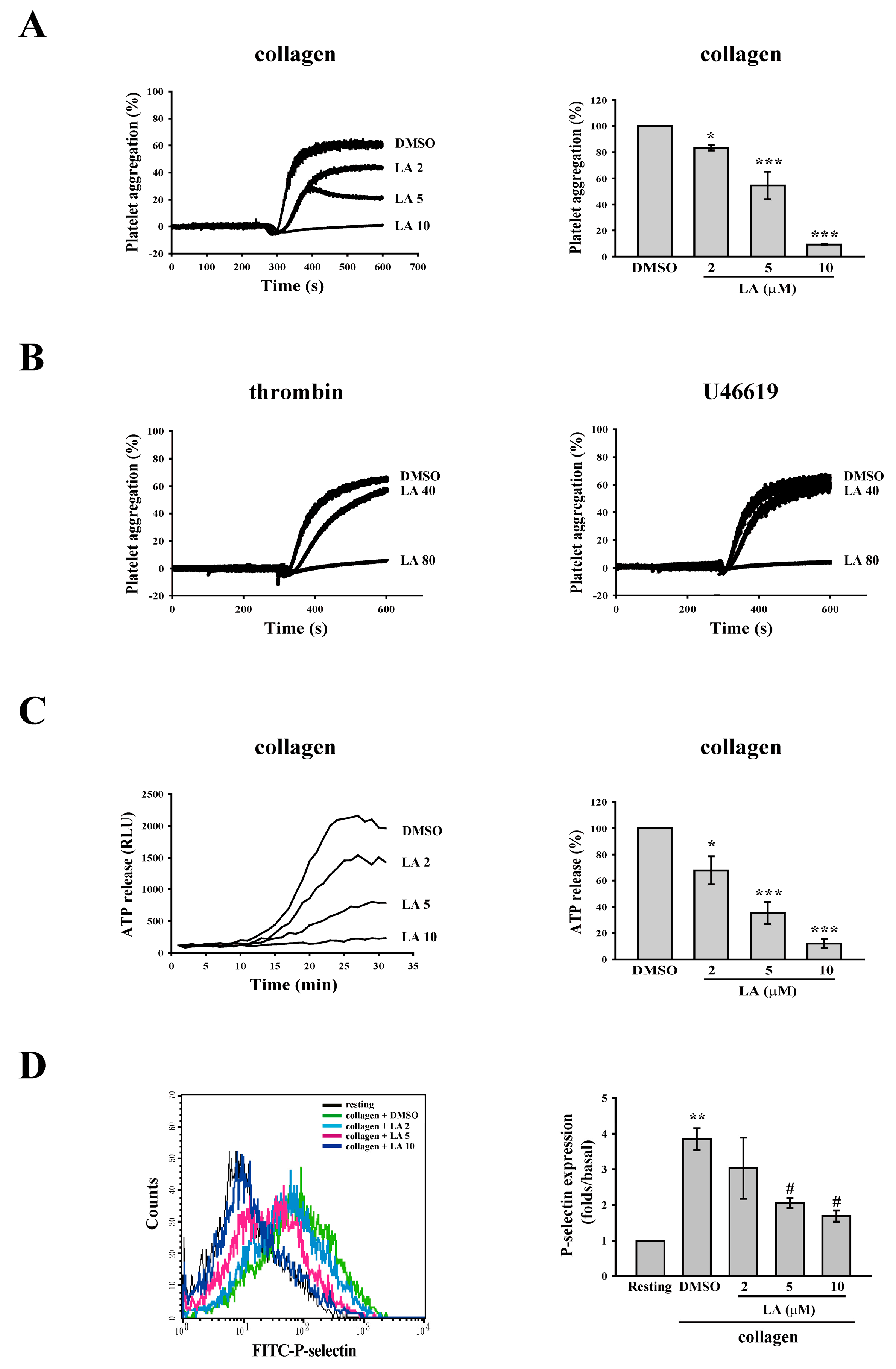
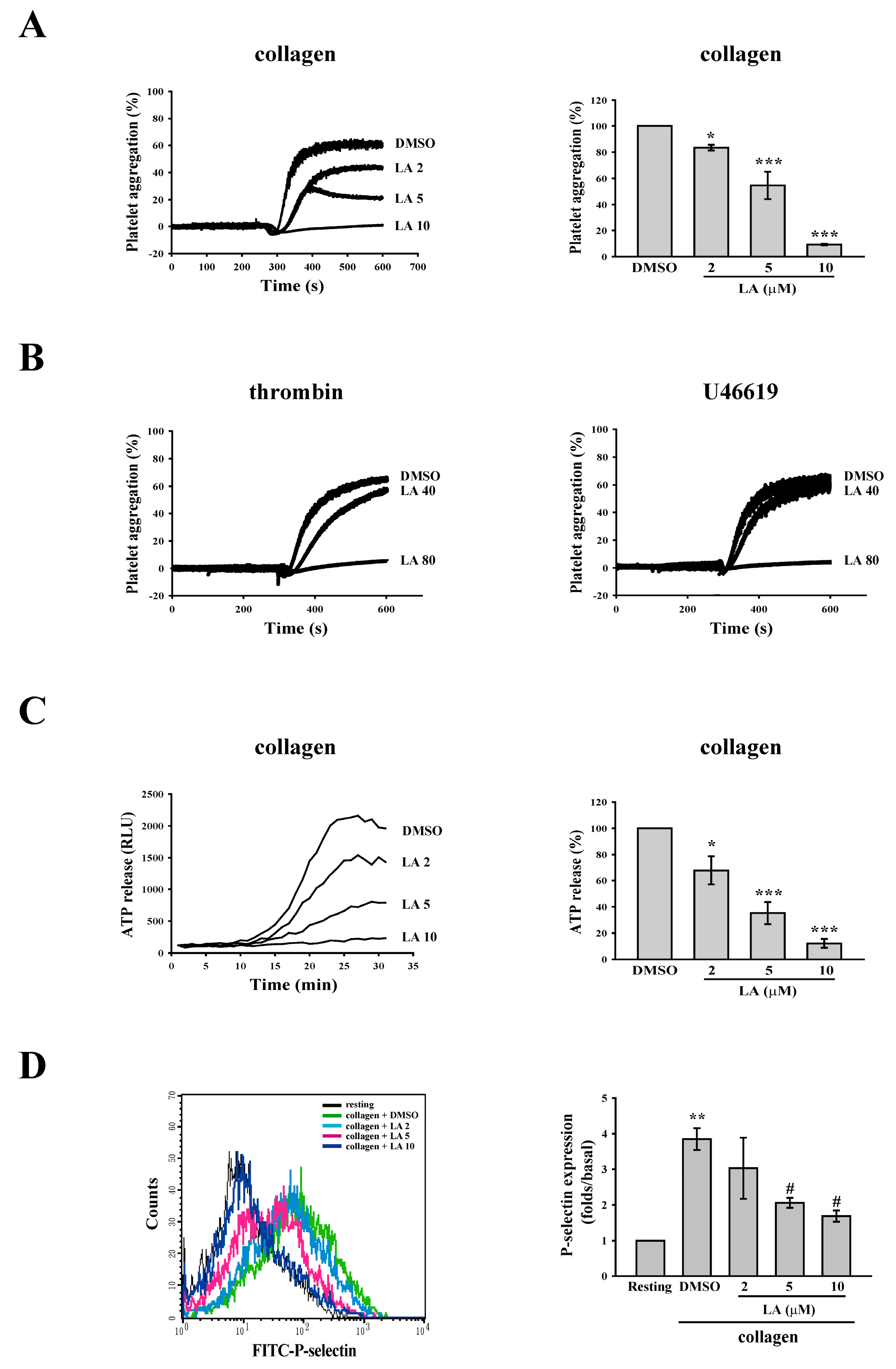
2.1. Licochalcone A (LA) Inhibited Collagen-Induced Platelet Aggregation
2.2. LA Inhibited Collagen-Mediated ATP Release and P-Selectin Secretion
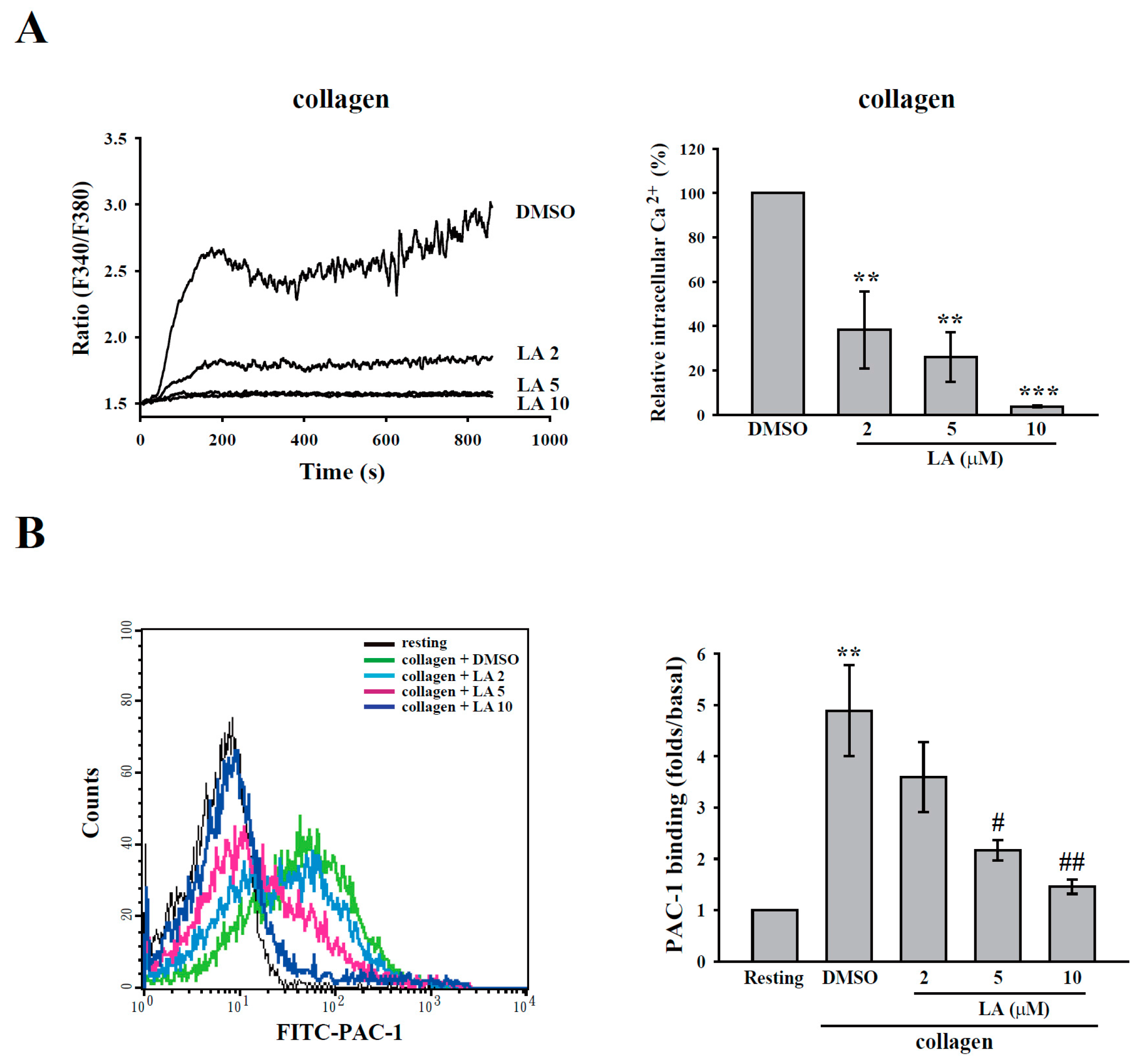
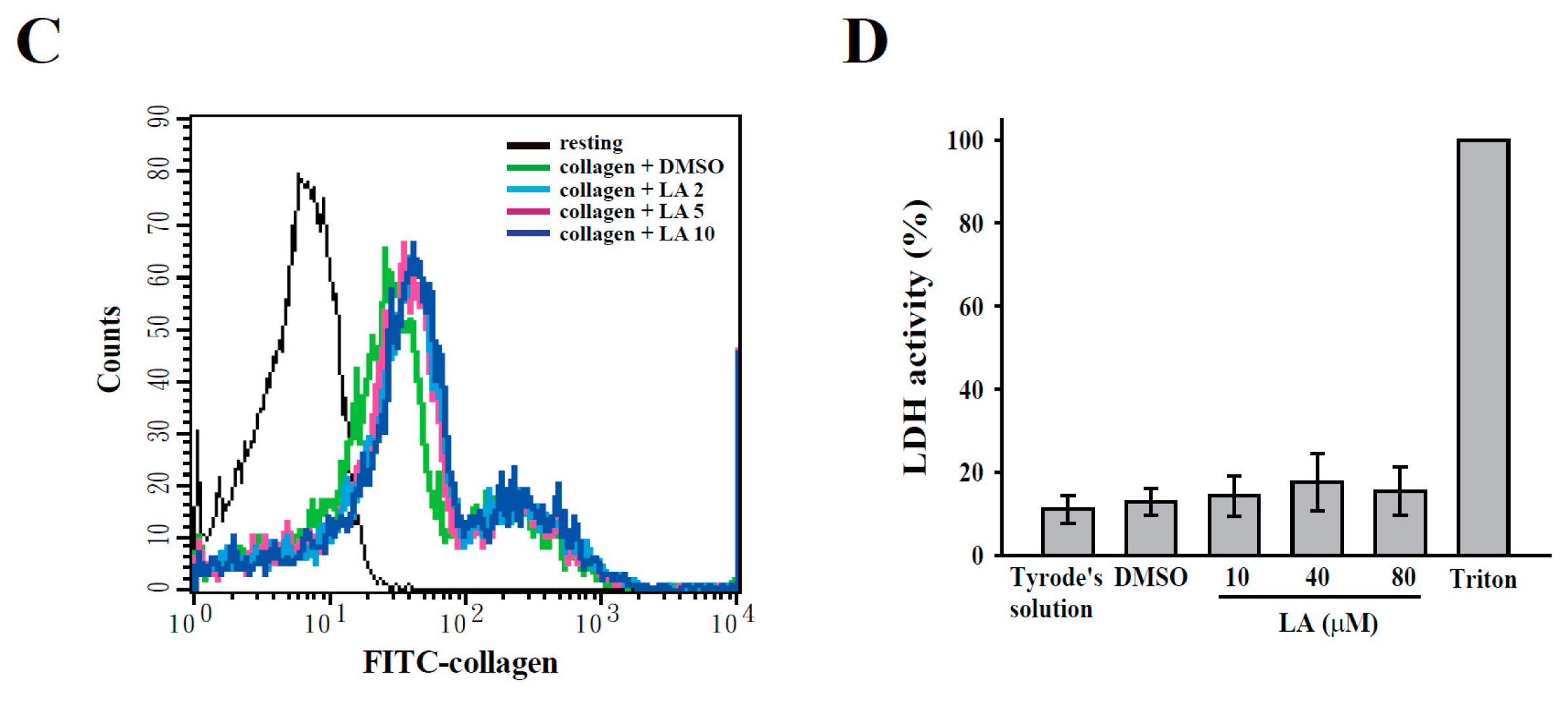
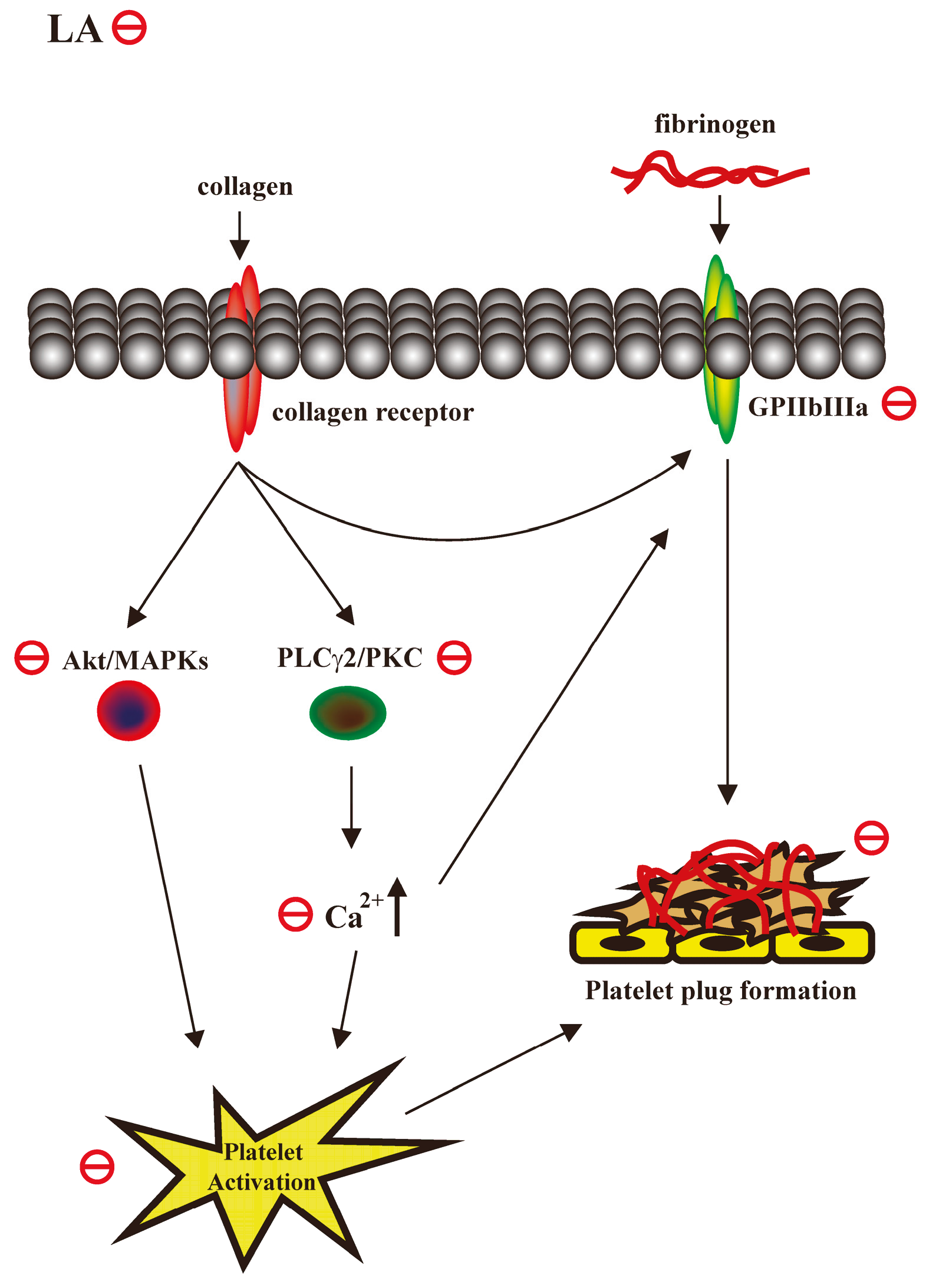
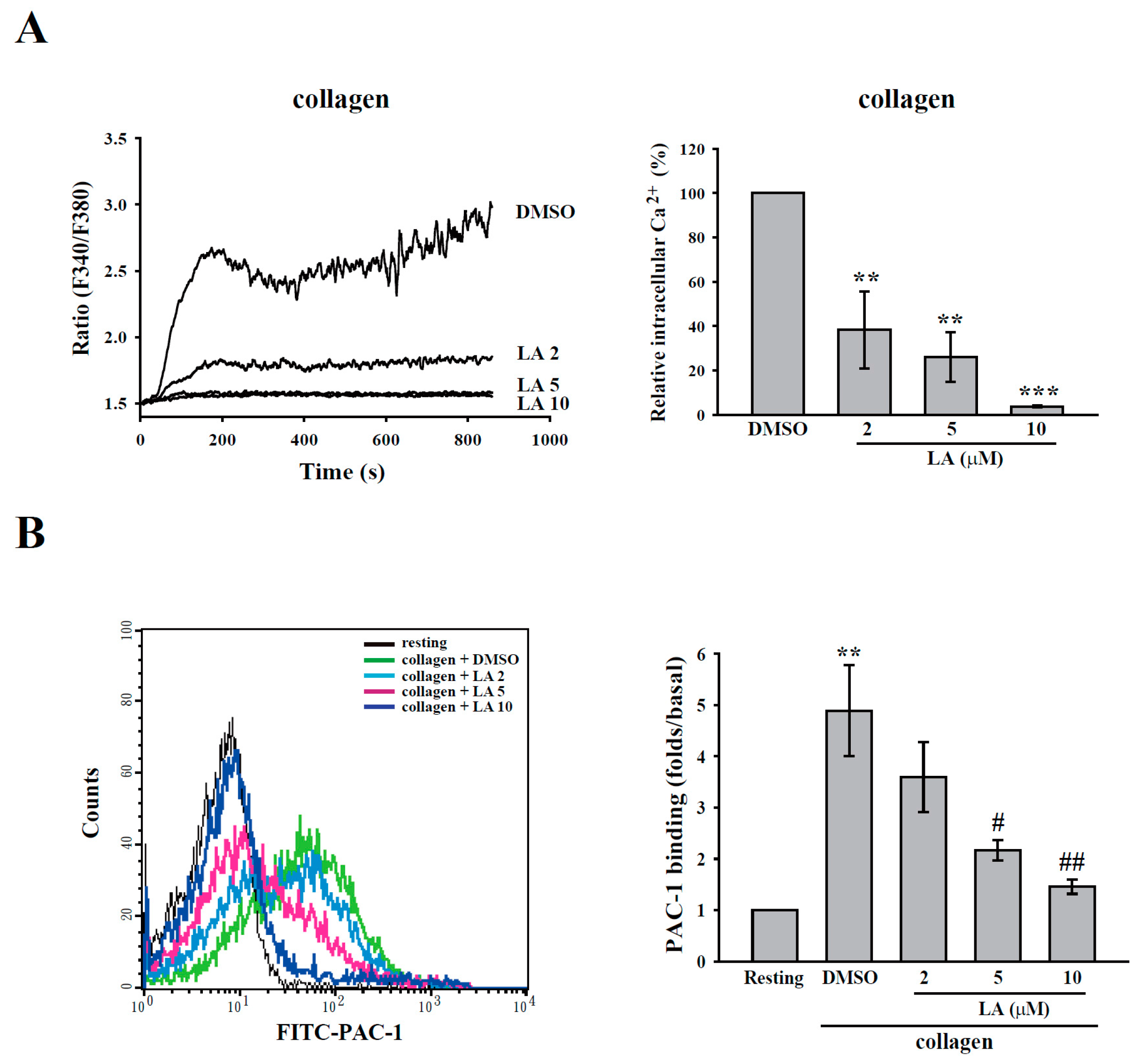
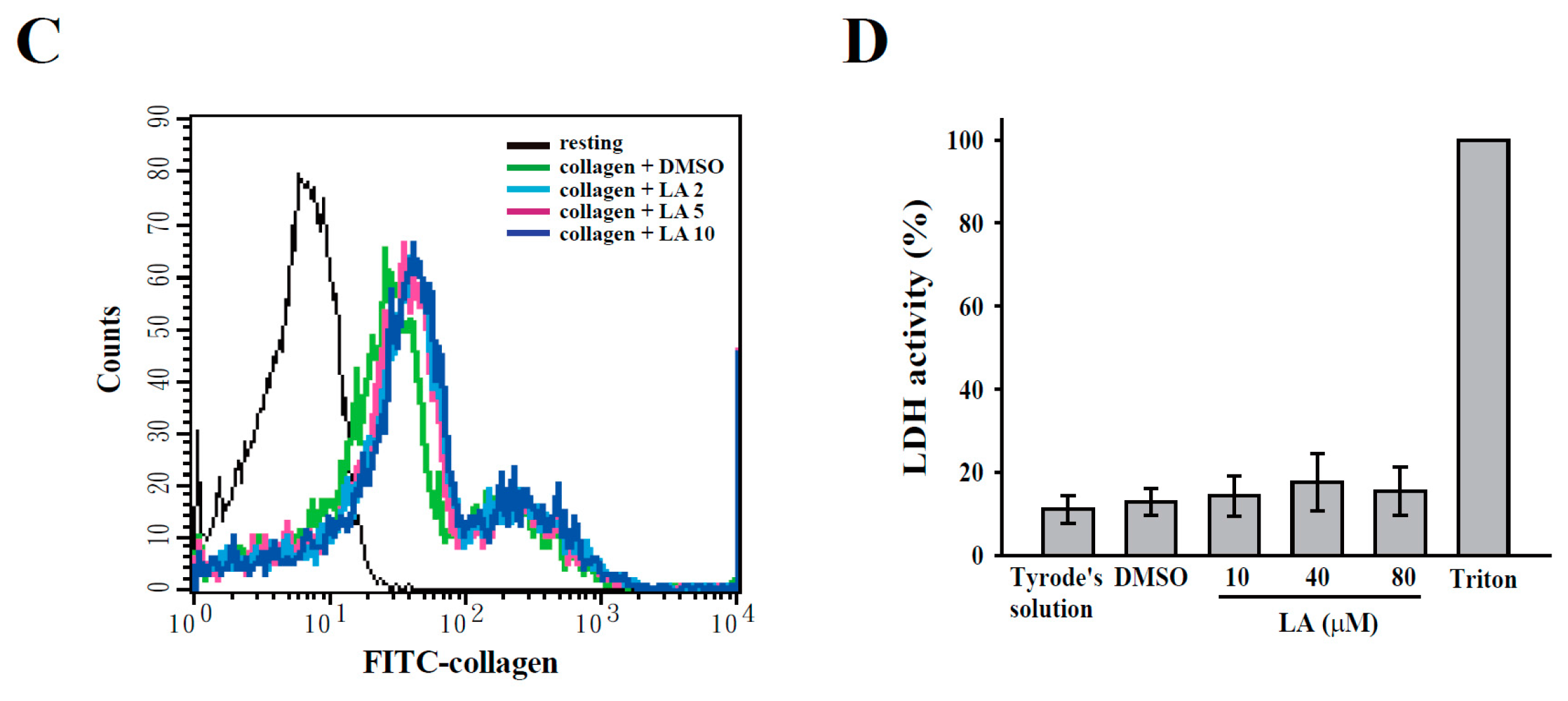
2.3. LA Inhibited Collagen-Mediated Calcium Mobilization and GPIIbIIIa Activation without Interfering with Collagen Receptors
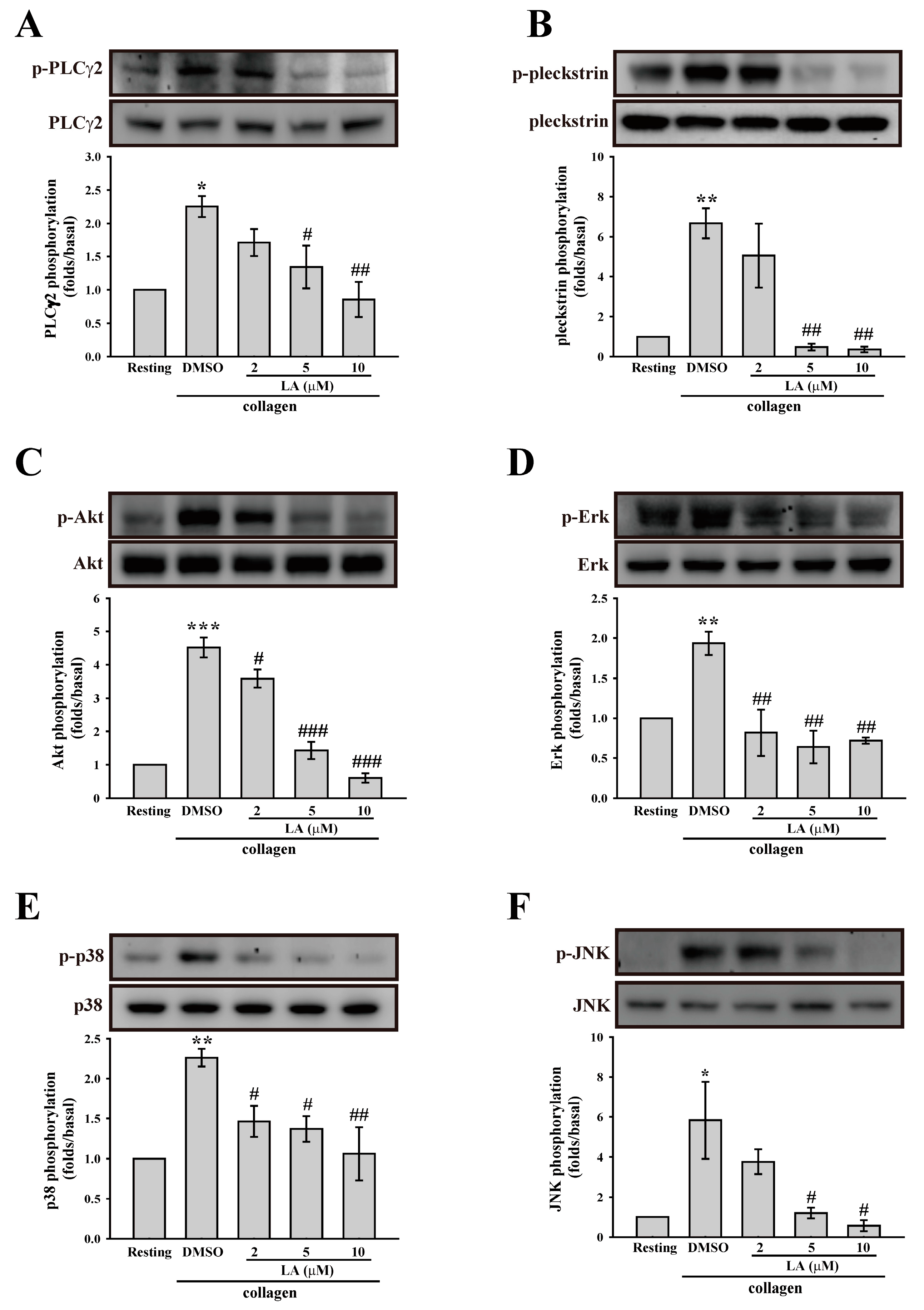
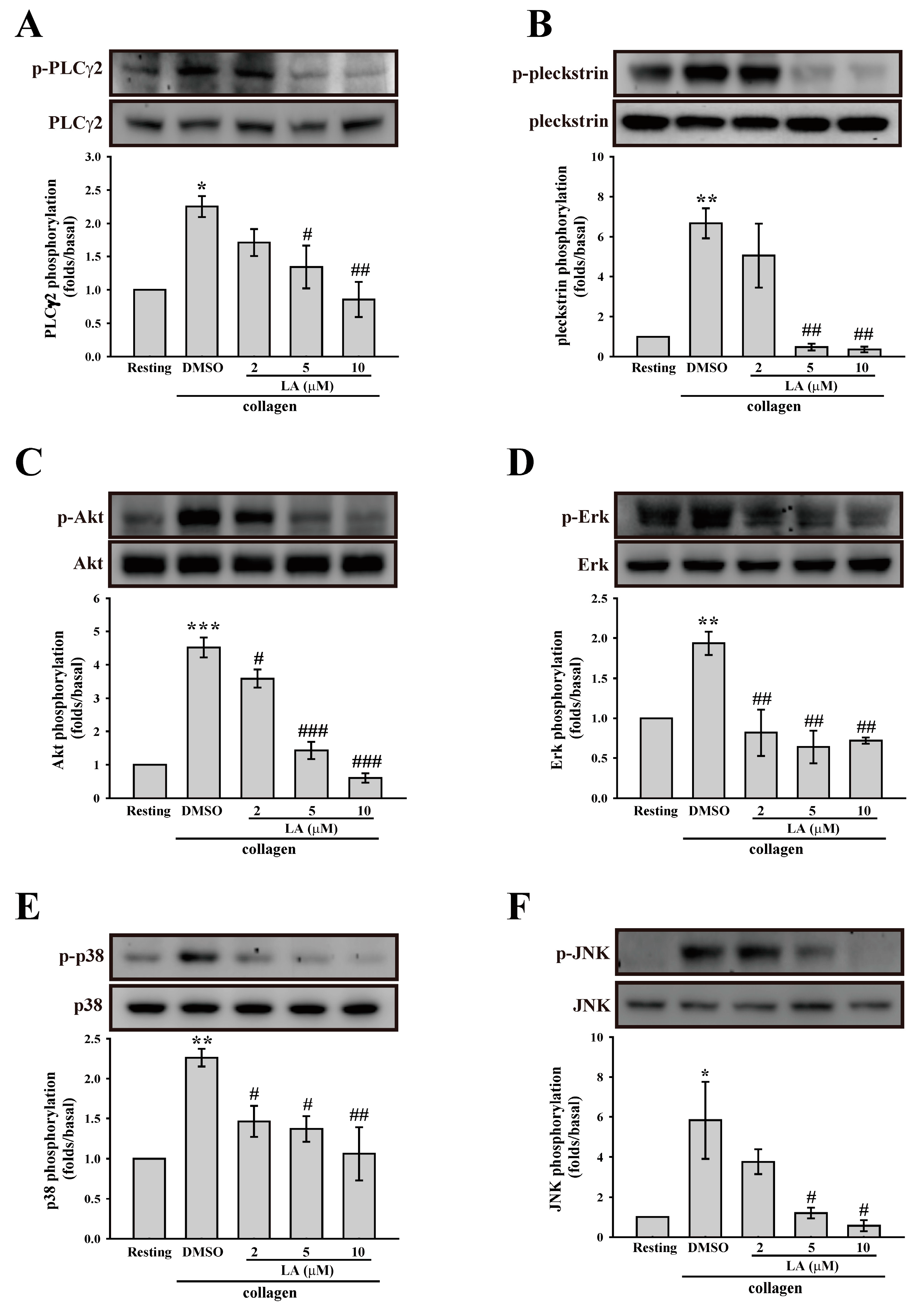
2.4. LA Inhibited Collagen-Mediated Platelet Activation Signaling
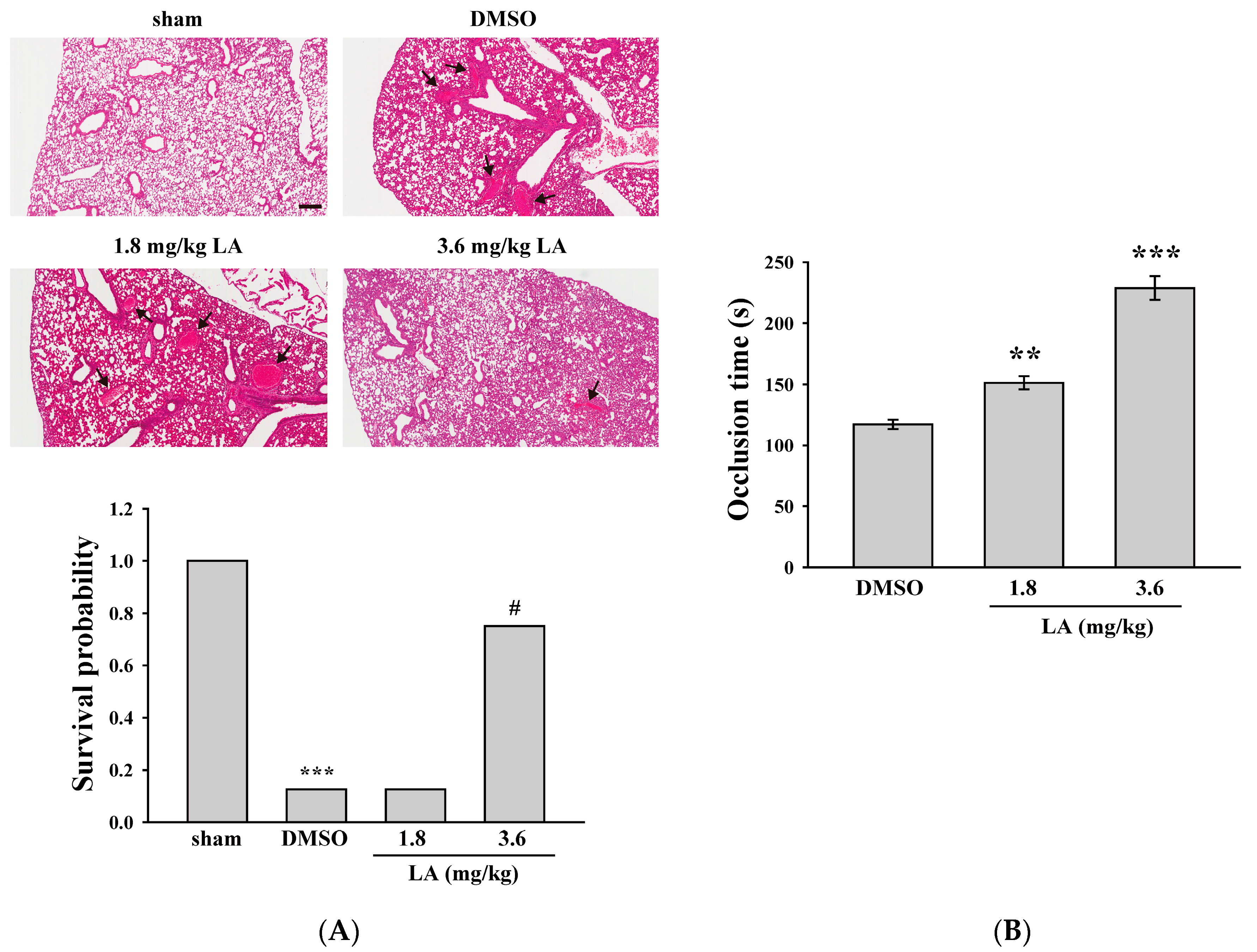
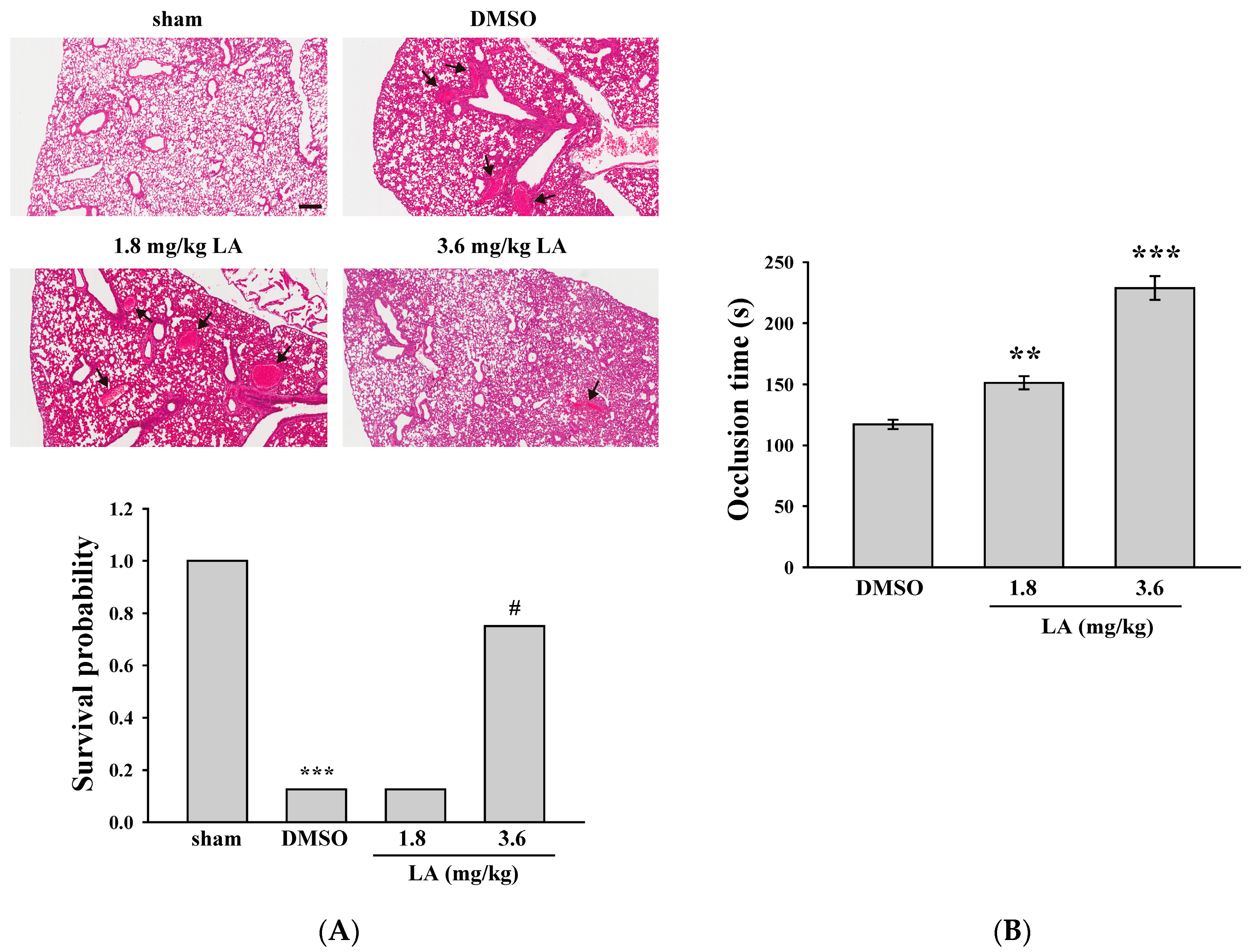
2.5. LA Alleviated ADP-Induced Pulmonary Thrombosis and Fluorescein Sodium-Induced Platelet Thrombus Formation in the Mesenteric Microvessels of Mice
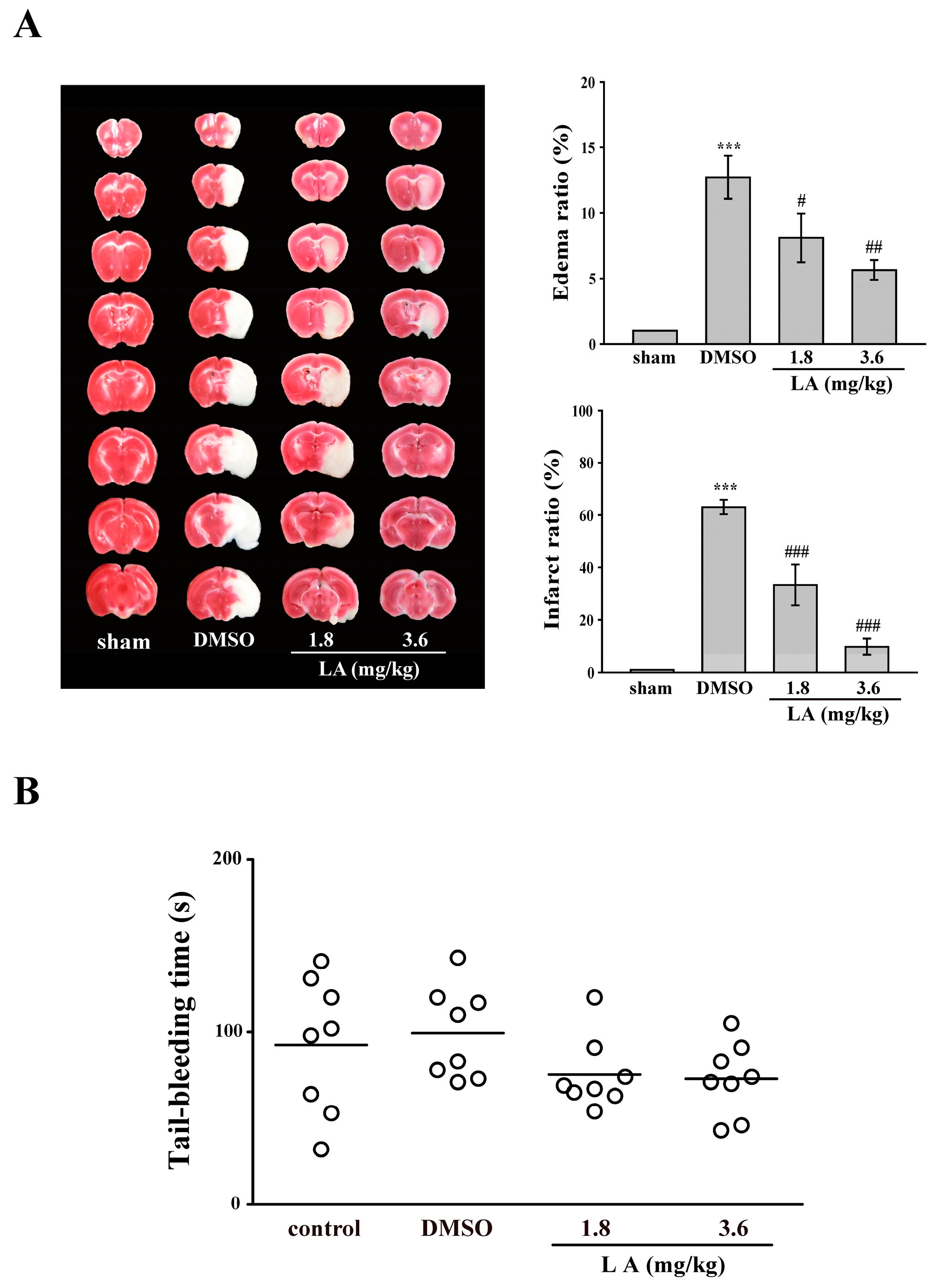
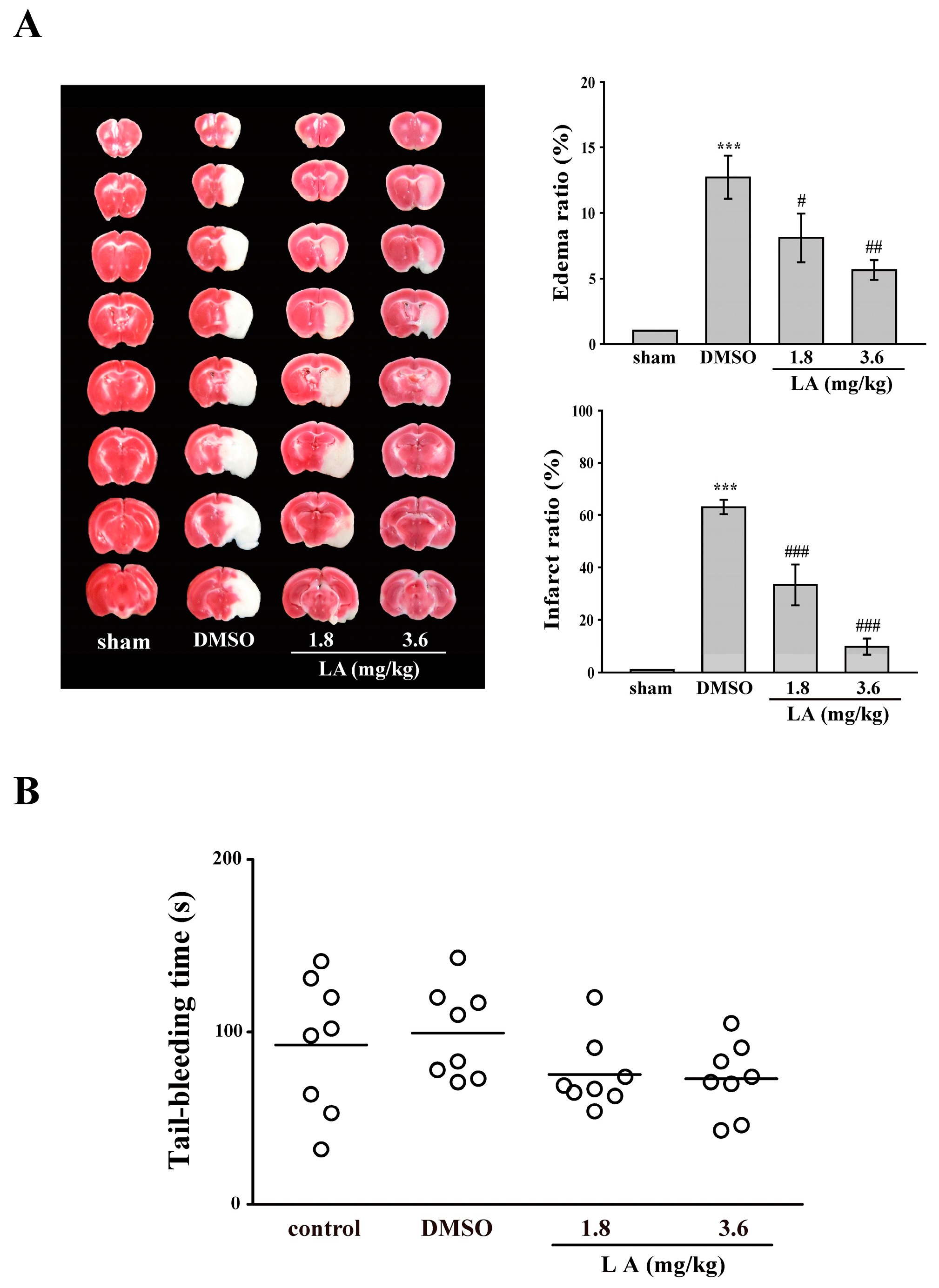
2.6. LA Protected against Middle Cerebral Artery Occlusion/Reperfusion-Induced Brain Injury without Affecting Normal Hemostasis
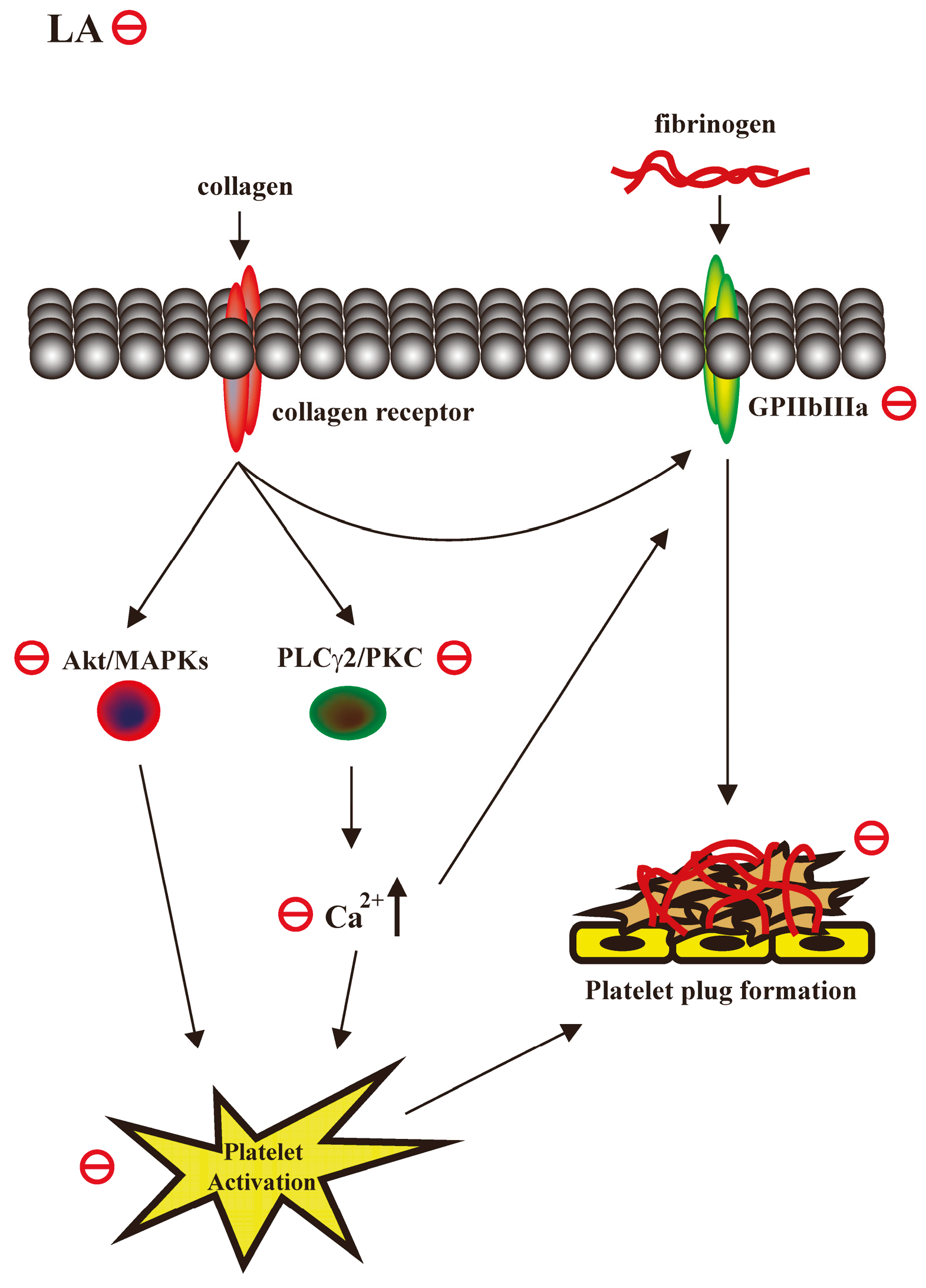
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Platelet Aggregation
4.3. ATP Release Measured Using a Microplate Reader
4.4. P-Selectin Secretion and GPIIbIIIa Activation
4.5. Calcium Mobilization
4.6. Determination of Lactate Dehydrogenase (LDH)
4.7. Immunoblotting Study
4.8. Competitive Binding Assay of Collagen Receptors through Flow Cytometry
4.9. Animals
4.10. ADP-Induced Acute Pulmonary Thrombosis in Mice
4.11. Fluorescein Sodium-Induced Platelet Thrombus Formation in Mesenteric Microvessels of Mice
4.12. Middle Cerebral Artery Occlusion/Reperfusion-Induced Brain Injury in Mice
4.13. Tail Bleeding Time
4.14. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GPIIbIIIa | Glycoprotein IIbIIIa |
| GPVI | Glycoprotein VI |
| LA | Licochalcone A |
| MAPK | Mitogen-activated protein kinase |
| MCAO | Middle cerebral artery occlusion |
| PKC | Protein kinase C |
| PLCγ2 | Phospholipase Cγ2 |
| ROS | Reactive oxygen species |
References
- Lindemann, S.; Kramer, B.; Seizer, P.; Gawaz, M. Platelets, inflammation and atherosclerosis. J. Thromb. Haemost. 2007, 5, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Pleines, I.; Bender, M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J. Thromb. Haemost. 2011, 9, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Dutting, S.; Bender, M.; Nieswandt, B. Platelet GPVI: A target for antithrombotic therapy?! Trends Pharmacol. Sci. 2012, 33, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; Meade, T.; et al. Aspirin in the primary and secondary prevention of vascular disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009, 373, 1849–1860. [Google Scholar] [PubMed]
- Adianti, M.; Aoki, C.; Komoto, M.; Deng, L.; Shoji, I.; Wahyuni, T.S.; Lusida, M.I.; Soetjipto; Fuchino, H.; Kawahara, N.; et al. Anti-hepatitis C virus compounds obtained from Glycyrrhiza uralensis and other Glycyrrhiza species. Microbiol. Immunol. 2014, 58, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, J.; Li, Y.J.; Zheng, Y.F.; Li, P. Antioxidant and anti-inflammatory activities of six flavonoids separated from licorice. Food Chem. 2013, 141, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Hui, W.; Liu, P.; Lv, Q.; Zeng, X.; Jiang, H.; Wang, Y.; Zheng, X.; Zheng, Y.; Li, J.; et al. Effect of licochalcone A on growth and properties of Streptococcus suis. PLoS ONE 2013, 8, e67728. [Google Scholar] [CrossRef]
- Messier, C.; Grenier, D. Effect of licorice compounds licochalcone A, glabridin and glycyrrhizic acid on growth and virulence properties of Candida albicans. Mycoses 2011, 54, e801–e806. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.H.; Chen, X.; Wang, Z.Y.; Chai, K.; Wang, Y.F.; Xu, X.H.; Wang, X.W.; Lu, J.H.; Wang, Y.T.; Chen, X.P.; et al. Induction of C/EBP homologous protein-mediated apoptosis and autophagy by licochalcone A in non-small cell lung cancer cells. Sci. Rep. 2016, 6, 26241. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.Y.; Hao, M.; Yang, X.Y.; Ba, Q.; Li, M.; Ni, S.J.; Wang, L.S.; Du, X. Licochalcone A inhibits growth of gastric cancer cells by arresting cell cycle progression and inducing apoptosis. Cancer Lett. 2011, 302, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Ci, X.; Wei, M.; Yang, X.; Cao, Q.; Guan, M.; Li, H.; Deng, Y.; Feng, H.; Deng, X. Licochalcone a inhibits lipopolysaccharide-induced inflammatory response in vitro and in vivo. J. Agric. Food Chem. 2012, 60, 3947–3954. [Google Scholar] [CrossRef] [PubMed]
- Fontes, L.B.; Dos Santos Dias, D.; de Carvalho, L.S.; Mesquita, H.L.; da Silva Reis, L.; Dias, A.T.; Da Silva Filho, A.A.; do Amaral Correa, J.O. Immunomodulatory effects of licochalcone A on experimental autoimmune encephalomyelitis. J. Pharm. Pharmacol. 2014, 66, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, J. Licochalcone a attenuates lipopolysaccharide-induced acute kidney injury by inhibiting nf-κ activation. Inflammation 2016, 39, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Okuda-Tanino, A.; Sugawara, D.; Tashiro, T.; Iwashita, M.; Obara, Y.; Moriya, T.; Tsushima, C.; Saigusa, D.; Tomioka, Y.; Ishii, K.; et al. Licochalcones extracted from Glycyrrhiza inflata inhibit platelet aggregation accompanied by inhibition of COX-1 activity. PLoS ONE 2017, 12, e0173628. [Google Scholar] [CrossRef] [PubMed]
- Suo, T.; Liu, J.; Chen, X.; Yu, H.; Wang, T.; Li, C.; Wang, Y.; Wang, C.; Li, Z. Combining chemical profiling and network analysis to investigate the pharmacology of complex prescriptions in traditional chinese medicine. Sci. Rep. 2017, 13, 40529. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.; Bachelot, C.; Chen, C.S.; Falck, J.R.; Hartwig, J.H.; Cantley, L.C.; Kovacsovics, T.J. Phosphorylation of the platelet p47 phosphoprotein is mediated by the lipid products of phosphoinositide 3-kinase. J. Biol. Chem. 1995, 270, 29525–29531. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Mangin, P.; Dangelmaier, C.; Lillian, R.; Jackson, S.P.; Daniel, J.L.; Kunapuli, S.P. Role of phosphoinositide 3-kinase β in glycoprotein VI-mediated Akt activation in platelets. J. Biol. Chem. 2009, 284, 33763–33772. [Google Scholar] [CrossRef] [PubMed]
- Adam, F.; Kauskot, A.; Rosa, J.P.; Bryckaert, M. Mitogen-activated protein kinases in hemostasis and thrombosis. J. Thromb. Haemost. 2008, 6, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, M.; Tuma, R.F.; Kunapuli, S.P. Deficiency of PAR4 attenuates cerebral ischemia/reperfusion injury in mice. J. Cereb. Blood Flow Metab. 2010, 30, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-peptide receptor 2/3/lipoxin a4 receptor regulates neutrophil-platelet aggregation and attenuates cerebral inflammation: Impact for therapy in cardiovascular disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Cooper, D.; Arumugam, T.V.; Zhang, J.H.; Nanda, A.; Granger, D.N. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J. Cereb. Blood Flow Metab. 2004, 24, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Research and future trends in the pharmaceutical development of medicinal herbs from Chinese medicine. Public Health Nutr. 2000, 3, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The traditional medicine and modern medicine from natural products. Molecules 2016, 21, E559. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; De, S.; Damron, D.S.; Chen, W.S.; Hay, N.; Byzova, T.V. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood 2004, 104, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Adam, F.; Kauskot, A.; Nurden, P.; Sulpice, E.; Hoylaerts, M.F.; Davis, R.J.; Rosa, J.P.; Bryckaert, M. Platelet JNK1 is involved in secretion and thrombus formation. Blood 2010, 115, 4083–4092. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Jiang, L.; Wei, M.; Yang, X.; Guan, M.; Xie, X.; Wei, J.; Liu, D.; Wang, D. Attenuation of allergic airway inflammation in a murine model of asthma by Licochalcone A. Immunopharmacol. Immunotoxicol. 2013, 35, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Park, J.H.; Kim, D.H.; Kim, Y.H.; Shin, H.K.; Kim, J.K. Licochalcone A isolated from licorice suppresses lipopolysaccharide-stimulated inflammatory reactions in RAW264.7 cells and endotoxin shock in mice. J. Mol. Med. 2008, 86, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Ren, H.; Wang, L.; Chen, W.; Ci, X. Lico a enhances nrf2-mediated defense mechanisms against t-bhp-induced oxidative stress and cell death via akt and erk activation in raw 264.7 cells. Oxid. Med. Cell. Longev. 2015, 2015, 709845. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Hsiao, G.; Shih, C.M.; Chou, D.S.; Sheu, J.R. Mechanisms of resveratrol-induced platelet apoptosis. Cardiovasc. Res. 2009, 83, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Chen, F.Y.; Hsu, J.F.; Fu, R.H.; Chang, C.M.; Chang, C.T.; Liu, C.H.; Wu, J.R.; Lee, A.S.; Chan, H.C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, D.; Theoret, J.F.; Villeneuve, L.; Abou-Saleh, H.; Mourad, W.; Allen, B.G.; Merhi, Y. Essential role of protein kinase C delta in platelet signaling, α IIb β 3 activation, and thromboxane A2 release. J. Biol. Chem. 2006, 281, 30024–30035. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lin, K.H.; Hsu, M.J.; Chou, D.S.; Hsiao, G.; Sheu, J.R. Suppression of NF-κB signaling by andrographolide with a novel mechanism in human platelets: Regulatory roles of the p38 MAPK-hydroxyl radical-ERK2 cascade. Biochem. Pharmacol. 2012, 84, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lee, J.J.; Chou, D.S.; Jayakumar, T.; Fong, T.H.; Hsiao, G.; Sheu, J.R. A novel role of andrographolide, an NF-κB inhibitor, on inhibition of platelet activation: The pivotal mechanisms of endothelial nitric oxide synthase/cyclic GMP. J. Mol. Med. 2011, 89, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Kuo, J.R.; Lu, W.J.; Chung, C.L.; Chou, D.S.; Huang, S.Y.; Lee, H.C.; Sheu, J.R. Hinokitiol inhibits platelet activation ex vivo and thrombus formation in vivo. Biochem. Pharmacol. 2013, 85, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.L.; Chen, R.J.; Jayakumar, T.; Lu, W.J.; Hsieh, C.Y.; Hsu, M.J.; Yang, C.H.; Chang, C.C.; Lin, Y.K.; Lin, K.H.; et al. Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 2016, 170, 57–72. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lien, L.-M.; Lin, K.-H.; Huang, L.-T.; Tseng, M.-F.; Chiu, H.-C.; Chen, R.-J.; Lu, W.-J. Licochalcone A Prevents Platelet Activation and Thrombus Formation through the Inhibition of PLCγ2-PKC, Akt, and MAPK Pathways. Int. J. Mol. Sci. 2017, 18, 1500. https://doi.org/10.3390/ijms18071500
Lien L-M, Lin K-H, Huang L-T, Tseng M-F, Chiu H-C, Chen R-J, Lu W-J. Licochalcone A Prevents Platelet Activation and Thrombus Formation through the Inhibition of PLCγ2-PKC, Akt, and MAPK Pathways. International Journal of Molecular Sciences. 2017; 18(7):1500. https://doi.org/10.3390/ijms18071500
Chicago/Turabian StyleLien, Li-Ming, Kuan-Hung Lin, Li-Ting Huang, Mei-Fang Tseng, Hou-Chang Chiu, Ray-Jade Chen, and Wan-Jung Lu. 2017. "Licochalcone A Prevents Platelet Activation and Thrombus Formation through the Inhibition of PLCγ2-PKC, Akt, and MAPK Pathways" International Journal of Molecular Sciences 18, no. 7: 1500. https://doi.org/10.3390/ijms18071500
APA StyleLien, L.-M., Lin, K.-H., Huang, L.-T., Tseng, M.-F., Chiu, H.-C., Chen, R.-J., & Lu, W.-J. (2017). Licochalcone A Prevents Platelet Activation and Thrombus Formation through the Inhibition of PLCγ2-PKC, Akt, and MAPK Pathways. International Journal of Molecular Sciences, 18(7), 1500. https://doi.org/10.3390/ijms18071500