Identification of Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by Structure-Based Virtual Screening and Molecular Docking Approach


Abstract
:
1. Introduction
2. Results and Discussion
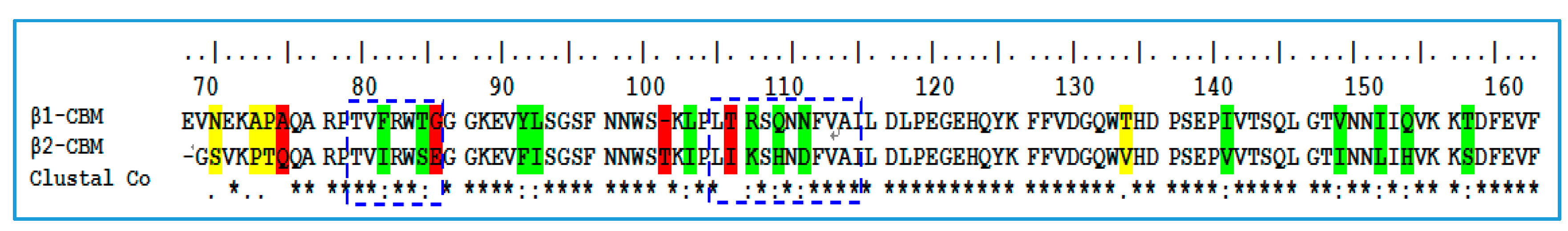
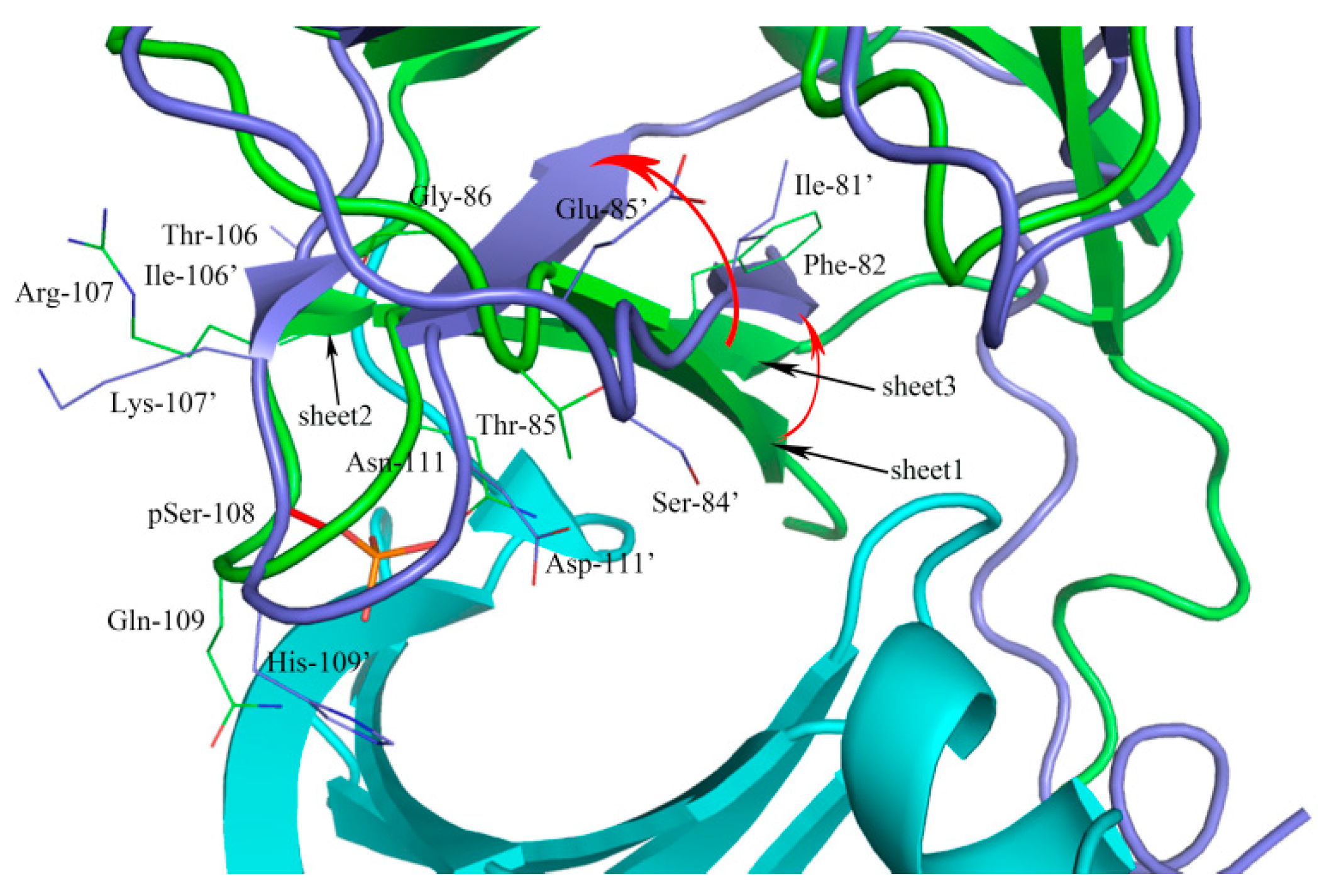
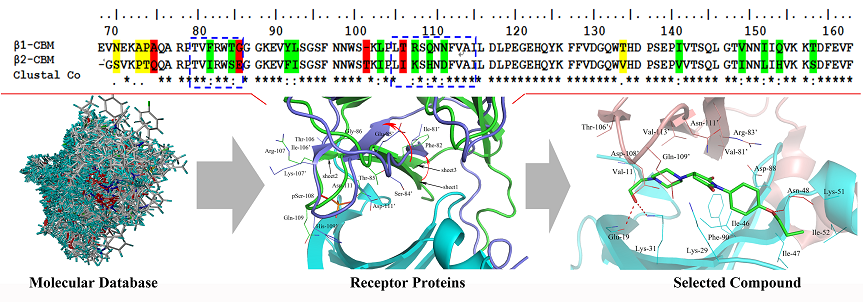
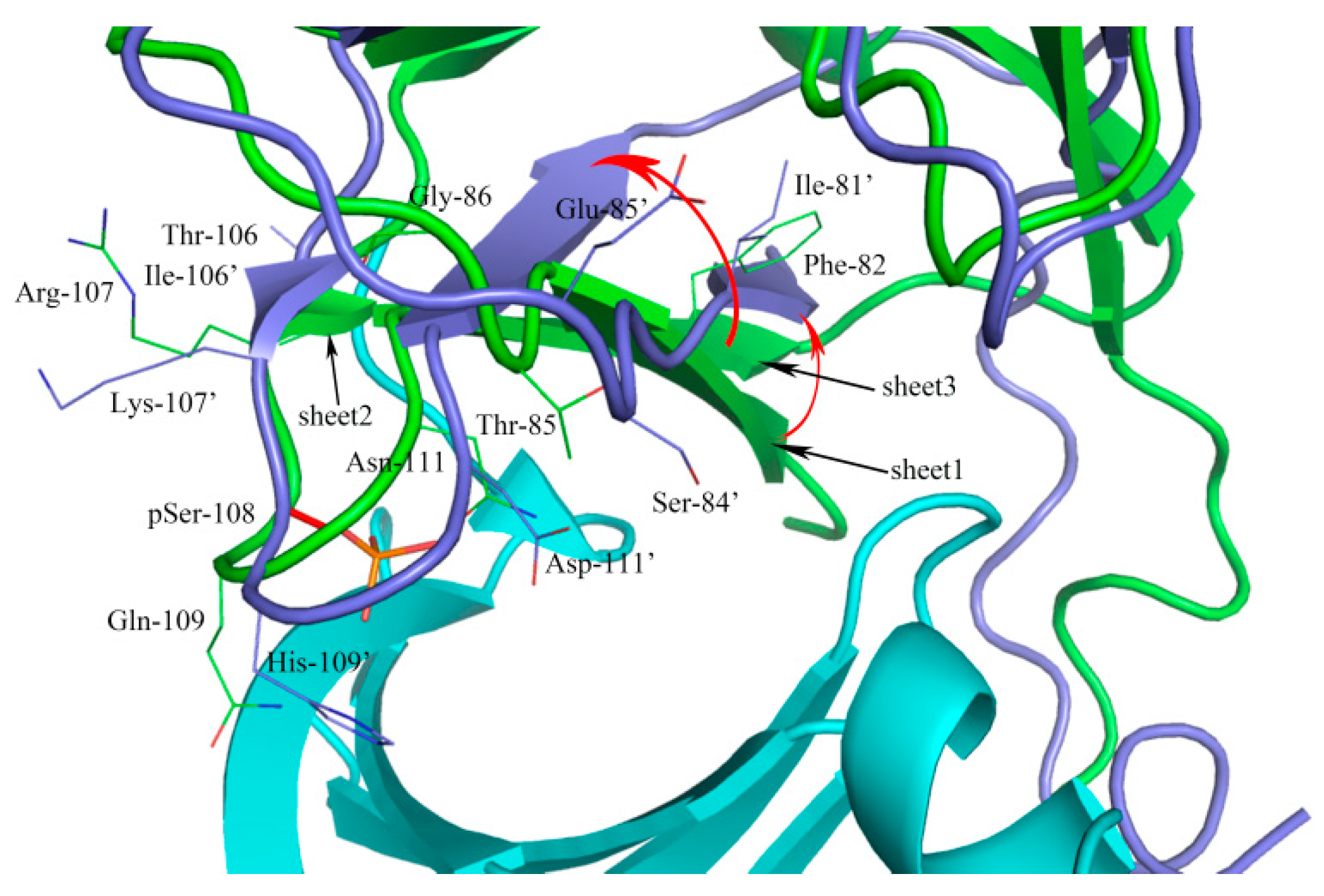
2.1. Sequence Alignment and Structural Comparison
2.2. Parameter Setting for Molecular Docking
2.3. High-Throughput Virtual Screening Procedure
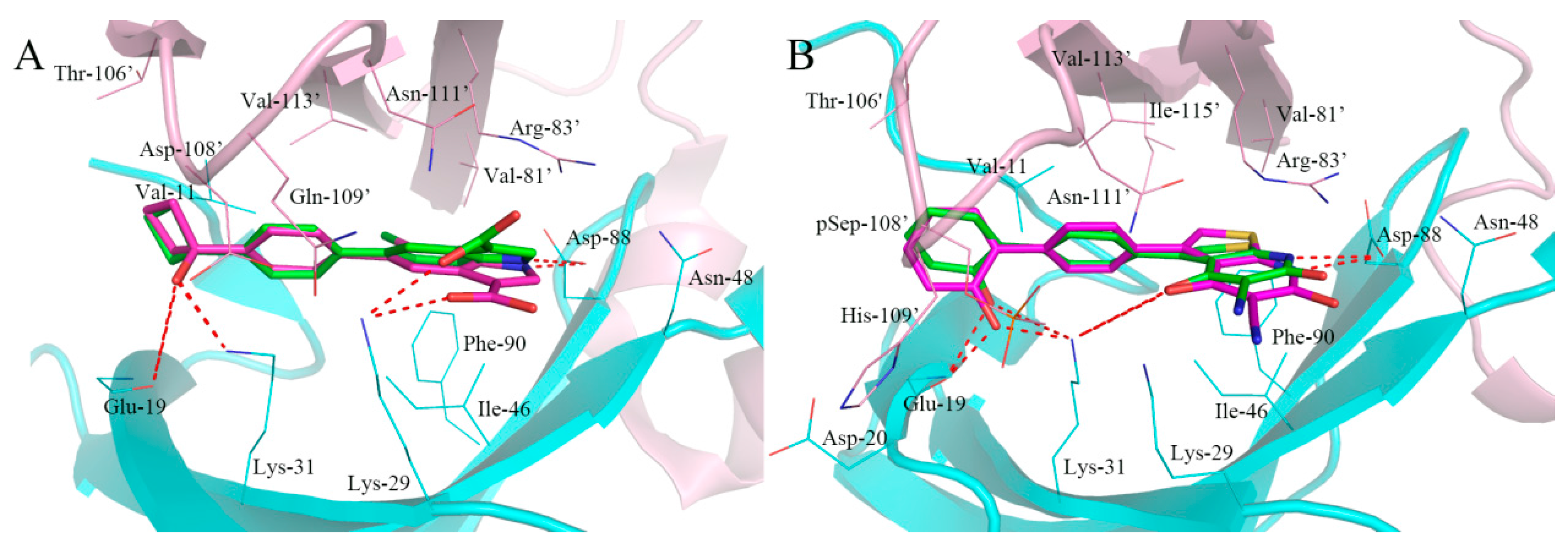
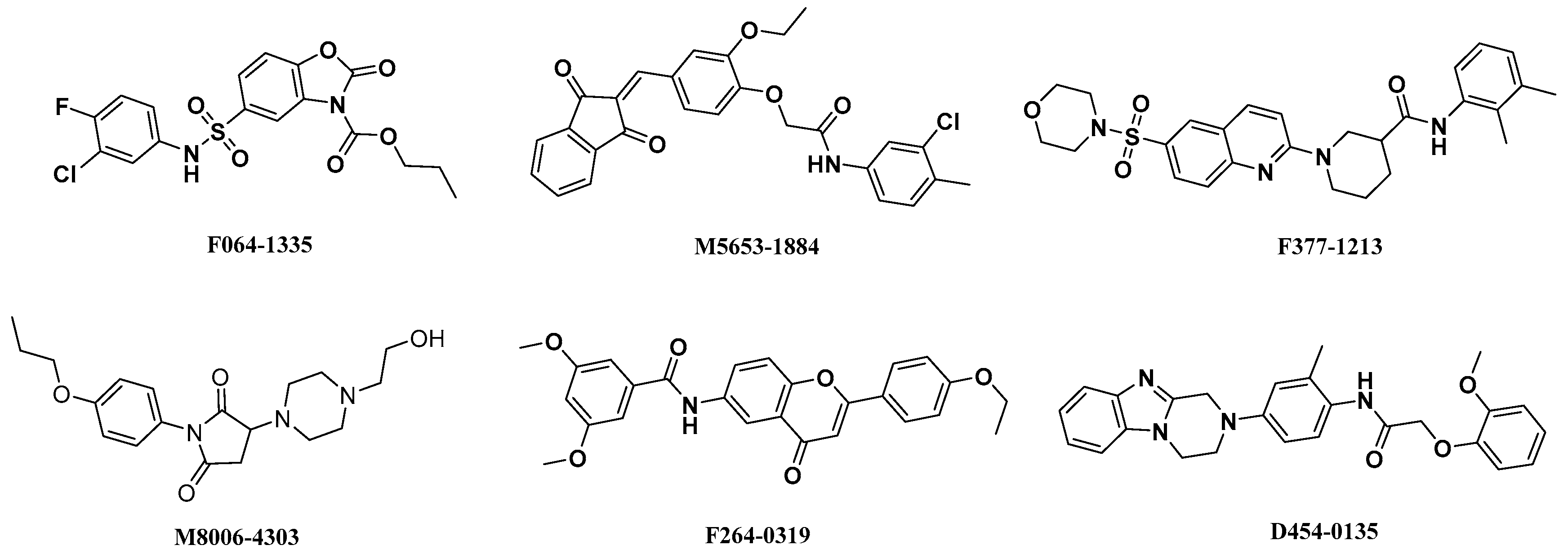
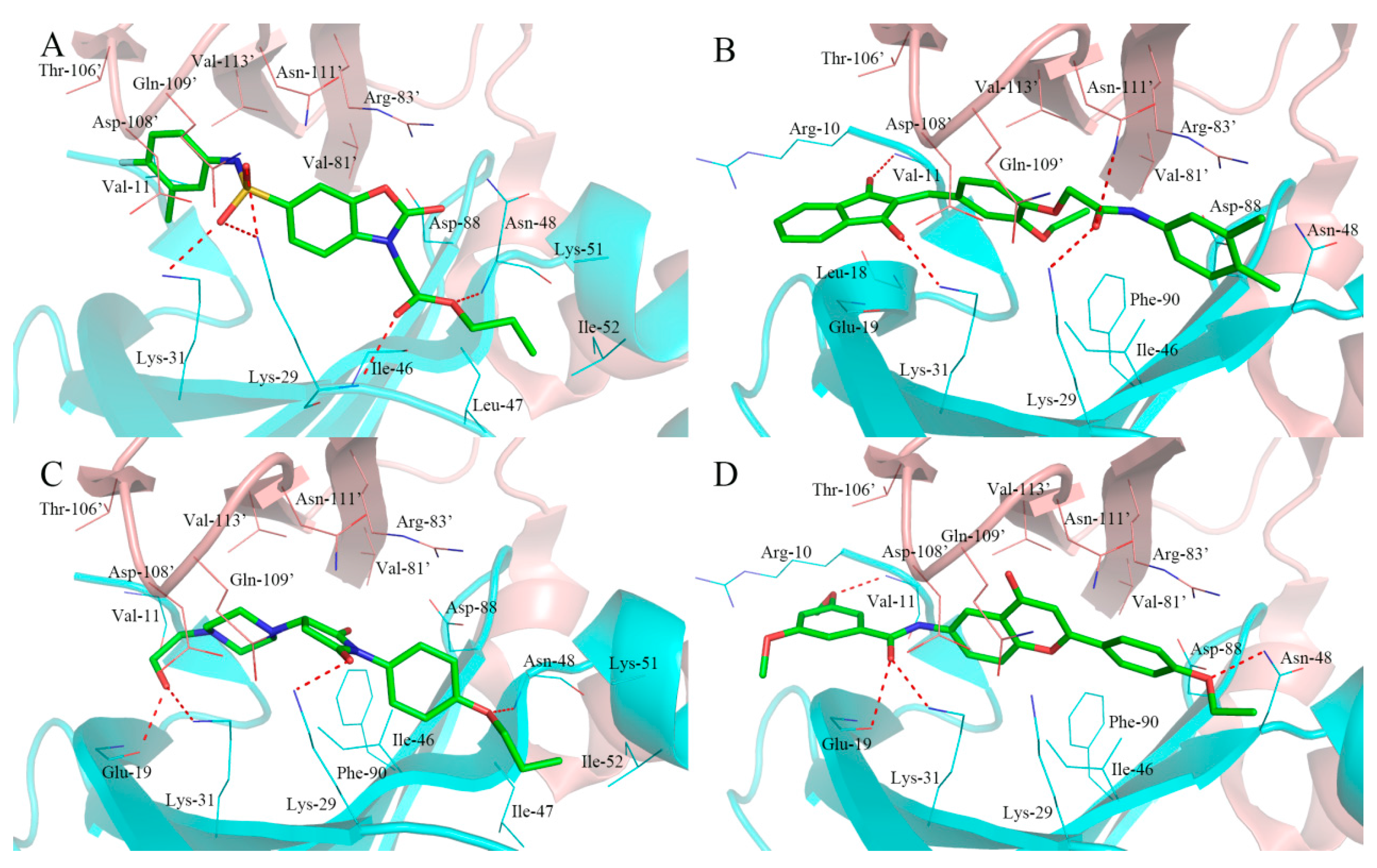
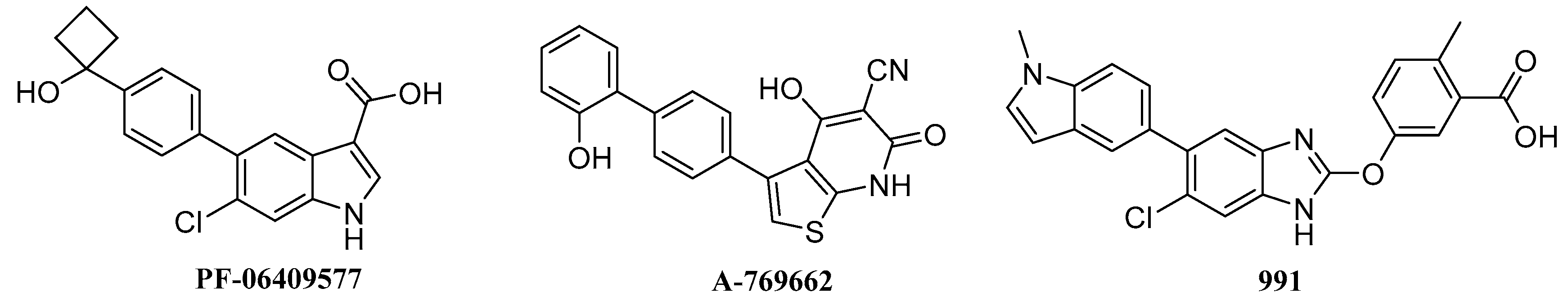
2.4. Analysis of Binding Mode of Activators for α1β1γ1 AMPK Isoform
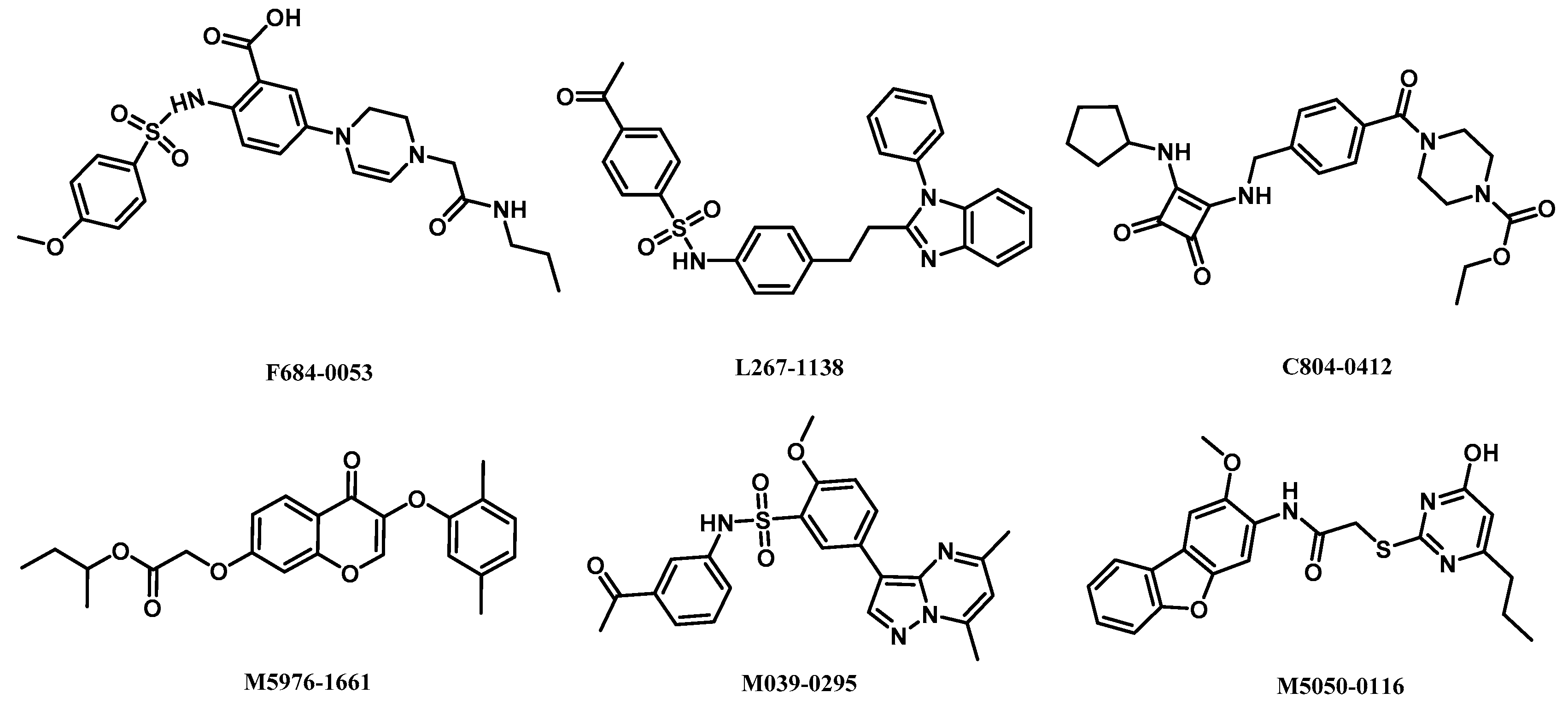
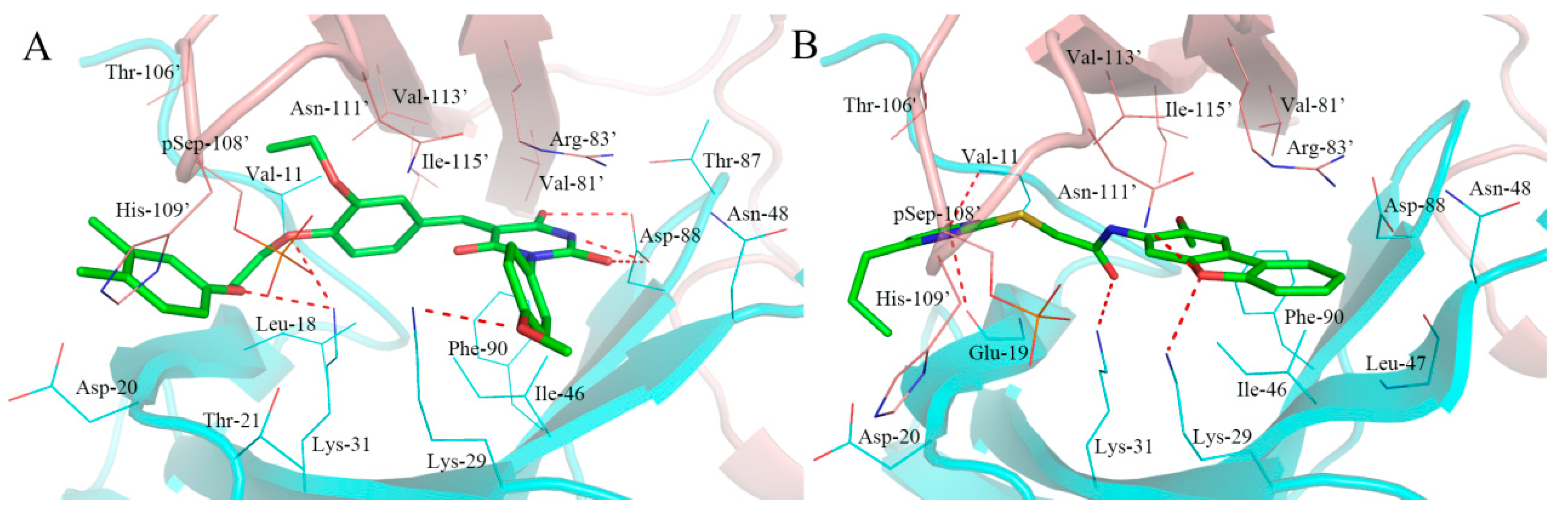
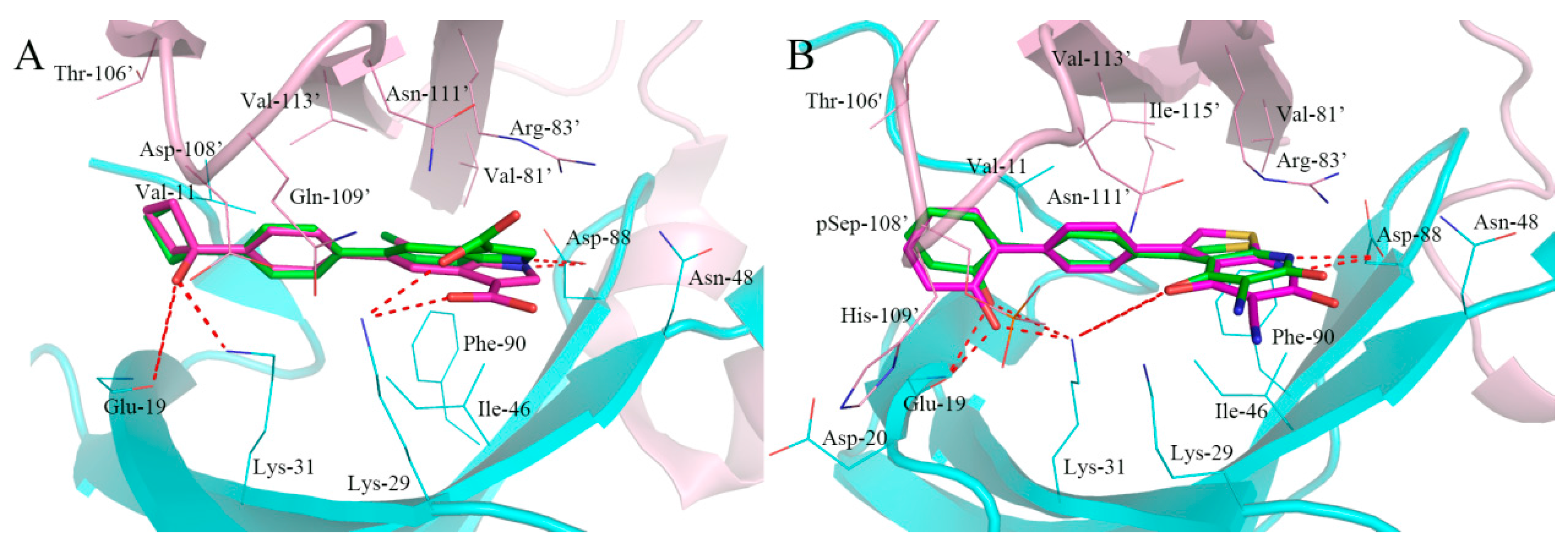
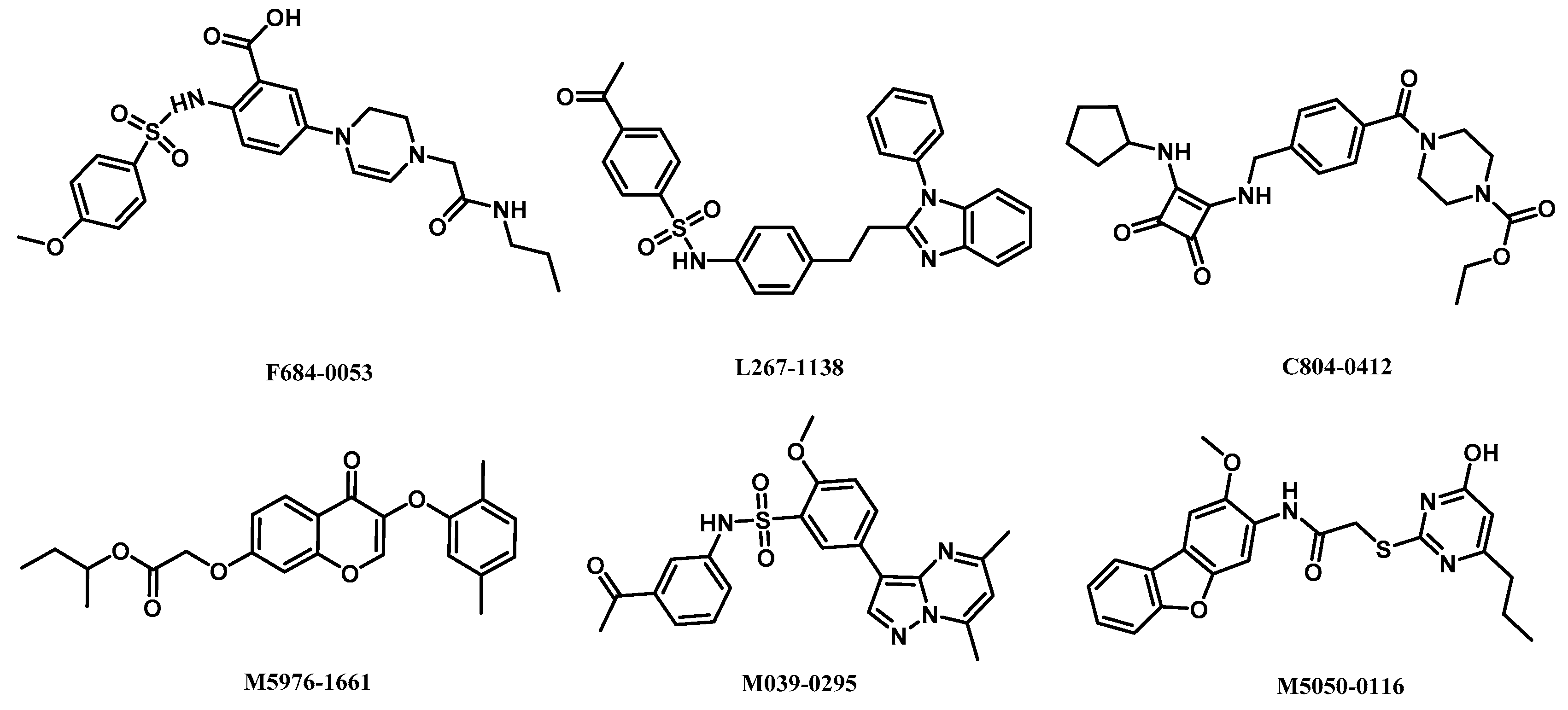
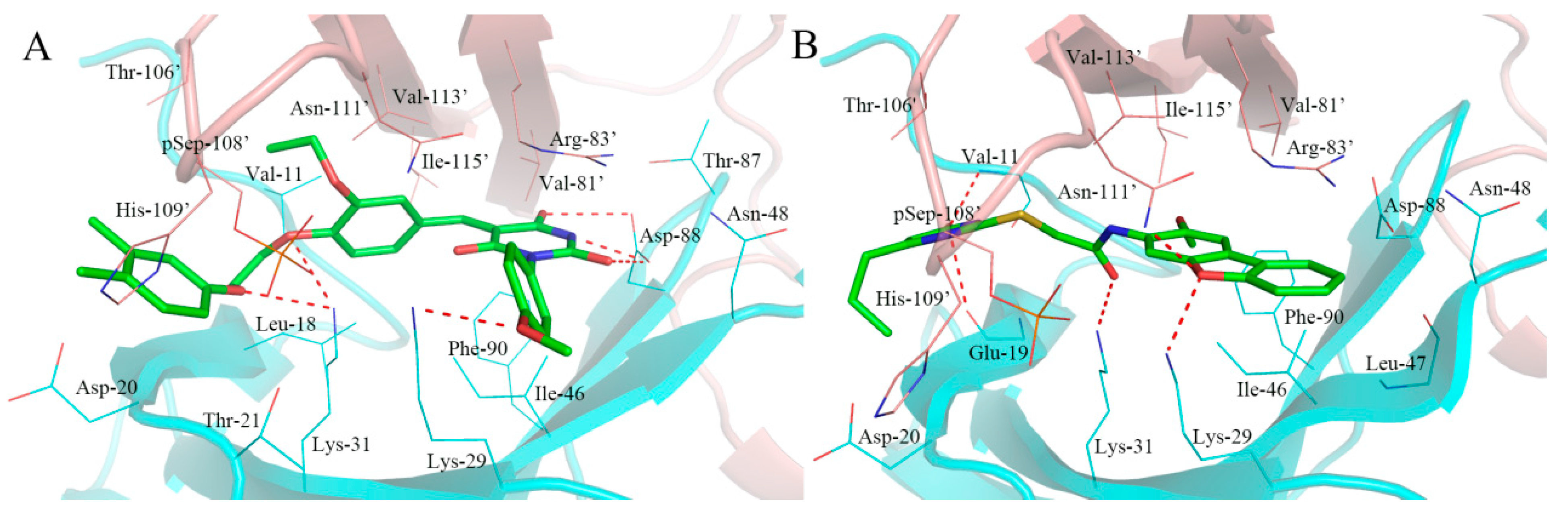
2.5. Analysis of Binding Mode of Activators for α2β1γ1 AMPK Isoform
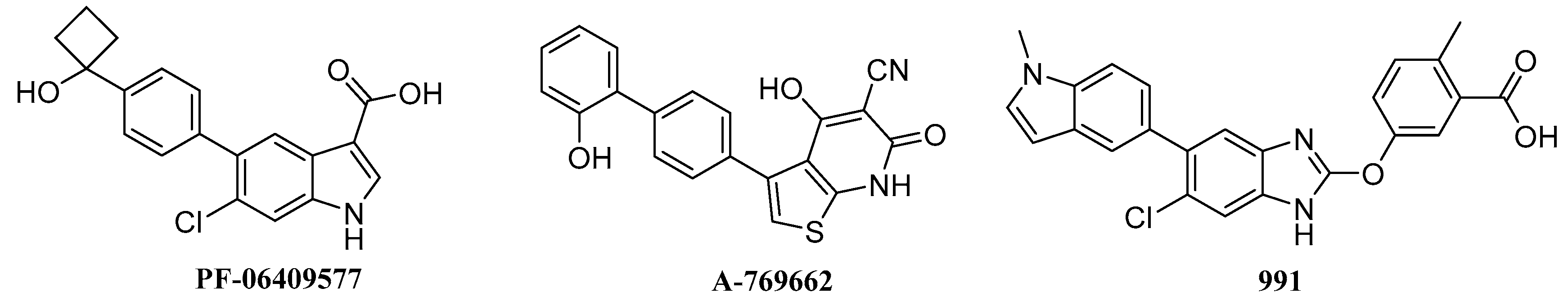
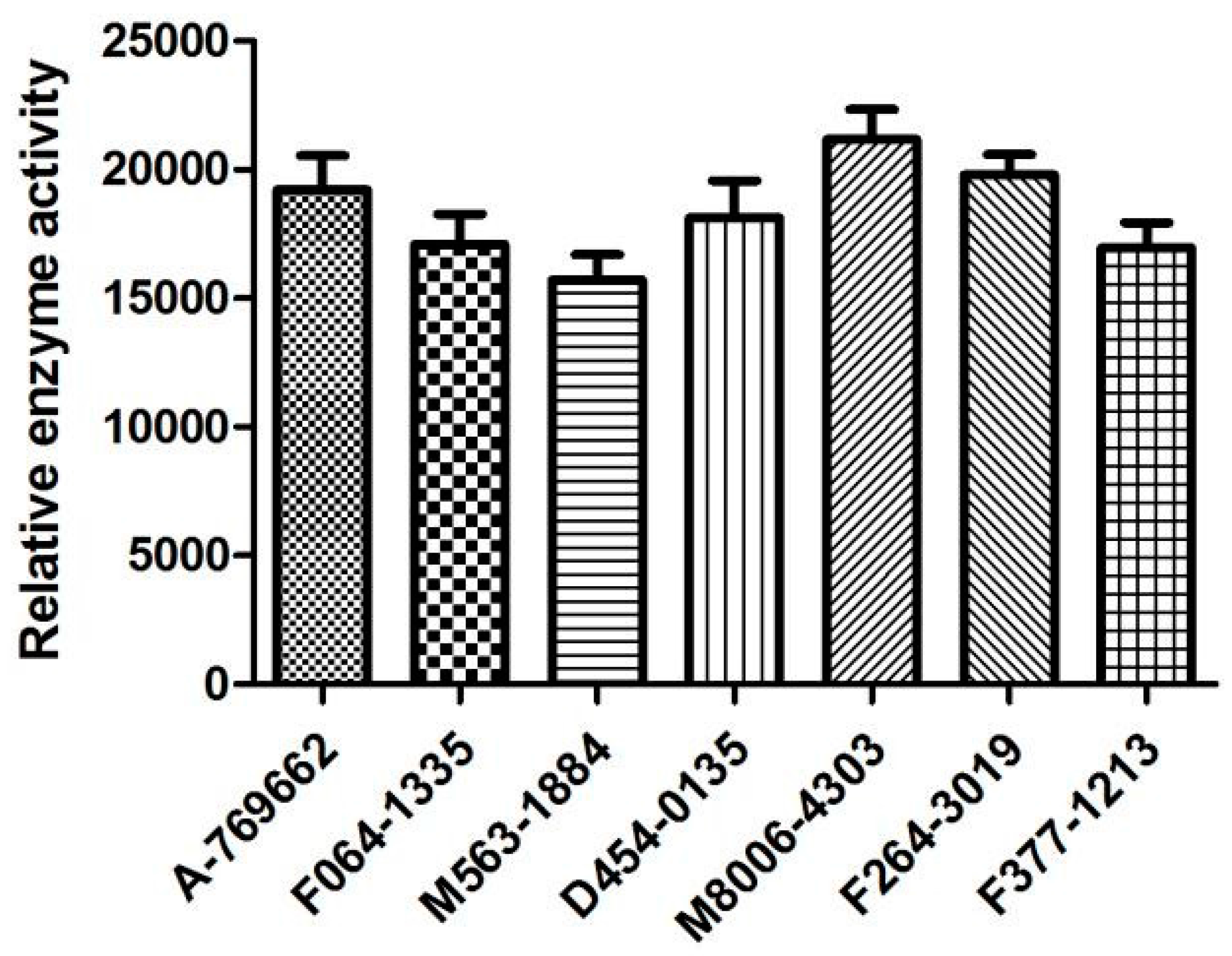
2.6. Biological Activities
3. Materials and Methods
3.1. Sequence Alignment and Structural Comparison
3.2. Molecular Docking
3.3. High-Throughput Virtual Screening
3.4. The In Vitro Activation Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gallagher, H.; Suckling, R.J. Diabetic nephropathy: Where are we on the journey from pathophysiology to treatment? Diabetes Obes. Metab. 2016, 18, 641–647. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas—7th Edition; IDF: Brussels, Belgium, 2015; Available online: http://www.diabetesatlas.org (accessed on 9 June 2016).
- Quiroga, B.; Arroyo, D.; de Arriba, G. Present and future in the treatment of diabetic kidney disease. J. Diabetes Res. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.C.; Tang, S.C. Diabetic nephropathy: Landmark clinical trials and tribulations. Nephrol. Dial. Transpl. 2016, 31, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Perezgomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Martἱn-Cleary, C.; Ruiz-Ortega, M.; Egido, J.; Navarro-González, J.F.; Ortiz, A.; Fernandez-Fernandez, B. Horizon 2020 in diabetic kidney disease: The clinical trial pipeline for add-on therapies on top of renin angiotensin system blockade. J. Clin. Med. 2015, 4, 1325–1347. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.O.; Kung, D.W.; Kalgutkar, A.S.; Kurumbail, R.G.; Miller, R.; Salatto, C.T.; Ward, J.; Withka, J.M.; Bhattacharya, S.K.; Boehm, M.; et al. Discovery and preclinical characterization of 6-chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic Acid (PF-06409577), a direct activator of adenosine monophosphate-activated protein kinase (AMPK), for the potential treatment of diabetic nephropathy. J. Med. Chem. 2016, 59, 8068–8081. [Google Scholar] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Wang, L.L.; Zhou, X.E.; Ke, J.Y.; Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Blowers, E.C.; Natarajan, A. Small molecule adenosine 5′-monophosphate activated protein kinase (AMPK) modulators and human diseases. J. Med. Chem. 2014, 58, 2–29. [Google Scholar] [CrossRef] [PubMed]
- Miglianico, M.; Nicolaes, G.A.F.; Neumann, D. Pharmacological targeting of AMP-activated protein kinase and opportunities for computer-aided drug design: Miniperspective. J. Med. Chem. 2016, 59, 2879–2893. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural basis for AMPK activation: Natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 2014, 22, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.O.; Kurumbail, R.G. Recent progress in the identification of adenosine monophosphate-activated protein kinase (AMPK) activators. Bioorg. Med. Chem. Lett. 2016, 26, 5139–5148. [Google Scholar] [CrossRef] [PubMed]
- Langendorf, C.G.; Kemp, B.E. Choreography of AMPK activation. Cell Res. 2015, 25, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Karis, D. Direct AMP-activated protein kinase activators: A review of evidence from the patent literature. Expert. Opin. Ther. Pat. 2012, 22, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Wlaker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Zuegg, J.; Cooper, M.A. Drug-Likeness and Increased Hydrophobicity of Commercially Available Compound Libraries for Drug Screening. Curr. Top. Med. Chem. 2012, 12, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Ling, N.M.; Issa, S.M.A.; Dite, T.A.; O’Brien, M.T.; Chen, Z.P.; Galic, S.; Langendorf, C.G.; Steinberg, G.R.; Kemp, B.E.; et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem. Biol. 2014, 21, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.J.; Ali, Z.S.; Hegarty, B.D.; Heath, R.; Snowden, M.A.; Carling, D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 2007, 282, 32539–32548. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.; Sarhan, A.; Alhajj, R. Multiple sequence alignment with affine gap by using multi-objective genetic algorithm. Comput. Methods Progr. Biomed. 2014, 114, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hu, Q.H.; Guo, M.Z.; Wang, G.H. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015, 31, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, X.B.; Jiang, W.R.; Lin, Z.Y.; Li, G.L.; Chen, K. Survey of MapReduce frame operation in bioinformatics. Brief. Bioinform. 2014, 15, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Gladue, D.P.; Baker-Bransetter, R.; Holinka, L.G.; Fernandez-Sainz, I.J.; O’Donnell, V.; Fletcher, P.; Lu, Z.Q.; Borca, M.V. Interaction of CSFV E2 protein with swine host factors as detected by yeast two-hybrid system. PLoS ONE 2014, 9, e85324. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Asokan, R.; Nagesha, S.N.; Manamohan, M.; Krishnakumar, N.K.; Mahadevaswamy, H.M.; Rebijith, K.B.; Prakash, M.N.; Sharath Chandra, G. Molecular diversity of Helicoverpa armigera Hubner (Noctuidae: Lepidoptera) in India. Orient. Insects 2012, 46, 130–143. [Google Scholar] [CrossRef]
- Dammganamet, K.L.; Bembenek, S.D.; Venable, J.W.; Castro, G.G.; Mangelschots, L.; Peeterst, D.C.G.; Mcallister, H.M.; Edwards, J.P.; Disepio, D.; Mirzadegan, T. A prospective virtual screening study: Enriching hit rates and designing focus libraries to find inhibitors of PI3Kδ and PI3Kγ. J. Med. Chem. 2016, 59, 4302–4313. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Drug-Likeness and Molecular Property Prediction. Available online: http://www.molsoft.com/mprop/ (accessed on 9 June 2017).
- Kashem, M.A.; Nelson, R.M.; Yingling, J.D.; Pullen, S.S.; Prokopowicz, A.S., III; Jones, J.W.; Wolak, J.P.; Rogers, G.R.; Morelock, M.M.; Snow, R.J.; et al. Three Mechanistically Distinct Kinase Assays Compared: Measurement of Intrinsic ATPase Activity Identified the Most Comprehensive Set of ITK Inhibitors. J. Biomol. Screen. 2007, 12, 70–83. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoforms | Compound No. | Total-Score | Crash | Polar | Similarity | Number of HBA/HBD | MolLog P | Drug-Likeness Model Score |
|---|---|---|---|---|---|---|---|---|
| AMPK (α1β1γ1) activators | F064-1335 | 10.50 | −2.35 | 3.54 | 0.44 | 6/1 | 3.79 | −0.16 |
| M5653-1884 | 10.24 | −1.43 | 2.90 | 0.50 | 5/1 | 6.07 | 0.22 | |
| D454-0135 | 10.20 | −1.58 | 3.41 | 0.44 | 6/2 | 4.03 | −0.31 | |
| M8006-4303 | 10.07 | −1.83 | 4.29 | 0.58 | 6/1 | 1.01 | 1.02 | |
| F264-3019 | 9.93 | −1.39 | 3.19 | 0.46 | 6/1 | 5.44 | 1.00 | |
| F377-1213 | 10.03 | −1.43 | 1.32 | 0.44 | 6/1 | 4.12 | 0.16 | |
| PF-06409577 | 7.29 | −0.07 | 3.08 | 0.93 | 3/3 | 3.80 | 0.71 | |
| AMPK (α2β1γ1) activators | L267-1138 | 10.96 | −2.46 | 2.78 | 0.52 | 4/1 | 6.05 | −0.08 |
| F684-0053 | 10.60 | −2.77 | 4.31 | 0.54 | 7/3 | 2.04 | 0.54 | |
| C804-0412 | 10.15 | −3.27 | 3.39 | 0.54 | 5/2 | 2.53 | 1.00 | |
| M5976-1661 | 9.46 | −0.92 | 1.32 | 0.46 | 6/0 | 4.84 | 0.62 | |
| M039-0295 | 9.35 | −1.61 | 1.48 | 0.50 | 6/1 | 2.66 | −0.20 | |
| M5050-0116 | 9.27 | −1.35 | 2.82 | 0.49 | 7/2 | 5.13 | 0.38 | |
| A-769662 | 7.44 | −1.46 | 1.26 | 0.93 | 5/3 | 3.46 | 0.30 | |
| 991 | 8.38 | −0.96 | 4.08 | 0.73 | 4/2 | 5.38 | 0.41 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.; Sun, J.; Zhou, S.; Gao, J.; Liu, Y. Identification of Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by Structure-Based Virtual Screening and Molecular Docking Approach. Int. J. Mol. Sci. 2017, 18, 1408. https://doi.org/10.3390/ijms18071408
Huang T, Sun J, Zhou S, Gao J, Liu Y. Identification of Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by Structure-Based Virtual Screening and Molecular Docking Approach. International Journal of Molecular Sciences. 2017; 18(7):1408. https://doi.org/10.3390/ijms18071408
Chicago/Turabian StyleHuang, Tonghui, Jie Sun, Shanshan Zhou, Jian Gao, and Yi Liu. 2017. "Identification of Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by Structure-Based Virtual Screening and Molecular Docking Approach" International Journal of Molecular Sciences 18, no. 7: 1408. https://doi.org/10.3390/ijms18071408
APA StyleHuang, T., Sun, J., Zhou, S., Gao, J., & Liu, Y. (2017). Identification of Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK) by Structure-Based Virtual Screening and Molecular Docking Approach. International Journal of Molecular Sciences, 18(7), 1408. https://doi.org/10.3390/ijms18071408