Macrophages and Phospholipases at the Intersection between Inflammation and the Pathogenesis of HIV-1 Infection

Abstract
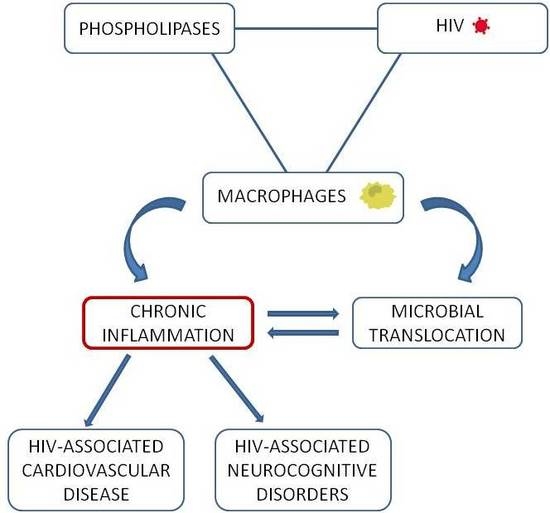
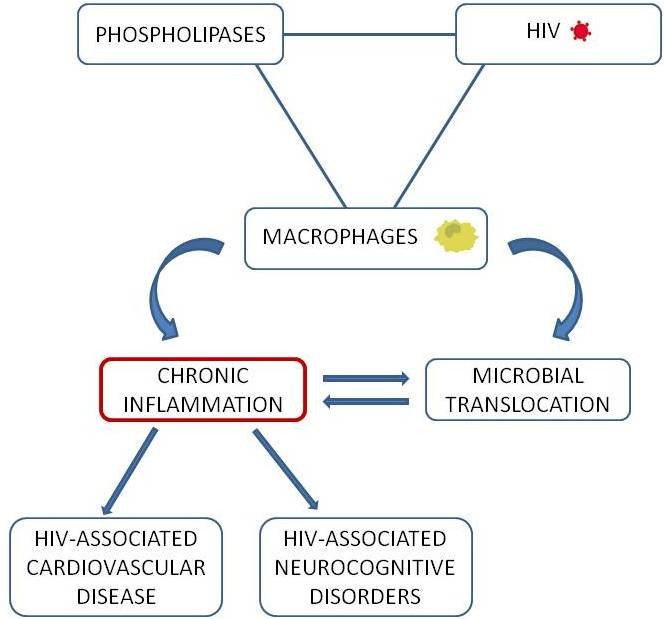
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Macrophage and Phospholipase Biology
2.1. Phospholipases
2.2. Role of Phospholipases in Regulating Differentiation, Polarization and Immune Functions of Monocytes/Macrophages
2.3. A Loop Intersecting Phospholipases, Macrophages and Chronic Inflammation
3. Macrophage and Phospholipase Contribution to Immune Activation and Inflammation in HIV-1 Infection
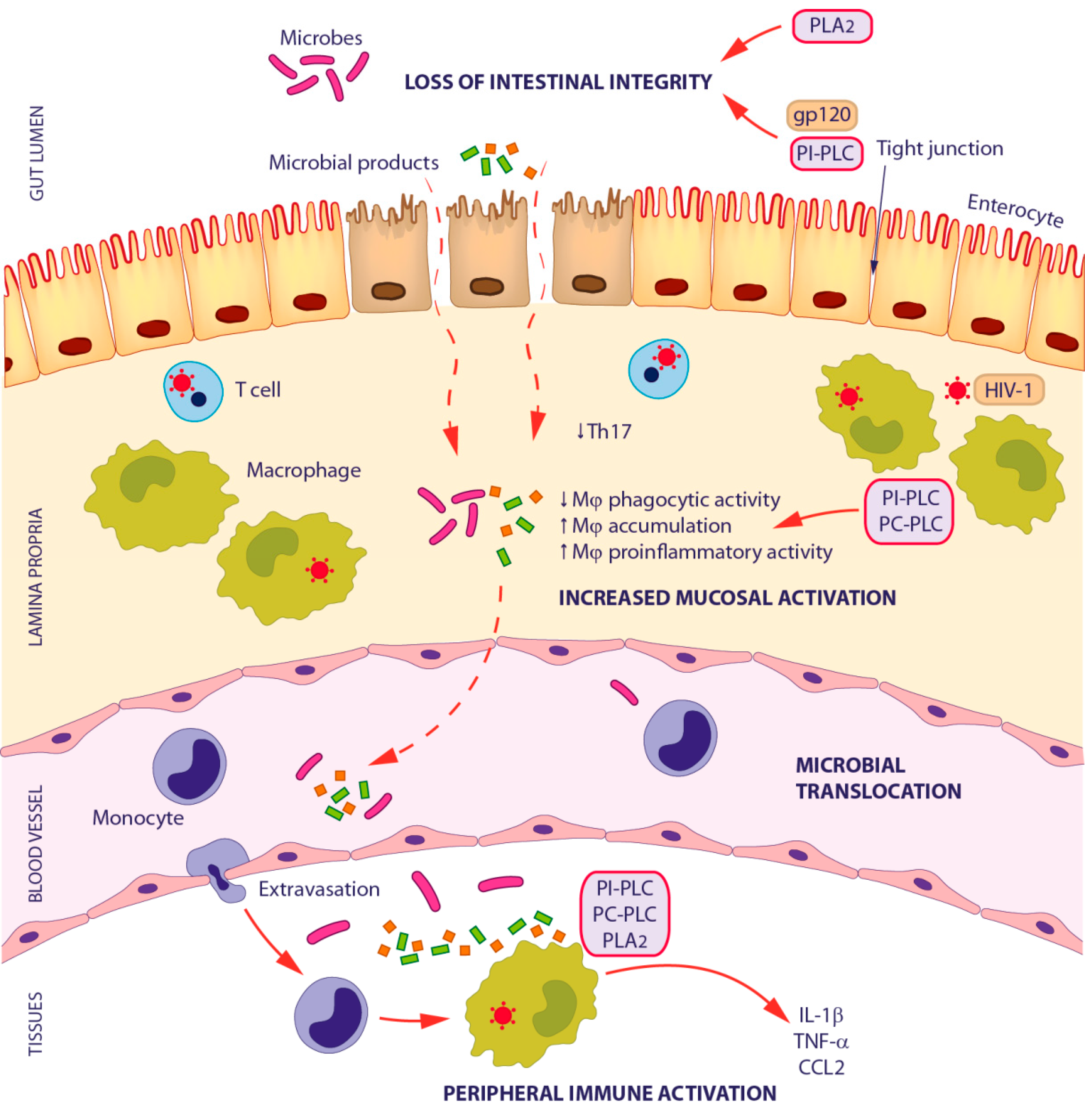
3.1. Microbial Translocation in HIV-1 Infection
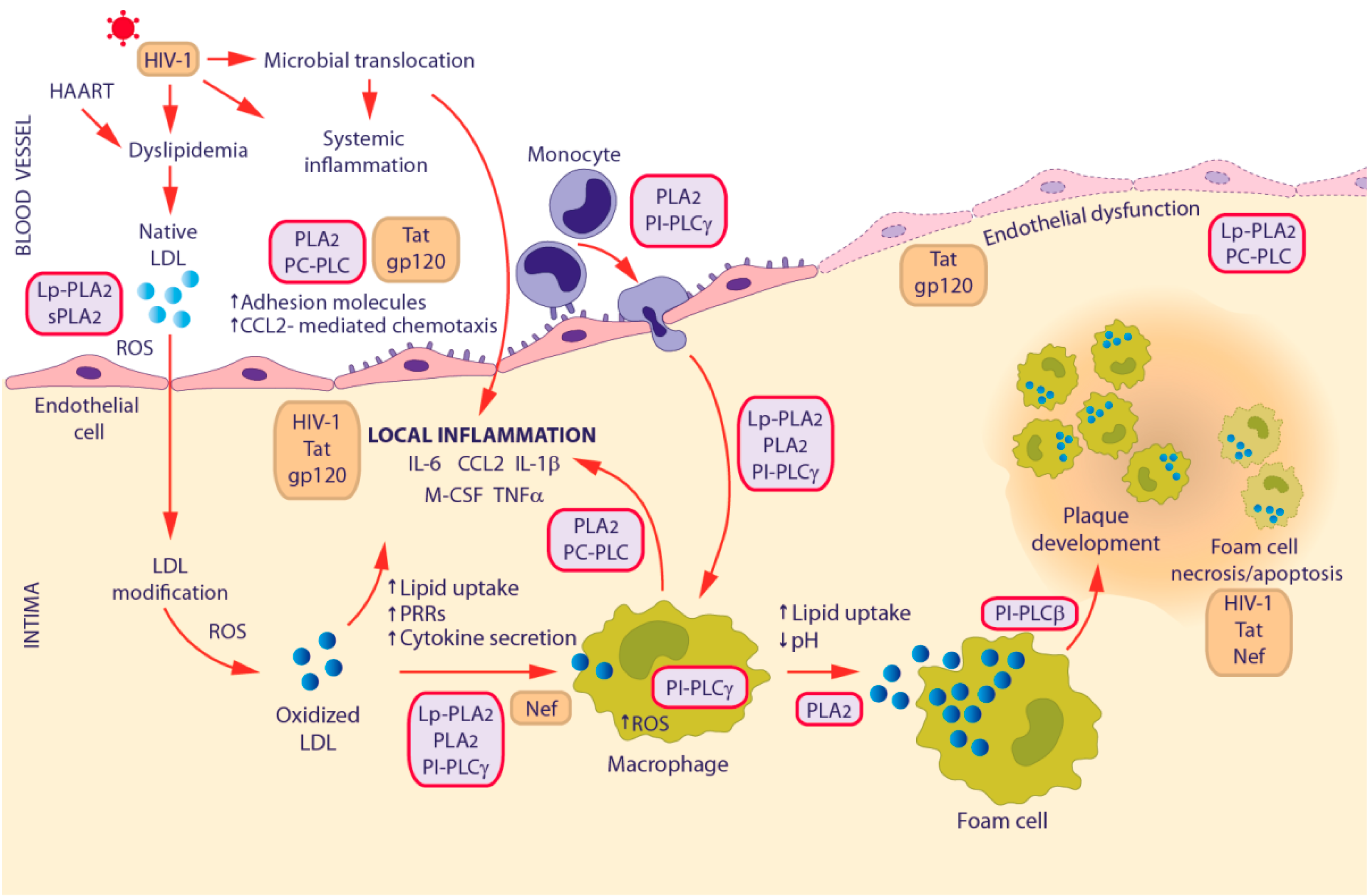
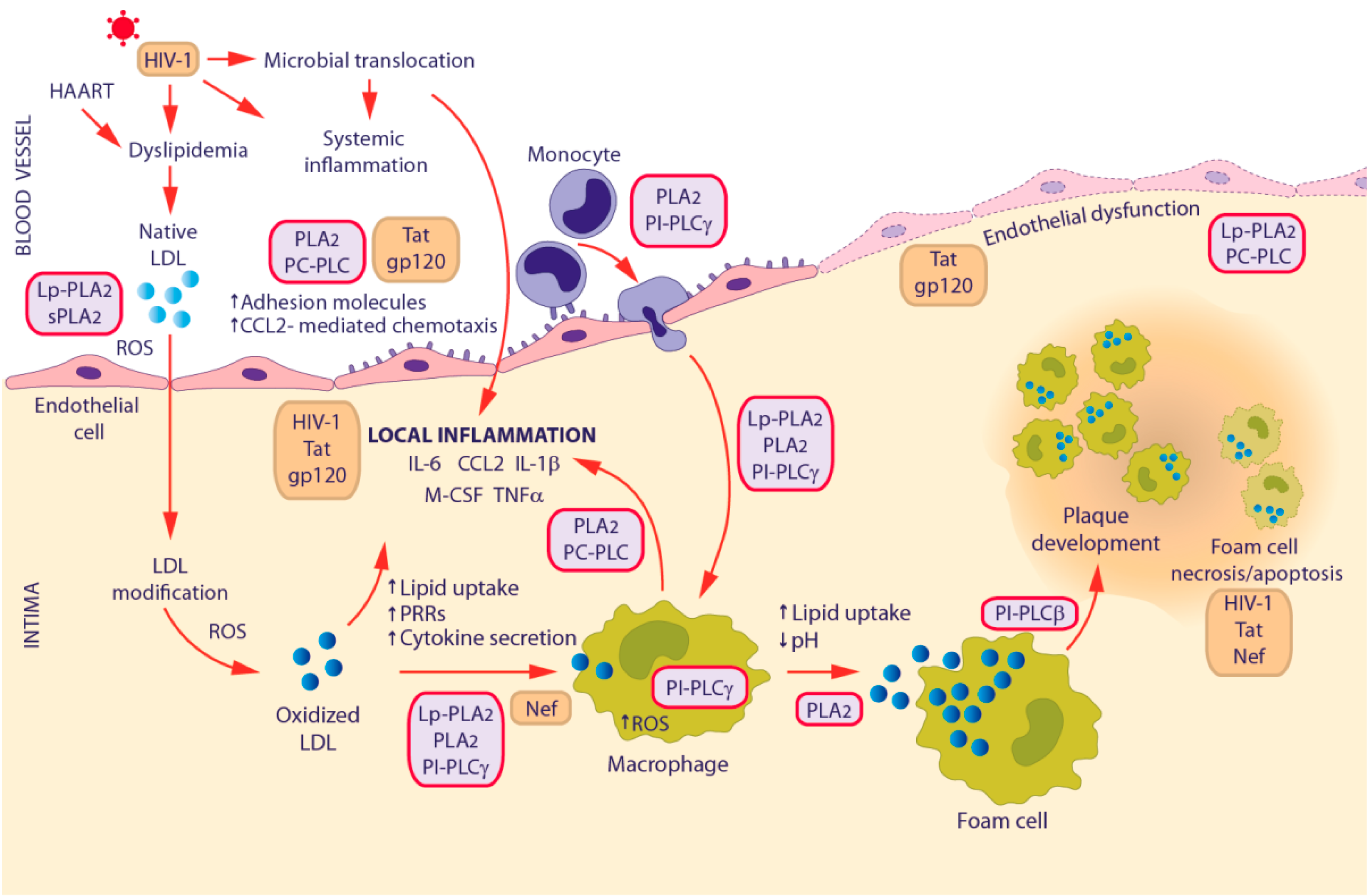
3.2. HIV-1-Associated Cardiovascular Disease
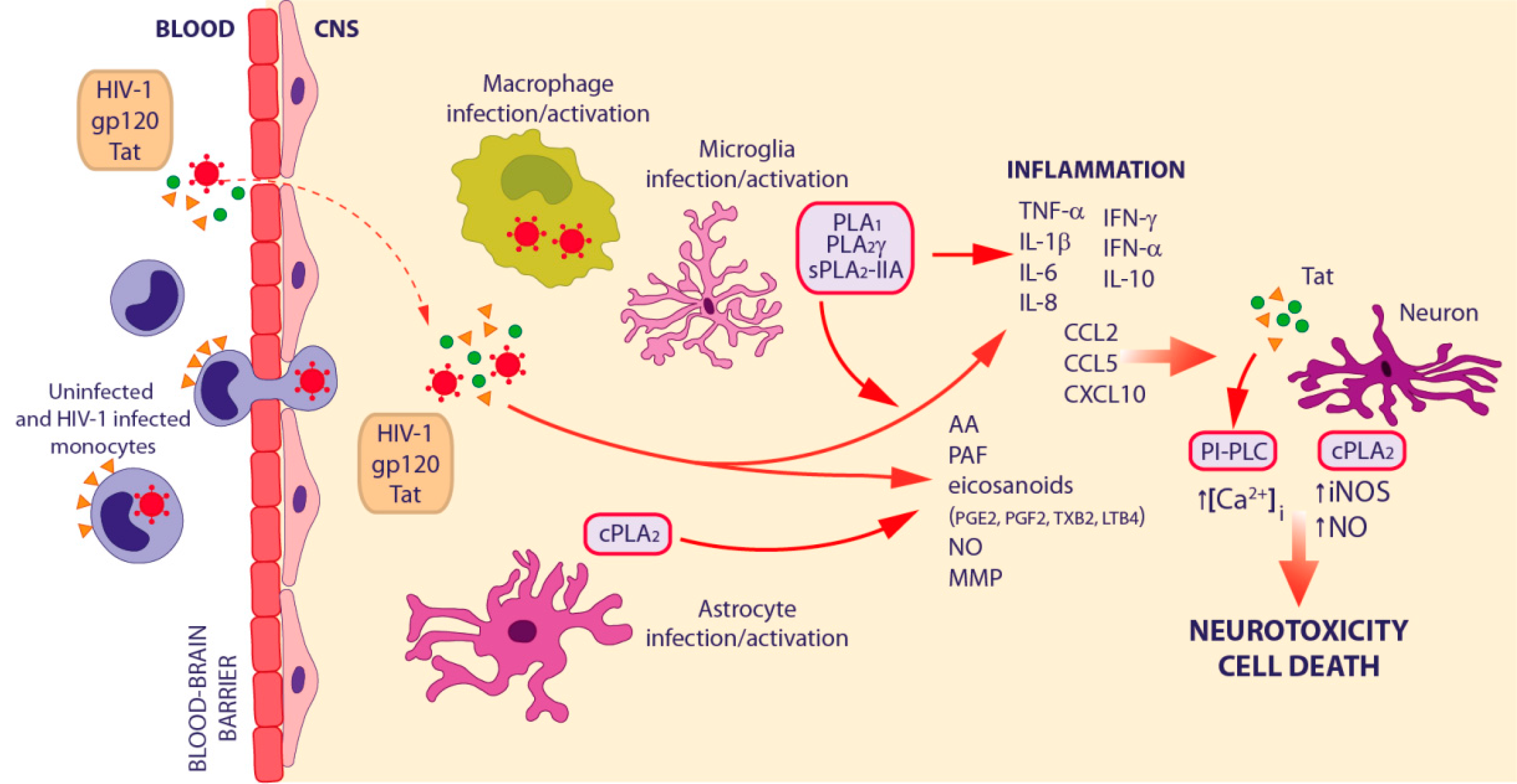
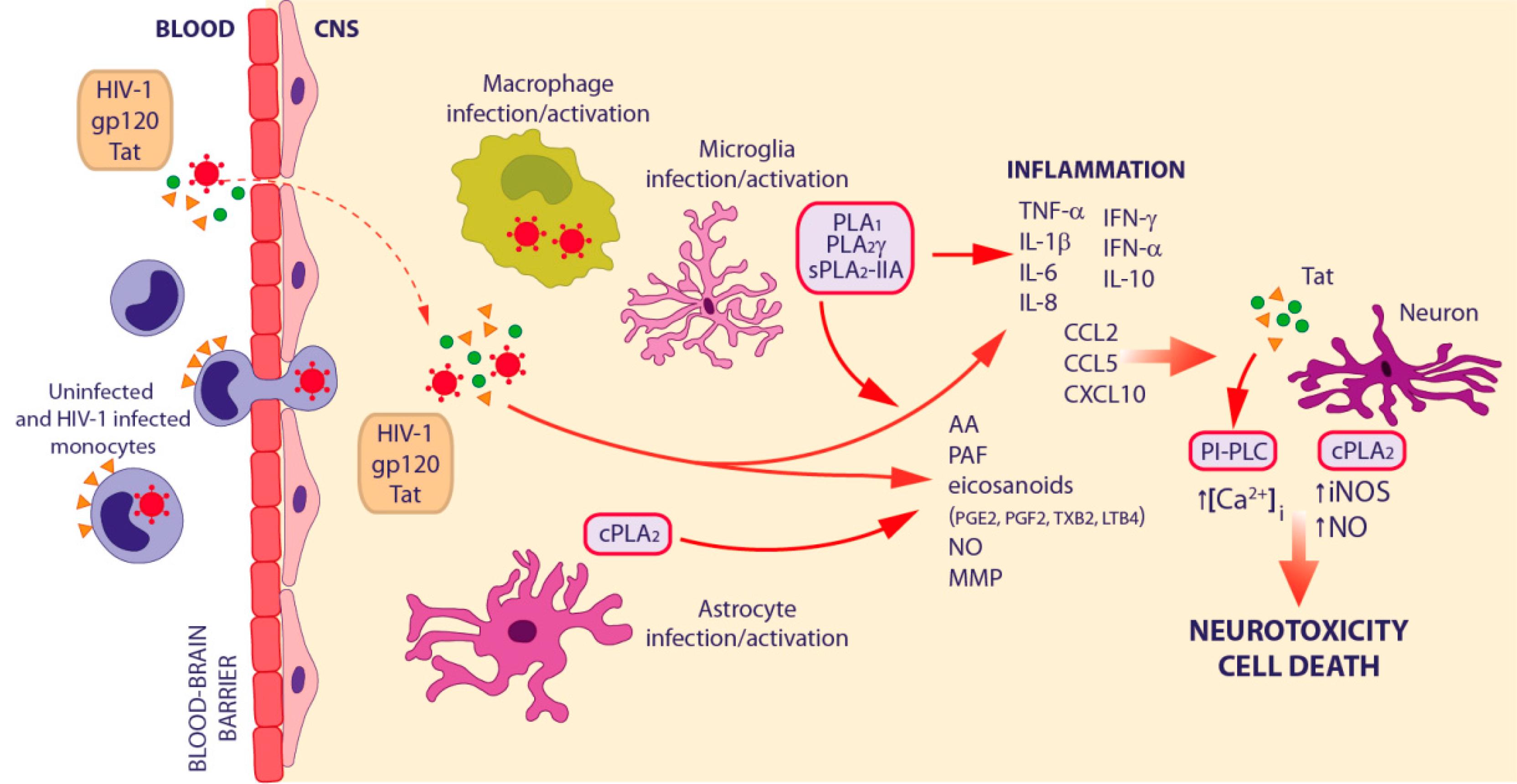
3.3. HIV-1-Associated Neurocognitive Disorders
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| AIDS | acquired immunodeficiency syndrome |
| BBB | blood–brain barrier |
| CCL2 | C-C motif chemokine ligand 2 |
| CNS | central nervous system |
| CVD | cardiovascular disease |
| DAG | diacylglycerol |
| FA | fatty acid |
| HAART | highly active antiretroviral therapy |
| HAND | HIV-associated neurocognitive disorder |
| HIV-1 | human immunodeficiency virus type I |
| IP3 | inositol 1,4,5-trisphosphate |
| LDL | low-density lipoprotein |
| LPA | lysophosphatidate |
| LPC | lysophosphatidylcholine |
| Lp-PLA2 | lipoprotein-associated phospholipase A2 |
| LPS | lipopolysaccharide |
| LT | leukotriene |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | nitric oxide |
| PAF | platelet-activating factor |
| PC | phosphatidylcholine |
| PC-PLC | phosphatidylcholine specific phospholipase C |
| PG | prostaglandin |
| PI | phosphoinositide |
| PI-PLC | phosphoinositide-specific phospholipase C |
| PL | phospholipase |
| PLA2 | phospholipase A2 |
| cPLA2 | cytosolic PLA2 |
| iPLA2 | Ca2+-insensitive phospholipase A2 |
| sPLA2 | secretory phospholipase A2 |
| ROS | reactive oxygen species |
| SIV | simian immunodeficiency virus |
| TNF | tumor necrosis factor |
References
- Katlama, C.; Deeks, S.G.; Autran, B.; Martinez-Picado, J.; van Lunzen, J.; Rouzioux, C.; Miller, M.; Vella, S.; Schmitz, J.E.; Ahlers, J.; et al. Barriers to a Cure for HIV: New Ways to Target and Eradicate HIV-1 Reservoirs. Lancet 2013, 381, 2109–2117. [Google Scholar] [CrossRef]
- Shan, L.; Siliciano, R.F. From Reactivation of Latent HIV-1 to Elimination of the Latent Reservoir: The Presence of Multiple Barriers to Viral Eradication. Bioessays 2013, 35, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Massanella, M.; Fromentin, R.; Chomont, N. Residual Inflammation and Viral Reservoirs: Alliance against an HIV Cure. Curr. Opin. HIV AIDS 2016, 11, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Tincati, C.; Silvestri, G. Microbial Translocation in the Pathogenesis of HIV Infection and AIDS. Clin. Microbiol. Rev. 2013, 26, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Sandler, N.G.; Douek, D.C. Microbial Translocation in HIV Infection: Causes, Consequences and Treatment Opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, A.R.; Zheng, Y.; Bosch, R.J.; Krishnan, S.; Rodriguez, B.; Hunt, P.W.; Plants, J.; Seth, A.; Wilson, C.C.; Deeks, S.G.; et al. Soluble Markers of Inflammation and Coagulation but Not T-Cell Activation Predict Non-AIDS-Defining Morbid Events during Suppressive Antiretroviral Treatment. J. Infect. Dis. 2014, 210, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Kalayjian, R.C.; Machekano, R.N.; Rizk, N.; Robbins, G.K.; Gandhi, R.T.; Rodriguez, B.A.; Pollard, R.B.; Lederman, M.M.; Landay, A. Pretreatment Levels of Soluble Cellular Receptors and Interleukin-6 are Associated with HIV Disease Progression in Subjects Treated with Highly Active Antiretroviral Therapy. J. Infect. Dis. 2010, 201, 1796–1805. [Google Scholar] [CrossRef] [PubMed]
- Sandler, N.G.; Wand, H.; Roque, A.; Law, M.; Nason, M.C.; Nixon, D.E.; Pedersen, C.; Ruxrungtham, K.; Lewin, S.R.; Emery, S.; et al. Plasma Levels of Soluble CD14 Independently Predict Mortality in HIV Infection. J. Infect. Dis. 2011, 203, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, T.B.; Ertner, G.; Petersen, J.; Moller, H.J.; Moestrup, S.K.; Eugen-Olsen, J.; Kronborg, G.; Benfield, T. Plasma Soluble CD163 Level Independently Predicts all-Cause Mortality in HIV-1-Infected Individuals. J. Infect. Dis. 2016, 214, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Gras, G.; Khan, K.A.; Abbas, W. Macrophage Signaling in HIV-1 Infection. Retrovirology 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-Induced Cytokine Production in Human Monocytes and Macrophages. Crit. Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, S.; Spadaro, F.; Gessani, S.; Podo, F.; Fantuzzi, L. Phospholipases: At the Crossroads of the Immune System and the Pathogenesis of HIV-1 Infection. J. Leukoc. Biol. 2017, 101, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Aloulou, A.; Ali, Y.B.; Bezzine, S.; Gargouri, Y.; Gelb, M.H. Phospholipases: An Overview. Methods Mol. Biol. 2012, 861, 63–85. [Google Scholar] [PubMed]
- Li, Z.; Vance, D.E. Phosphatidylcholine and Choline Homeostasis. J. Lipid Res. 2008, 49, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 Enzymes: Physical Structure, Biological Function, Disease Implication, Chemical Inhibition, and Therapeutic Intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; de Camilli, P. Phosphoinositides in Cell Regulation and Membrane Dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.G. Phosphoinositide-Specific Phospholipase C in Health and Disease. J. Lipid Res. 2015, 56, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Okabe, Y.; Medzhitov, R. Tissue Biology Perspective on Macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Haldar, M.; Murphy, K.M. Origin, Development, and Homeostasis of Tissue-Resident Macrophages. Immunol. Rev. 2014, 262, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.M.; Bincoletto, C.; Barros, C.C.; Ferreira, A.T.; Paredes-Gamero, E.J. PLCgamma2 and PKC are Important to Myeloid Lineage Commitment Triggered by M-SCF and G-CSF. J. Cell. Biochem. 2014, 115, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, K.L.; Ueyama, T.; Michaud, T.M.; Kashiwagi, K.; Wang, D.; Flax, L.A.; Shirai, Y.; Loegering, D.J.; Saito, N.; Lennartz, M.R. Targeting of Protein Kinase C-Epsilon during Fcγ Receptor-Dependent Phagocytosis Requires the epsilonC1B Domain and Phospholipase C-γ1. Mol. Biol. Cell 2006, 17, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Bohdanowicz, M.; Schlam, D.; Hermansson, M.; Rizzuti, D.; Fairn, G.D.; Ueyama, T.; Somerharju, P.; Du, G.; Grinstein, S. Phosphatidic Acid is Required for the Constitutive Ruffling and Macropinocytosis of Phagocytes. Mol. Biol. Cell 2013, 24, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, S.; Hasko, G.; Wu, D.; Leibovich, S.J. Suppression of PLCβ2 by Endotoxin Plays a Role in the Adenosine A2A Receptor-Mediated Switch of Macrophages from an Inflammatory to an Angiogenic Phenotype. Am. J. Pathol. 2009, 175, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Ando, T.; Wang, H.Y.; Kawakami, Y.; Kawakami, T. Lyn- and PLC-β3-Dependent Regulation of SHP-1 Phosphorylation Controls Stat5 Activity and Myelomonocytic Leukemia-Like Disease. Blood 2010, 116, 6003–6013. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.W.; Hancock, W.D.; Nelson, A.J.; Bone, R.N.; Tse, H.M.; Wohltmann, M.; Turk, J.; Ramanadham, S. Polarization of Macrophages Toward M2 Phenotype is Favored by Reduction in iPLA2β (Group VIA Phospholipase A2). J. Biol. Chem. 2016, 291, 23268–23281. [Google Scholar] [CrossRef] [PubMed]
- Di Raimo, T.; Leopizzi, M.; Mangino, G.; Rocca, C.D.; Businaro, R.; Longo, L.; Lo Vasco, V.R. Different Expression and Subcellular Localization of Phosphoinositide-Specific Phospholipase C Enzymes in Differently Polarized Macrophages. J. Cell. Commun. Signal. 2016, 10, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.M.; Vadali, S.; He, B.; Ware, J.; Kelly, T.; Post, S.R. Prostaglandins Produced during Class A Scavenger Receptor-Mediated Macrophage Adhesion Differentially Regulate Cytokine Production. J. Leukoc. Biol. 2015, 97, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Suram, S.; Silveira, L.J.; Mahaffey, S.; Brown, G.D.; Bonventre, J.V.; Williams, D.L.; Gow, N.A.; Bratton, D.L.; Murphy, R.C.; Leslie, C.C. Cytosolic Phospholipase A(2)α and Eicosanoids Regulate Expression of Genes in Macrophages Involved in Host Defense and Inflammation. PLoS ONE 2013, 8, e69002. [Google Scholar] [CrossRef] [PubMed]
- Andrei, C.; Margiocco, P.; Poggi, A.; Lotti, L.V.; Torrisi, M.R.; Rubartelli, A. Phospholipases C and A2 Control Lysosome-Mediated IL-1 β Secretion: Implications for Inflammatory Processes. Proc. Natl. Acad. Sci. USA 2004, 101, 9745–9750. [Google Scholar] [CrossRef] [PubMed]
- Balboa, M.A.; Perez, R.; Balsinde, J. Calcium-Independent Phospholipase A2 Mediates Proliferation of Human Promonocytic U937 Cells. FEBS J. 2008, 275, 1915–1924. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.M.; Gong, M.C.; Turk, J.; Post, S.R. Class A Scavenger Receptor-Mediated Macrophage Adhesion Requires Coupling of Calcium-Independent Phospholipase A(2) and 12/15-Lipoxygenase to Rac and Cdc42 Activation. J. Biol. Chem. 2007, 282, 33405–33411. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Balboa, M.A.; Balsinde, J. Involvement of Group VIA Calcium-Independent Phospholipase A2 in Macrophage Engulfment of Hydrogen Peroxide-Treated U937 Cells. J. Immunol. 2006, 176, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.K. Signal-Activated Phospholipase Regulation of Leukocyte Chemotaxis. J. Lipid Res. 2009, 50, S231–S236. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Lee, H.Y.; Jung, Y.S.; Lee, M.; Suh, P.G. Phospholipase Cγ in Toll-Like Receptor-Mediated Inflammation and Innate Immunity. Adv. Biol. Regul. 2017, 63, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.Z.; Libby, P. Obesity, Inflammation, and Atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Cuschieri, J.; Billgren, J.; Maier, R.V. Phosphatidylcholine-Specific Phospholipase C (PC-PLC) is Required for LPS-Mediated Macrophage Activation through CD14. J. Leukoc. Biol. 2006, 80, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Carter, A.B.; Gudmundsson, G.; Mallampalli, R.; Powers, L.S.; Hunninghake, G.W. A Phosphatidylcholine-Specific Phospholipase C Regulates Activation of p42/44 Mitogen-Activated Protein Kinases in Lipopolysaccharide-Stimulated Human Alveolar Macrophages. J. Immunol. 1999, 162, 3005–3012. [Google Scholar] [PubMed]
- Schutze, S.; Machleidt, T.; Kronke, M. The Role of Diacylglycerol and Ceramide in Tumor Necrosis Factor and Interleukin-1 Signal Transduction. J. Leukoc. Biol. 1994, 56, 533–541. [Google Scholar] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Quach, N.D.; Arnold, R.D.; Cummings, B.S. Secretory Phospholipase A2 Enzymes as Pharmacological Targets for Treatment of Disease. Biochem. Pharmacol. 2014, 90, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-Independent Phospholipases A2 and their Roles in Biological Processes and Diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Speranza, F.; Mahankali, M.; Henkels, K.M.; Gomez-Cambronero, J. The Molecular Basis of Leukocyte Adhesion Involving Phosphatidic Acid and Phospholipase D. J. Biol. Chem. 2014, 289, 28885–28897. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.; Yin, D.; Zhang, Y.; Miao, J. Targeting Phosphatidylcholine-Specific Phospholipase C for Atherogenesis Therapy. Trends Cardiovasc. Med. 2010, 20, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Zanni, M.V.; Schouten, J.; Grinspoon, S.K.; Reiss, P. Risk of Coronary Heart Disease in Patients with HIV Infection. Nat. Rev. Cardiol. 2014, 11, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Oorni, K.; Rajamaki, K.; Nguyen, S.D.; Lahdesmaki, K.; Plihtari, R.; Lee-Rueckert, M.; Kovanen, P.T. Acidification of the Intimal Fluid: The Perfect Storm for Atherogenesis. J. Lipid Res. 2015, 56, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Coll, B.; Alonso-Villaverde, C.; Joven, J. Monocyte Chemoattractant Protein-1 and Atherosclerosis: Is there Room for an Additional Biomarker? Clin. Chim. Acta 2007, 383, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages and Immune Cells in Atherosclerosis: Recent Advances and Novel Concepts. Basic Res. Cardiol. 2015, 110, 34. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory Processes in Cardiovascular Disease: A Route to Targeted Therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Tellis, C.C.; Tselepis, A.D. The Role of Lipoprotein-Associated Phospholipase A2 in Atherosclerosis may Depend on its Lipoprotein Carrier in Plasma. Biochim. Biophys. Acta 2009, 1791, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, B.; Wang, P.; Dong, X.; Fernandez-Hernando, C.; Li, Z.; Hla, T.; Li, Z.; Claffey, K.; Smith, J.D.; et al. Phospholipase Cβ3 Deficiency Leads to Macrophage Hypersensitivity to Apoptotic Induction and Reduction of Atherosclerosis in Mice. J. Clin. Investig. 2008, 118, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Serbulea, V.; de Weese, D.; Leitinger, N. The Effect of Oxidized Phospholipids on Phenotypic Polarization and Function of Macrophages. Free Radic. Biol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Oestvang, J.; Anthonsen, M.W.; Johansen, B. LysoPC and PAF Trigger Arachidonic Acid Release by Divergent Signaling Mechanisms in Monocytes. J. Lipids 2011, 2011, 532145. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Bisogni, V.; Rossitto, G.; Rossi, G.P. Lipoprotein-Associated Phospholipase A2 Prognostic Role in Atherosclerotic Complications. World J. Cardiol. 2015, 7, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Lee, J.H.; Choi, S.H.; Kim, S.; Almazan, F.; Witztum, J.L.; Miller, Y.I. Macrophages Generate Reactive Oxygen Species in Response to Minimally Oxidized Low-Density Lipoprotein: Toll-Like Receptor 4- and Spleen Tyrosine Kinase-Dependent Activation of NADPH Oxidase 2. Circ. Res. 2009, 104, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Gelb, M.H. Secretory Phospholipase A2: A Multifaceted Family of Proatherogenic Enzymes. Curr. Cardiol. Rep. 2009, 11, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Q.; Zhong, C.Y.; Sun, W.W.; Xiao, H.; Zhu, P.; Lin, Y.Z.; Zhang, C.L.; Gao, H.; Song, Z.Y. Elevated Type II Secretory Phospholipase A2 Increases the Risk of Early Atherosclerosis in Patients with Newly Diagnosed Metabolic Syndrome. Sci. Rep. 2016, 6, 34929. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Stafforini, D.M.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Expression of Plasma Platelet-Activating Factor Acetylhydrolase is Transcriptionally Regulated by Mediators of Inflammation. J. Biol. Chem. 1998, 273, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Macphee, C.H. Lipoprotein-Associated Phospholipase A2: A Potential New Risk Factor for Coronary Artery Disease and a Therapeutic Target. Curr. Opin. Pharmacol. 2001, 1, 121–125. [Google Scholar] [CrossRef]
- Ongen, B.; Kalkan Ucar, S.; Levent, E.; Azarsiz, E.; Kologlu, T.; Coker, M.; Sozmen, E.; Sagin, F.G. Lipoprotein-Associated Phospholipase A2: A New Marker to Determine Cardiovascular Risk in Hypercholesterolemic Dyslipidaemic Children. Ann. Clin. Biochem. 2016. [Google Scholar] [CrossRef]
- Goebeler, M.; Gillitzer, R.; Kilian, K.; Utzel, K.; Brocker, E.B.; Rapp, U.R.; Ludwig, S. Multiple Signaling Pathways Regulate NF-κB-Dependent Transcription of the Monocyte Chemoattractant Protein-1 Gene in Primary Endothelial Cells. Blood 2001, 97, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, H.Y.; Li, H.; Zhao, J.; Su, L.; Zhang, Y.; Zhang, S.L.; Miao, J.Y. Lipopolysaccharide Activated Phosphatidylcholine-Specific Phospholipase C and Induced IL-8 and MCP-1 Production in Vascular Endothelial Cells. J. Cell. Physiol. 2011, 226, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Tariq, M.; Iqbal, M.; Kumar, A.; Herbein, G. Eradication of HIV-1 from the Macrophage Reservoir: An Uncertain Goal? Viruses 2015, 7, 1578–1598. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.H.; Hearps, A.C.; Martin, G.E.; Williams, K.C.; Crowe, S.M. The Importance of Monocytes and Macrophages in HIV Pathogenesis, Treatment, and Cure. AIDS 2014, 28, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Eugenin, E.A.; Calderon, T.M.; Berman, J.W. Monocyte Maturation, HIV Susceptibility, and Transmigration Across the Blood Brain Barrier are Critical in HIV Neuropathogenesis. J. Leukoc. Biol. 2012, 91, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Gama, L.; Shirk, E.N.; Russell, J.N.; Carvalho, K.I.; Li, M.; Queen, S.E.; Kalil, J.; Zink, M.C.; Clements, J.E.; Kallas, E.G. Expansion of a Subset of CD14highCD16negCCR2low/Neg Monocytes Functionally Similar to Myeloid-Derived Suppressor Cells during SIV and HIV Infection. J. Leukoc. Biol. 2012, 91, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Cassetta, L.; Alfano, M.; Poli, G. Macrophage Polarization and HIV-1 Infection. J. Leukoc. Biol. 2010, 87, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Gerngross, L.; Lehmicke, G.; Belkadi, A.; Fischer, T. Role for cFMS in Maintaining Alternative Macrophage Polarization in SIV Infection: Implications for HIV Neuropathogenesis. J. Neuroinflamm. 2015, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.E.; Babas, T.; Mankowski, J.L.; Suryanarayana, K.; Piatak, M., Jr.; Tarwater, P.M.; Lifson, J.D.; Zink, M.C. The Central Nervous System as a Reservoir for Simian Immunodeficiency Virus (SIV): Steady-State Levels of SIV DNA in Brain from Acute through Asymptomatic Infection. J. Infect. Dis. 2002, 186, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.A.; Gama, L.; Dudaronek, J.M.; Voelker, T.; Tarwater, P.M.; Clements, J.E. Mechanism for the Establishment of Transcriptional HIV Latency in the Brain in a Simian Immunodeficiency Virus-Macaque Model. J. Infect. Dis. 2006, 193, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain Cell Reservoirs of Latent Virus in Presymptomatic HIV-Infected Individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An Unhealthy Constellation. Cell. Host Microbe 2016, 19, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, L.; Borghi, P.; Ciolli, V.; Pavlakis, G.; Belardelli, F.; Gessani, S. Loss of CCR2 Expression and Functional Response to Monocyte Chemotactic Protein (MCP-1) during the Differentiation of Human Monocytes: Role of Secreted MCP-1 in the Regulation of the Chemotactic Response. Blood 1999, 94, 875–883. [Google Scholar] [PubMed]
- Covino, D.A.; Sabbatucci, M.; Fantuzzi, L. The CCL2/CCR2 Axis in the Pathogenesis of HIV-1 Infection: A New Cellular Target for Therapy? Curr. Drug Targets 2016, 17, 76–110. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, L.; Spadaro, F.; Vallanti, G.; Canini, I.; Ramoni, C.; Vicenzi, E.; Belardelli, F.; Poli, G.; Gessani, S. Endogenous CCL2 (Monocyte Chemotactic Protein-1) Modulates Human Immunodeficiency Virus Type-1 Replication and Affects Cytoskeleton Organization in Human Monocyte-Derived Macrophages. Blood 2003, 102, 2334–2337. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, L.; Canini, I.; Belardelli, F.; Gessani, S. HIV-1 gp120 Stimulates the Production of Beta-Chemokines in Human Peripheral Blood Monocytes through a CD4-Independent Mechanism. J. Immunol. 2001, 166, 5381–5387. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, L.; Spadaro, F.; Purificato, C.; Cecchetti, S.; Podo, F.; Belardelli, F.; Gessani, S.; Ramoni, C. Phosphatidylcholine-Specific Phospholipase C Activation is Required for CCR5-Dependent, NF-κB-Driven CCL2 Secretion Elicited in Response to HIV-1 gp120 in Human Primary Macrophages. Blood 2008, 111, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, F.; Cecchetti, S.; Purificato, C.; Sabbatucci, M.; Podo, F.; Ramoni, C.; Gessani, S.; Fantuzzi, L. Nuclear Phosphoinositide-Specific Phospholipase Cβ1 Controls Cytoplasmic CCL2 mRNA Levels in HIV-1 gp120-Stimulated Primary Human Macrophages. PLoS ONE 2013, 8, e59705. [Google Scholar] [CrossRef] [PubMed]
- Sabbatucci, M.; Covino, D.A.; Purificato, C.; Mallano, A.; Federico, M.; Lu, J.; Rinaldi, A.O.; Pellegrini, M.; Bona, R.; Michelini, Z.; et al. Endogenous CCL2 Neutralization Restricts HIV-1 Replication in Primary Human Macrophages by Inhibiting Viral DNA Accumulation. Retrovirology 2015, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Arenzana-Seisdedos, F.; Fernandez, B.; Dominguez, I.; Jacque, J.M.; Thomas, D.; Diaz-Meco, M.T.; Moscat, J.; Virelizier, J.L. Phosphatidylcholine Hydrolysis Activates NF-κB and Increases Human Immunodeficiency Virus Replication in Human Monocytes and T Lymphocytes. J. Virol. 1993, 67, 6596–6604. [Google Scholar] [PubMed]
- Allers, K.; Fehr, M.; Conrad, K.; Epple, H.J.; Schurmann, D.; Geelhaar-Karsch, A.; Schinnerling, K.; Moos, V.; Schneider, T. Macrophages Accumulate in the Gut Mucosa of Untreated HIV-Infected Patients. J. Infect. Dis. 2014, 209, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Sawai, T.; Usui, N.; Dwaihy, J.; Drongowski, R.A.; Abe, A.; Coran, A.G.; Harmon, C.M. The Effect of Phospholipase A2 on Bacterial Translocation in a Cell Culture Model. Pediatr. Surg. Int. 2000, 16, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Clayton, F.; Kotler, D.P.; Kuwada, S.K.; Morgan, T.; Stepan, C.; Kuang, J.; Le, J.; Fantini, J. Gp120-Induced Bob/GPR15 Activation: A Possible Cause of Human Immunodeficiency Virus Enteropathy. Am. J. Pathol. 2001, 159, 1933–1939. [Google Scholar] [CrossRef]
- Merlini, E.; Tincati, C.; Biasin, M.; Saulle, I.; Cazzaniga, F.A.; d’Arminio Monforte, A.; Cappione, A.J., 3rd; Snyder-Cappione, J.; Clerici, M.; Marchetti, G.C. Stimulation of PBMC and Monocyte-Derived Macrophages Via Toll-Like Receptor Activates Innate Immune Pathways in HIV-Infected Patients on Virally Suppressive Combination Antiretroviral Therapy. Front. Immunol. 2016, 7, 614. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Mallampalli, R.K.; Carter, A.B.; Flaherty, D.M.; McCoy, D.; Robeff, P.K.; Peterson, M.W.; Hunninghake, G.W. Ceramide Regulates Lipopolysaccharide-Induced Phosphatidylinositol 3-Kinase and Akt Activity in Human Alveolar Macrophages. J. Immunol. 2001, 167, 5977–5985. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.B.; Monick, M.M.; Hunninghake, G.W. Lipopolysaccharide-Induced NF-kappaB Activation and Cytokine Release in Human Alveolar Macrophages is PKC-Independent and TK- and PC-PLC-Dependent. Am. J. Respir. Cell Mol. Biol. 1998, 18, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, C.; Ma, Y.; Ding, X.; Zhu, G.; Zhu, Q. Anti-Inflammatory Activities of Phospholipase C Inhibitor U73122: Inhibition of Monocyte-to-Macrophage Transformation and LPS-Induced Pro-Inflammatory Cytokine Expression. Int. Immunopharmacol. 2015, 29, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Kitsiouli, E.; Antoniou, G.; Gotzou, H.; Karagiannopoulos, M.; Basagiannis, D.; Christoforidis, S.; Nakos, G.; Lekka, M.E. Effect of Azithromycin on the LPS-Induced Production and Secretion of Phospholipase A2 in Lung Cells. Biochim. Biophys. Acta 2015, 1852, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid Induction of Inflammatory Lipid Mediators by the Inflammasome in Vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Svard, J.; Paquin-Proulx, D.; Buggert, M.; Noyan, K.; Barqasho, B.; Sonnerborg, A.; Nowak, P. Role of Translocated Bacterial Flagellin in Monocyte Activation among Individuals with Chronic HIV-1 Infection. Clin. Immunol. 2015, 161, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Glesby, M.J. Cardiovascular Complications of HIV Infection. Top. Antivir. Med. 2017, 24, 127–131. [Google Scholar]
- Shrestha, S.; Irvin, M.R.; Grunfeld, C.; Arnett, D.K. HIV, Inflammation, and Calcium in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hsue, P.Y.; Deeks, S.G.; Hunt, P.W. Immunologic Basis of Cardiovascular Disease in HIV-Infected Adults. J. Infect. Dis. 2012, 205, S375–S382. [Google Scholar] [CrossRef] [PubMed]
- Crowe, S.M.; Westhorpe, C.L.; Mukhamedova, N.; Jaworowski, A.; Sviridov, D.; Bukrinsky, M. The Macrophage: The Intersection between HIV Infection and Atherosclerosis. J. Leukoc. Biol. 2010, 87, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.T.; Mayne, E.; Sieg, S.F.; Asaad, R.; Jiang, W.; Kalinowska, M.; Luciano, A.A.; Stevens, W.; Rodriguez, B.; Brenchley, J.M.; et al. Increased Tissue Factor Expression on Circulating Monocytes in Chronic HIV Infection: Relationship to in Vivo Coagulation and Immune Activation. Blood 2010, 115, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Kendall, M.A.; Yang, O.O.; Hodis, H.N.; Currier, J.S. Biomarkers of Microbial Translocation and Macrophage Activation: Association with Progression of Subclinical Atherosclerosis in HIV-1 Infection. J. Infect. Dis. 2012, 206, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Elbim, C.; Pillet, S.; Prevost, M.H.; Preira, A.; Girard, P.M.; Rogine, N.; Matusani, H.; Hakim, J.; Israel, N.; Gougerot-Pocidalo, M.A. Redox and Activation Status of Monocytes from Human Immunodeficiency Virus-Infected Patients: Relationship with Viral Load. J. Virol. 1999, 73, 4561–4566. [Google Scholar] [PubMed]
- Kulkarni, M.; Bowman, E.; Gabriel, J.; Amburgy, T.; Mayne, E.; Zidar, D.A.; Maierhofer, C.; Turner, A.N.; Bazan, J.A.; Koletar, S.L.; et al. Altered Monocyte and Endothelial Cell Adhesion Molecule Expression is Linked to Vascular Inflammation in Human Immunodeficiency Virus Infection. Open Forum. Infect. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Harezlak, J.; Buchthal, S.; Taylor, M.; Schifitto, G.; Zhong, J.; Daar, E.; Alger, J.; Singer, E.; Campbell, T.; Yiannoutsos, C.; et al. Persistence of HIV-Associated Cognitive Impairment, Inflammation, and Neuronal Injury in Era of Highly Active Antiretroviral Treatment. AIDS 2011, 25, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Eugenin, E.A. Mechanisms of HIV Neuropathogenesis: Role of Cellular Communication Systems. Curr. HIV. Res. 2016, 14, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Bardi, G.; Sengupta, R.; Khan, M.Z.; Patel, J.P.; Meucci, O. Human Immunodeficiency Virus gp120-Induced Apoptosis of Human Neuroblastoma Cells in the Absence of CXCR4 Internalization. J. Neurovirol. 2006, 12, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Lannuzel, A.; Lledo, P.M.; Lamghitnia, H.O.; Vincent, J.D.; Tardieu, M. HIV-1 Envelope Proteins gp120 and gp160 Potentiate NMDA-Induced [Ca2+]i Increase, Alter [Ca2+]i Homeostasis and Induce Neurotoxicity in Human Embryonic Neurons. Eur. J. Neurosci. 1995, 7, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- King, J.E.; Eugenin, E.A.; Hazleton, J.E.; Morgello, S.; Berman, J.W. Mechanisms of HIV-Tat-Induced Phosphorylation of N-Methyl-d-Aspartate Receptor Subunit 2A in Human Primary Neurons: Implications for neuroAIDS Pathogenesis. Am. J. Pathol. 2010, 176, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Catani, M.V.; Corasaniti, M.T.; Navarra, M.; Nistico, G.; Finazzi-Agro, A.; Melino, G. gp120 Induces Cell Death in Human Neuroblastoma Cells through the CXCR4 and CCR5 Chemokine Receptors. J. Neurochem. 2000, 74, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Holden, C.P.; Nath, A.; Geiger, J.D. Involvement of Inositol 1,4,5-Trisphosphate-Regulated Stores of Intracellular Calcium in Calcium Dysregulation and Neuron Cell Death Caused by HIV-1 Protein Tat. J. Neurochem. 1999, 73, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Toborek, M.; Lee, Y.W.; Flora, G.; Pu, H.; Andras, I.E.; Wylegala, E.; Hennig, B.; Nath, A. Mechanisms of the Blood-Brain Barrier Disruption in HIV-1 Infection. Cell. Mol. Neurobiol. 2005, 25, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Pu, H.; Hayashi, K.; Andras, I.E.; Eum, S.Y.; Hennig, B.; Toborek, M. Limited Role of COX-2 in HIV Tat-Induced Alterations of Tight Junction Protein Expression and Disruption of the Blood-Brain Barrier. Brain Res. 2007, 1184, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Nakamuta, S.; Endo, H.; Higashi, Y.; Kousaka, A.; Yamada, H.; Yano, M.; Kido, H. Human Immunodeficiency Virus Type 1 gp120-Mediated Disruption of Tight Junction Proteins by Induction of Proteasome-Mediated Degradation of Zonula Occludens-1 and -2 in Human Brain Microvascular Endothelial Cells. J. Neurovirol. 2008, 14, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Shiu, C.; Barbier, E.; Di Cello, F.; Choi, H.J.; Stins, M. HIV-1 gp120 as Well as Alcohol Affect Blood-Brain Barrier Permeability and Stress Fiber Formation: Involvement of Reactive Oxygen Species. Alcohol. Clin. Exp. Res. 2007, 31, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Kanmogne, G.D.; Schall, K.; Leibhart, J.; Knipe, B.; Gendelman, H.E.; Persidsky, Y. HIV-1 gp120 Compromises Blood-Brain Barrier Integrity and Enhances Monocyte Migration Across Blood-Brain Barrier: Implication for Viral Neuropathogenesis. J. Cereb. Blood Flow Metab. 2007, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Calderon, T.M.; Lopez, L.; Carvallo-Torres, L.; Gaskill, P.J.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Mechanisms of HIV Entry into the CNS: Increased Sensitivity of HIV Infected CD14+CD16+ Monocytes to CCL2 and Key Roles of CCR2, JAM-A, and ALCAM in Diapedesis. PLoS ONE 2013, 8, e69270. [Google Scholar] [CrossRef] [PubMed]
- Ancuta, P.; Kamat, A.; Kunstman, K.J.; Kim, E.Y.; Autissier, P.; Wurcel, A.; Zaman, T.; Stone, D.; Mefford, M.; Morgello, S.; et al. Microbial Translocation is Associated with Increased Monocyte Activation and Dementia in AIDS Patients. PLoS ONE 2008, 3, e2516. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.; Lyons, J.L.; Misra, V.; Uno, H.; Morgello, S.; Singer, E.J.; Gabuzda, D. Monocyte Activation Markers in Cerebrospinal Fluid Associated with Impaired Neurocognitive Testing in Advanced HIV Infection. J. Acquir. Immune Defic. Syndr. 2012, 60, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, L. Cognitive Consequences of a Sustained Monocyte Type 1 IFN Response in HIV-1 Infection. Curr. HIV. Res. 2014, 12, 77–84. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Sharer, L.R.; Chao, W.; Gu, C.J.; Borjabad, A.; Hadas, E.; Kelschenbach, J.; Ichiyama, K.; Do, M.; Potash, M.J.; et al. Enhanced Human Immunodeficiency Virus Type 1 Expression and Neuropathogenesis in Knockout Mice Lacking Type I Interferon Responses. J. Neuropathol. Exp. Neurol. 2014, 73, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Lotzerich, M.; Schaefer, A.; Werner, T.; Frankenberger, M.; Benkhart, E. IFN-α Induces the Human IL-10 Gene by Recruiting both IFN Regulatory Factor 1 and Stat3. J. Immunol. 2003, 171, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kim, Y.J.; Broxmeyer, H.E. Macrophage Colony-Stimulating Factor Drives Cord Blood Monocyte Differentiation into IL-10HighIL-12absent Dendritic Cells with Tolerogenic Potential. J. Immunol. 2005, 174, 4706–4717. [Google Scholar] [CrossRef] [PubMed]
- Zaritsky, L.A.; Gama, L.; Clements, J.E. Canonical Type I IFN Signaling in Simian Immunodeficiency Virus-Infected Macrophages is Disrupted by Astrocyte-Secreted CCL2. J. Immunol. 2012, 188, 3876–3885. [Google Scholar] [CrossRef] [PubMed]
- Lake, J.E.; Currier, J.S. Metabolic Disease in HIV Infection. Lancet Infect. Dis. 2013, 13, 964–975. [Google Scholar] [CrossRef]
- Glass, J.D.; Fedor, H.; Wesselingh, S.L.; McArthur, J.C. Immunocytochemical Quantitation of Human Immunodeficiency Virus in the Brain: Correlations with Dementia. Ann. Neurol. 1995, 38, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, E.; Morrison, D.; Sullivan, P.; Morgello, S.; Fischer, T. Brain Inflammation is a Common Feature of HIV-Infected Patients without HIV Encephalitis or Productive Brain Infection. Curr. HIV. Res. 2014, 12, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Carroll-Anzinger, D.; Kumar, A.; Adarichev, V.; Kashanchi, F.; Al-Harthi, L. Human Immunodeficiency Virus-Restricted Replication in Astrocytes and the Ability of γ Interferon to Modulate this Restriction are Regulated by a Downstream Effector of the Wnt Signaling Pathway. J. Virol. 2007, 81, 5864–5871. [Google Scholar] [CrossRef] [PubMed]
- Ton, H.; Xiong, H. Astrocyte Dysfunctions and HIV-1 Neurotoxicity. J. AIDS. Clin. Res. 2013, 4, 255. [Google Scholar] [PubMed]
- Moore, D.J.; Masliah, E.; Rippeth, J.D.; Gonzalez, R.; Carey, C.L.; Cherner, M.; Ellis, R.J.; Achim, C.L.; Marcotte, T.D.; Heaton, R.K.; et al. Cortical and Subcortical Neurodegeneration is Associated with HIV Neurocognitive Impairment. AIDS 2006, 20, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.M.; Chang, W.L.; Wang, S.M.; Su, M.J.; Wu, M.L. Arachidonic Acid Induces both Na+ and Ca2+ Entry Resulting in Apoptosis. J. Neurochem. 2008, 104, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Kempster, S.; Williams, A. Platelet-Activating Factor Antagonists Protect Amyloid-β Damaged Neurons from Microglia-Mediated Death. Neuropharmacology 2006, 51, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Kempster, S.; Williams, A. Prostaglandin D2 Mediates Neuronal Damage by Amyloid-β Or Prions which Activates Microglial Cells. Neuropharmacology 2006, 50, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, M.U.; Mokhtarzadeh, M.; Stichtenoth, D.O.; Skardelly, M.; Klinge, P.M.; Gutzki, F.M.; Samii, M.; Brinker, T. Temporal Profiles of Cerebrospinal Fluid Leukotrienes, Brain Edema and Inflammatory Response Following Experimental Brain Injury. Neurol. Res. 2003, 25, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Guillemin, G.J.; Pemberton, L.; Kerr, S.; Nath, A.; Smythe, G.A.; Brew, B.J. Quinolinic Acid is Produced by Macrophages Stimulated by Platelet Activating Factor, Nef and Tat. J. Neurovirol. 2001, 7, 56–60. [Google Scholar] [PubMed]
- Perry, S.W.; Hamilton, J.A.; Tjoelker, L.W.; Dbaibo, G.; Dzenko, K.A.; Epstein, L.G.; Hannun, Y.; Whittaker, J.S.; Dewhurst, S.; Gelbard, H.A. Platelet-Activating Factor Receptor Activation. An Initiator Step in HIV-1 Neuropathogenesis. J. Biol. Chem. 1998, 273, 17660–17664. [Google Scholar] [CrossRef] [PubMed]
- Del Sorbo, L.; DeMartino, A.; Biancone, L.; Bussolati, B.; Conaldi, P.G.; Toniolo, A.; Camussi, G. The Synthesis of Platelet-Activating Factor Modulates Chemotaxis of Monocytes Induced by HIV-1 Tat. Eur. J. Immunol. 1999, 29, 1513–1521. [Google Scholar] [CrossRef]
- Ryan, S.D.; Harris, C.S.; Carswell, C.L.; Baenziger, J.E.; Bennett, S.A. Heterogeneity in the Sn-1 Carbon Chain of Platelet-Activating Factor Glycerophospholipids Determines Pro- Or Anti-Apoptotic Signaling in Primary Neurons. J. Lipid Res. 2008, 49, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Ishii, S.; Kita, Y.; Toda, A.; Shimada, A.; Shimizu, T. Dual Phase Regulation of Experimental Allergic Encephalomyelitis by Platelet-Activating Factor. J. Exp. Med. 2005, 202, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Tomkowicz, B.; Freedman, B.D.; Collman, R.G. HIV-1 gp120-Induced TNF-α Production by Primary Human Macrophages is Mediated by Phosphatidylinositol-3 (PI-3) Kinase and Mitogen-Activated Protein (MAP) Kinase Pathways. J. Leukoc. Biol. 2005, 78, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, A.A.; McLeod, E.; Wahle, K.W.; Arthur, J.R. Cytokine-Induced Monocyte Adhesion to Endothelial Cells Involves Platelet-Activating Factor: Suppression by Conjugated Linoleic Acid. Biochim. Biophys. Acta 2006, 1761, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Khan, K.A. Is HIV Infection a TNF Receptor Signalling-Driven Disease? Trends Immunol. 2008, 29, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.G.; Moreira, L.; Paes-Leme, J.; Barreto-de-Souza, V.; Castro-Faria-Neto, H.C.; Bozza, P.T.; Bou-Habib, D.C. Interaction of Macrophages with Apoptotic Cells Enhances HIV Type 1 Replication through PGE2, PAF, and Vitronectin Receptor. AIDS Res. Hum. Retrovir. 2006, 22, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Limoges, J.; Rasmussen, J.; Zheng, J.; Gearing, A.; Gendelman, H.E. Reduction in Glial Immunity and Neuropathology by a PAF Antagonist and an MMP and TNFα Inhibitor in SCID Mice with HIV-1 Encephalitis. J. Neuroimmunol. 2001, 114, 57–68. [Google Scholar] [CrossRef]
- Eggert, D.; Dash, P.K.; Serradji, N.; Dong, C.Z.; Clayette, P.; Heymans, F.; Dou, H.; Gorantla, S.; Gelbard, H.A.; Poluektova, L.; et al. Development of a Platelet-Activating Factor Antagonist for HIV-1 Associated Neurocognitive Disorders. J. Neuroimmunol. 2009, 213, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E.; Wesselingh, S.L.; McArthur, J.C. Elevated Central Nervous System Prostaglandins in Human Immunodeficiency Virus-Associated Dementia. Ann. Neurol. 1994, 35, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Pendyala, G.; Siuzdak, G.; Fox, H.S. Metabolomic Analysis of the Cerebrospinal Fluid Reveals Changes in Phospholipase Expression in the CNS of SIV-Infected Macaques. J. Clin. Investig. 2008, 118, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The Orphan G Protein-Coupled Receptor GPR40 is Activated by Medium and Long Chain Fatty Acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.; Dreyer, C.; Medin, J.; Mahfoudi, A.; Ozato, K.; Wahli, W. Fatty Acids and Retinoids Control Lipid Metabolism through Activation of Peroxisome Proliferator-Activated Receptor-Retinoid X Receptor Heterodimers. Proc. Natl. Acad. Sci. USA 1993, 90, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Heringdorf, D.; Jakobs, K.H. Lysophospholipid Receptors: Signalling, Pharmacology and Regulation by Lysophospholipid Metabolism. Biochim. Biophys. Acta 2007, 1768, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, M.J.; Lu, S.M.; Masliah, E.; Gelbard, H.A. Synaptic Activity Becomes Excitotoxic in Neurons Exposed to Elevated Levels of Platelet-Activating Factor. J. Clin. Investig. 2005, 115, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Schilling, T.; Schwab, A.; Eder, C. Lysophosphatidylcholine Stimulates IL-1β Release from Microglia via a P2X7 Receptor-Independent Mechanism. J. Immunol. 2006, 177, 8560–8568. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Kim, H.W.; Kellom, M.; Greenstein, D.; Chen, M.; Kraft, A.D.; Harry, G.J.; Rapoport, S.I.; Basselin, M. Increased Neuroinflammatory and Arachidonic Acid Cascade Markers, and Reduced Synaptic Proteins, in Brain of HIV-1 Transgenic Rats. J. Neuroinflamm. 2011, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Ghorpade, A. Cytosolic Phospholipase A2 Regulates Alcohol-Mediated Astrocyte Inflammatory Responses in HIV-Associated Neurocognitive Disorders. Cell Death Discov. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Samikkannu, T.; Rao, K.V.; Ding, H.; Agudelo, M.; Raymond, A.D.; Yoo, C.; Nair, M.P. Immunopathogenesis of HIV Infection in Cocaine Users: Role of Arachidonic Acid. PLoS ONE 2014, 9, e106348. [Google Scholar] [CrossRef] [PubMed]
- Persichini, T.; Mastrantonio, R.; Del Matto, S.; Palomba, L.; Cantoni, O.; Colasanti, M. The Role of Arachidonic Acid in the Regulation of Nitric Oxide Synthase Isoforms by HIV gp120 Protein in Astroglial Cells. Free Radic. Biol. Med. 2014, 74, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H.; Lackner, A.; Williams, K.C. Monocyte/Macrophages and their Role in HIV Neuropathogenesis. Immunol. Rev. 2013, 254, 102–113. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spadaro, F.; Cecchetti, S.; Fantuzzi, L. Macrophages and Phospholipases at the Intersection between Inflammation and the Pathogenesis of HIV-1 Infection. Int. J. Mol. Sci. 2017, 18, 1390. https://doi.org/10.3390/ijms18071390
Spadaro F, Cecchetti S, Fantuzzi L. Macrophages and Phospholipases at the Intersection between Inflammation and the Pathogenesis of HIV-1 Infection. International Journal of Molecular Sciences. 2017; 18(7):1390. https://doi.org/10.3390/ijms18071390
Chicago/Turabian StyleSpadaro, Francesca, Serena Cecchetti, and Laura Fantuzzi. 2017. "Macrophages and Phospholipases at the Intersection between Inflammation and the Pathogenesis of HIV-1 Infection" International Journal of Molecular Sciences 18, no. 7: 1390. https://doi.org/10.3390/ijms18071390
APA StyleSpadaro, F., Cecchetti, S., & Fantuzzi, L. (2017). Macrophages and Phospholipases at the Intersection between Inflammation and the Pathogenesis of HIV-1 Infection. International Journal of Molecular Sciences, 18(7), 1390. https://doi.org/10.3390/ijms18071390